
Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics
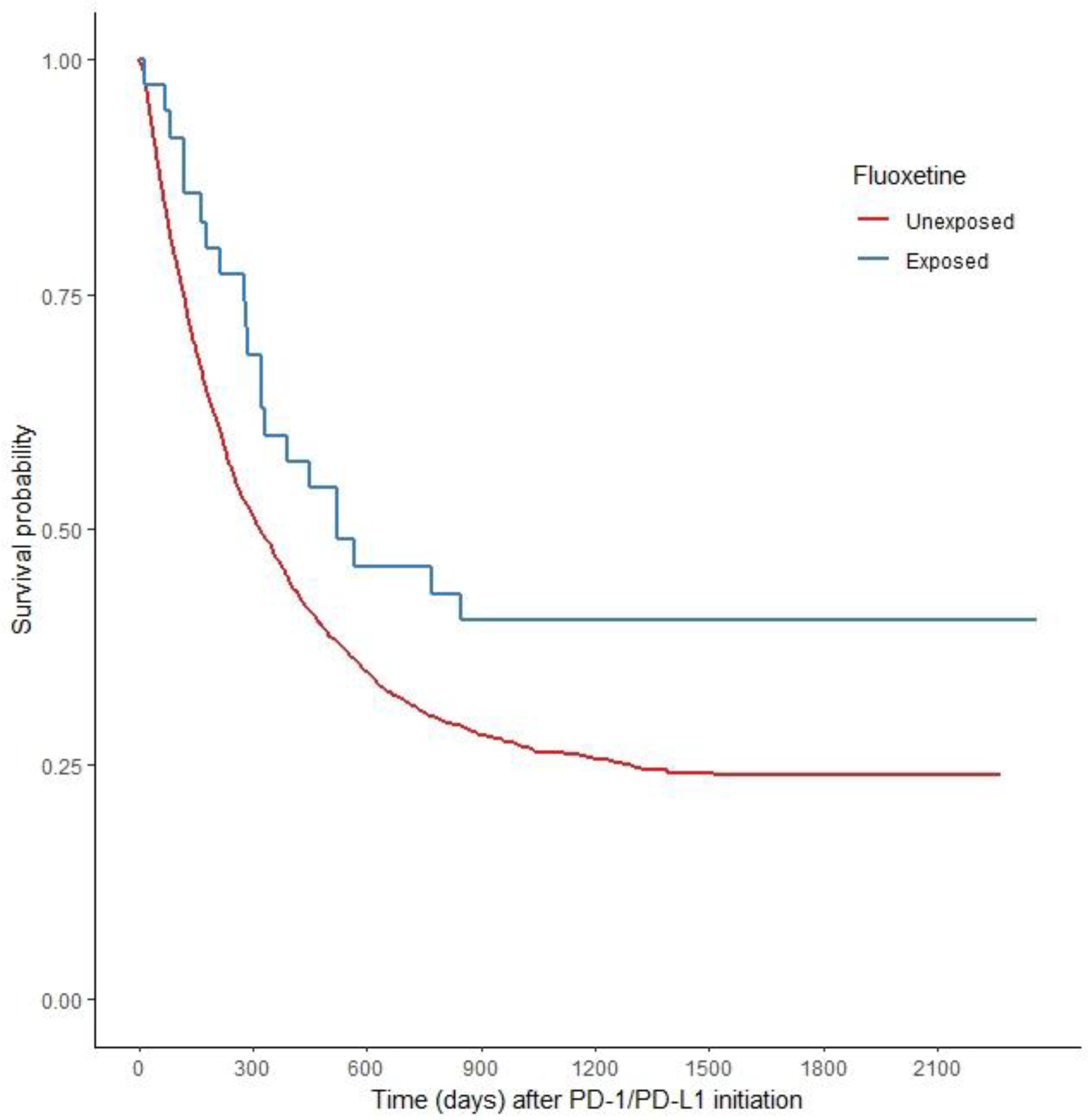
2.2. Fluoxetine and PD-1/PD-L1 Therapy Improves Overall Survival (OS) versus PD-1/PD-L1 Alone
2.3. Other Antidepressants Do Not Increase OS
2.4. Fluoxetine without PD-1/PD-L1 Therapy Reveals No Benefit on OS
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. Cohort Creation
4.3. Exposure Definition
4.4. Covariate Data
4.5. Outcomes
4.6. Statistical Analysis
4.6.1. Other Antidepressants and OS
4.6.2. Secondary Analysis: Fluoxetine without PD-1/PD-L1 Therapy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I Study of Single-Agent Anti–Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Drake, C.G.; Sznol, M.; Choueiri, T.K.; Powderly, J.D.; Smith, D.; Brahmer, J.R.; Carvajal, R.D.; Hammers, H.J.; Puzanov, I.; et al. Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. J. Clin. Oncol. 2015, 33, 2013–2020. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Singh, S.; Xiao, Z.; Bavisi, K.; Roszik, J.; Melendez, B.D.; Wang, Z.; Cantwell, M.J.; Davis, R.E.; Lizee, G.; Hwu, P.; et al. IL-1α Mediates Innate and Acquired Resistance to Immunotherapy in Melanoma. J. Immunol. 2021, 206, 1966–1975. [Google Scholar] [CrossRef]
- Mehta, A.; Kim, Y.J.; Robert, L.; Tsoi, J.; Comin-Anduix, B.; Berent-Maoz, B.; Cochran, A.J.; Economou, J.S.; Tumeh, P.C.; Puig-Saus, C.; et al. Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov. 2018, 8, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Brighi, N.; Cursano, M.C.; Gurioli, G.; Ravaglia, G.; Altavilla, A.; Burgio, S.L.; Testoni, S.; Menna, C.; Farolfi, A.; et al. Inflammatory Biomarkers as Predictors of Response to Immunotherapy in Urological Tumors. J. Oncol. 2019, 2019, 7317964. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology 2019, 159, 357–364. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti–PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef]
- Lu, F.; Zhao, Y.; Pang, Y.; Ji, M.; Sun, Y.; Wang, H.; Zou, J.; Wang, Y.; Li, G.; Sun, T.; et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 2020, 497, 178–189. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Klück, V.; Jansen, T.L.T.A.; Janssen, M.; Comarniceanu, A.; Efdé, M.; Tengesdal, I.W.; Schraa, K.; Cleophas, M.C.P.; Scribner, C.L.; Skouras, D.B.; et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020, 2, e270–e280. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; Brough, D.; Freeman, S. Inhibiting the NLRP3 Inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef]
- Chen, Q.-L.; Yin, H.-R.; He, Q.-Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef]
- Ambati, J.; Magagnoli, J.; Leung, H.; Wang, S.-B.; Andrews, C.A.; Fu, D.; Pandey, A.; Sahu, S.; Narendran, S.; Hirahara, S.; et al. Repurposing anti-inflammasome NRTIs for improving insulin sensitivity and reducing type 2 diabetes development. Nat. Commun. 2020, 11, 4737. [Google Scholar] [CrossRef]
