
Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD

 , , and
, , and
Abstract
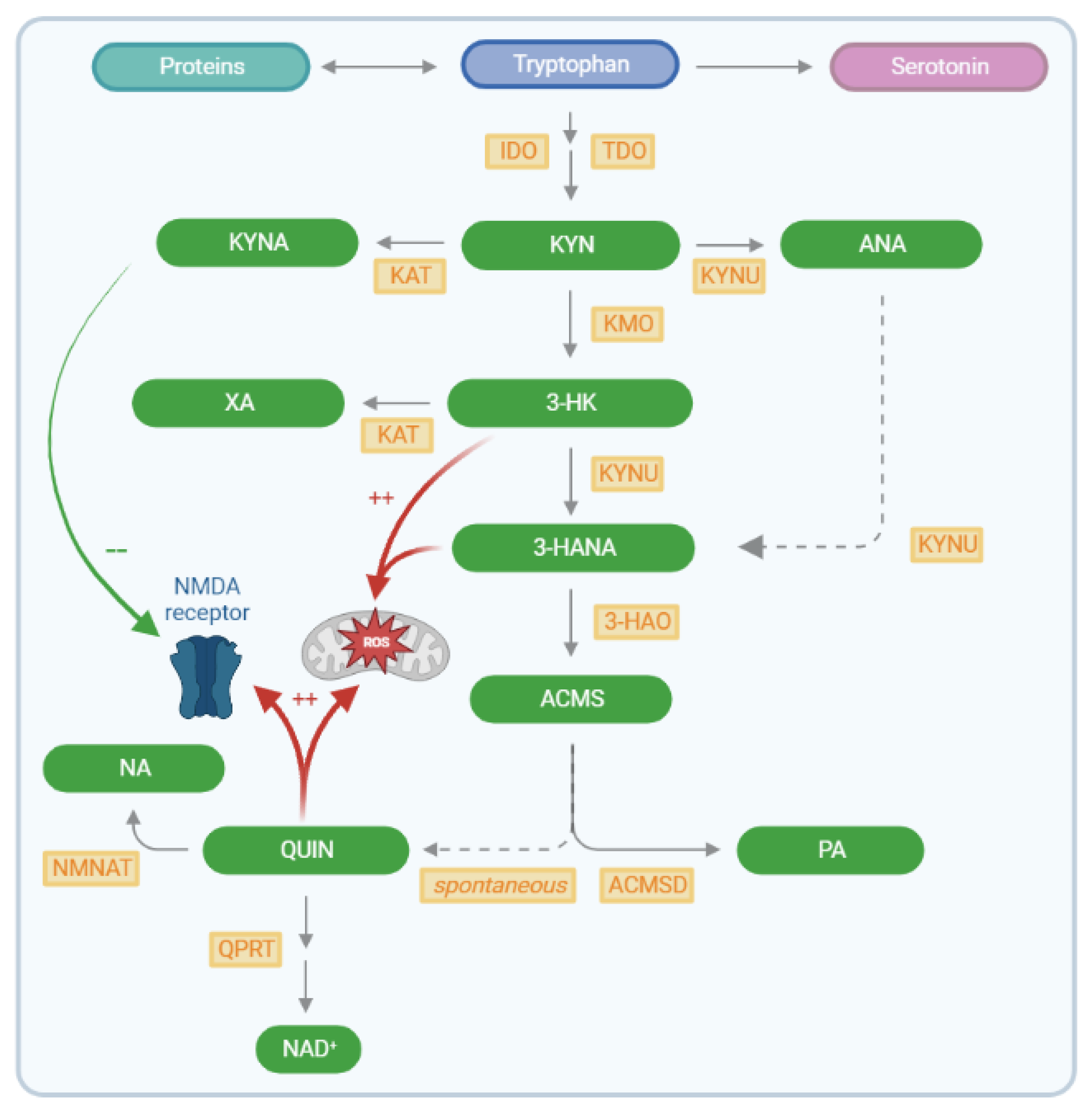
1. Introduction

2. Results
2.1. Demographic Information