- Ambati, M.; Apicella, I.; Wang, S.-B.; Narendran, S.; Leung, H.; Pereira, F.; Nagasaka, Y.; Huang, P.; Varshney, A.; Baker, K.L.; et al. Identification of fluoxetine as a direct NLRP3 inhibitor to treat atrophic macular degeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2102975118. [Google Scholar] [CrossRef]
- Du, R.-H.; Tan, J.; Sun, X.-Y.; Lu, M.; Ding, J.-H.; Hu, G. Fluoxetine Inhibits NLRP3 Inflammasome Activation: Implication in Depression. Int. J. Neuropsychopharmacol. 2016, 19, pyw037. [Google Scholar] [CrossRef]
- Li, J.-R.; Xu, H.-Z.; Nie, S.; Peng, Y.-C.; Fan, L.-F.; Wang, Z.-J.; Wu, C.; Yan, F.; Chen, J.-Y.; Gu, C.; et al. Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subarachnoid hemorrhage in rats. J. Neuroinflamm. 2017, 14, 186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Duan, J.; Wang, Y.; Chen, X.; Zhou, G.; Wang, R.; Fu, L.; Xu, F. Fluoxetine synergys with anticancer drugs to overcome multidrug resistance in breast cancer cells. Tumor Biol. 2012, 33, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-H.; Yang, S.-T.; Lin, Y.-K.; Lin, J.-W.; Lee, Y.-H.; Wang, J.-Y.; Hu, C.-J.; Lin, E.-Y.; Chen, S.-M.; Then, C.-K.; et al. Fluoxetine, an antidepressant, suppresses glioblastoma by evoking AMPAR-mediated calcium-dependent apoptosis. Oncotarget 2014, 6, 5088–5101. [Google Scholar] [CrossRef] [PubMed]
- Di Rosso, M.E.; Sterle, H.; Cremaschi, G.A.; Genaro, A.M. Beneficial Effect of Fluoxetine and Sertraline on Chronic Stress-Induced Tumor Growth and Cell Dissemination in a Mouse Model of Lymphoma: Crucial Role of Antitumor Immunity. Front. Immunol. 2018, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-C.; Tu, H.-F.; Hsu, F.-T.; Yueh, P.-F.; Chiang, I.-T. Beneficial effect of fluoxetine on anti-tumor progression on hepatocellular carcinoma and non-small cell lung cancer bearing animal model. Biomed. Pharmacother. 2020, 126, 110054. [Google Scholar] [CrossRef]
- Marigo, I.; Dolcetti, L.; Serafini, P.; Zanovello, P.; Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 2008, 222, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR− Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef]
- Weber, J.S.; Gibney, G.; Kudchadkar, R.R.; Yu, B.; Cheng, P.; Martinez, A.J.; Kroeger, J.; Richards, A.; McCormick, L.; Moberg, V.; et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol. Res. 2016, 4, 345–353. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- US Department of Veterans Affairs: Ascertaining Veterans’ Vital Status: Data Sources for Mortality Ascertainment and Cause of Death. Available online: https://www.hsrd.research.va.gov/for_researchers/cyber_seminars/archives/video_archive.cfm?SessionID=1242 (accessed on 18 October 2022).
- Setodji, C.M.; McCaffrey, D.F.; Burgette, L.F.; Almirall, D.; Griffin, B.A. The Right Tool for the Job: Choosing Between Covariate-balancing and Generalized Boosted Model Propensity Scores. Epidemiology 2017, 28, 802–811. [Google Scholar] [CrossRef]
- McCaffrey, D.F.; Griffin, B.A.; Almirall, D.; Slaughter, M.E.; Ramchand, R.; Burgette, L.F. A tutorial on propensity score estimation for multiple treatments using generalized boosted models. Stat. Med. 2013, 32, 3388–3414. [Google Scholar] [CrossRef]
- Ridgeway, G.; McCaffrey, D.; Morral, A.; Griffin, B.A.; Burgette, L.; Cefalu, M. Twang: Toolkit for Weighting and Analysis of Nonequivalent Groups. R Package Version 1.6. 2020. Available online: https://CRAN.R-project.org/package=twang (accessed on 28 August 2022).
- Uno, H.; Tian, L.; Horiguchi, M.; Cronin, A.; Battioui, C.; Bell, J. survRM2: Comparing Restricted Mean Survival Time Version 1.0-3. 2020. Available online: https://CRAN.R-project.org/package=survRM2 (accessed on 29 August 2022).
- Ho, D.; Imai, K.; King, G.; Stuart, E.A. MatchIt: Nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 2011, 42, 1–28. Available online: https://www.jstatsoft.org/v42/i08/ (accessed on 30 August 2022). [CrossRef]


{kind=link}
| Variable | All Follow Up HR (95% CI) | 1 yr HR (95% CI) | 2 yr HR (95% CI) |
|---|---|---|---|
| FLX +PD-1/L1 vs. PD-1/L1 alone | 0.59 (0.371–0.936) | 0.606 (0.362–1.015) | 0.6 (0.376–0.958) |
| Age | 1.002 (0.997–1.008) | 1.001 (0.995–1.008) | 1.002 (0.997–1.008) |
| Race: Other/unknown vs. Black | 1.569 (1.188–2.072) | 1.781 (1.331–2.382) | 1.587 (1.195–2.108) |
| White vs. Black | 1.282 (1.128–1.456) | 1.33 (1.137–1.555) | 1.258 (1.103–1.435) |
| Sex: Male vs. Female | 0.958 (0.703–1.306) | 1.056 (0.711–1.571) | 0.977 (0.703–1.357) |
| BMI: 18.5–24.9 vs. <18.5 | 0.779 (0.648–0.936) | 0.85 (0.678–1.066) | 0.809 (0.666–0.983) |
| 25–29.9 vs. <18.5 | 0.655 (0.543–0.79) | 0.691 (0.547–0.874) | 0.678 (0.554–0.829) |
| 30+ vs. <18.5 | 0.669 (0.55–0.814) | 0.687 (0.538–0.878) | 0.684 (0.554–0.844) |
| Missing vs. <18.5 | 1.498 (0.498–4.504) | 2.377 (0.991–5.704) | 1.724 (0.61–4.868) |
| Depression | 0.978 (0.869–1.101) | 0.988 (0.86–1.136) | 0.989 (0.876–1.117) |
| Charlson comorbidity index | 1.051 (1.036–1.067) | 1.06 (1.042–1.08) | 1.056 (1.04–1.072) |
| seer summary: localized vs. distant metastasis | 0.978 (0.841–1.137) | 0.841 (0.701–1.009) | 0.943 (0.804–1.106) |
| Regional vs. distant metastasis | 0.938 (0.837–1.052) | 0.863 (0.753–0.989) | 0.894 (0.793–1.007) |
| ECOG performance at diagnosis ECOG 1vs. 0 | 1.06 (0.953–1.179) | 1.088 (0.957–1.238) | 1.076 (0.963–1.203) |
| ECOG 2 vs. 0 | 1.277 (1.084–1.505) | 1.379 (1.14–1.668) | 1.281 (1.077–1.523) |
| ECOG 3 vs. 0 | 1.544 (1.134–2.102) | 1.748 (1.278–2.39) | 1.54 (1.118–2.12) |
| ECOG 4 vs. 0 | 1.971 (0.784–4.956) | 2.081 (0.805–5.383) | 2.194 (0.951–5.062) |
| Liver vs. Kidney/other urinary | 1.627 (1.212–2.185) | 1.696 (1.205–2.388) | 1.704 (1.269–2.287) |
| Lung vs. Kidney/other urinary | 1.631 (1.356–1.961) | 1.699 (1.341–2.151) | 1.668 (1.37–2.031) |
| Skin vs. Kidney/other urinary | 0.699 (0.538–0.909) | 0.861 (0.622–1.191) | 0.705 (0.531–0.937) |
| Throat vs. Kidney/other urinary | 1.487 (1.151–1.919) | 1.471 (1.067–2.026) | 1.529 (1.172–1.994) |
| Year of PD-1/PD-L1 start | 0.85 (0.811–0.892) | 0.924 (0.874–0.977) | 0.877 (0.834–0.922) |
| AVELUMAB | 0.8 (0.156–4.116) | 0.975 (0.209–4.545) | 0.882 (0.174–4.464) |
| DURVALUMAB | 0.241 (0.151–0.386) | 0.169 (0.092–0.309) | 0.252 (0.157–0.406) |
| NIVOLUMAB | 0.811 (0.591–1.113) | 0.739 (0.536–1.019) | 0.813 (0.59–1.12) |
| PEMBROLIZUMAB | 0.66 (0.476–0.914) | 0.588 (0.422–0.819) | 0.66 (0.475–0.918) |
| Total prior TX 2 vs. 1 | 1.153 (1.028–1.295) | 1.122 (0.979–1.286) | 1.154 (1.023–1.302) |
| 3 vs. 1 | 1.091 (0.812–1.465) | 1.12 (0.792–1.585) | 1.122 (0.824–1.526) |
| 0 vs. 1 | 0.928 (0.763–1.129) | 0.952 (0.755–1.201) | 0.938 (0.764–1.15) |
| Exposure | Restricted Mean Survival Times (95% CI) | Mean Survival Difference (95% CI) | p-Value | |
|---|---|---|---|---|
| All follow-up (1484-day truncation time) | FLX+PD-1/L1 | 794.6 (593.5–995.8) | 287 (48.1–525.8) | 0.019 |
| PD-1/L1 | 507.7 (378.8–636.5) | |||
| 1 year (365-day truncation time) | FLX+PD-1/L1 | 296.7 (261.5–332) | 62.6 (16.6–108.6) | 0.008 |
| PD-1/L1 | 234.1 (204.6–263.7) | |||
| 2 year (730-day truncation time) | FLX+PD-1/L1 | 482.2 (395.4–569) | 137.1 (29.7–244.4) | 0.012 |
| PD-1/L1 | 345.2 (282.1–408.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.H.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy. Pharmaceuticals 2023, 16, 640. https://doi.org/10.3390/ph16050640
Magagnoli J, Narendran S, Pereira F, Cummings TH, Hardin JW, Sutton SS, Ambati J. Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy. Pharmaceuticals. 2023; 16(5):640. https://doi.org/10.3390/ph16050640
Chicago/Turabian StyleMagagnoli, Joseph, Siddharth Narendran, Felipe Pereira, Tammy H. Cummings, James W. Hardin, S. Scott Sutton, and Jayakrishna Ambati. 2023. "Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy" Pharmaceuticals 16, no. 5: 640. https://doi.org/10.3390/ph16050640
APA StyleMagagnoli, J., Narendran, S., Pereira, F., Cummings, T. H., Hardin, J. W., Sutton, S. S., & Ambati, J. (2023). Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy. Pharmaceuticals, 16(5), 640. https://doi.org/10.3390/ph16050640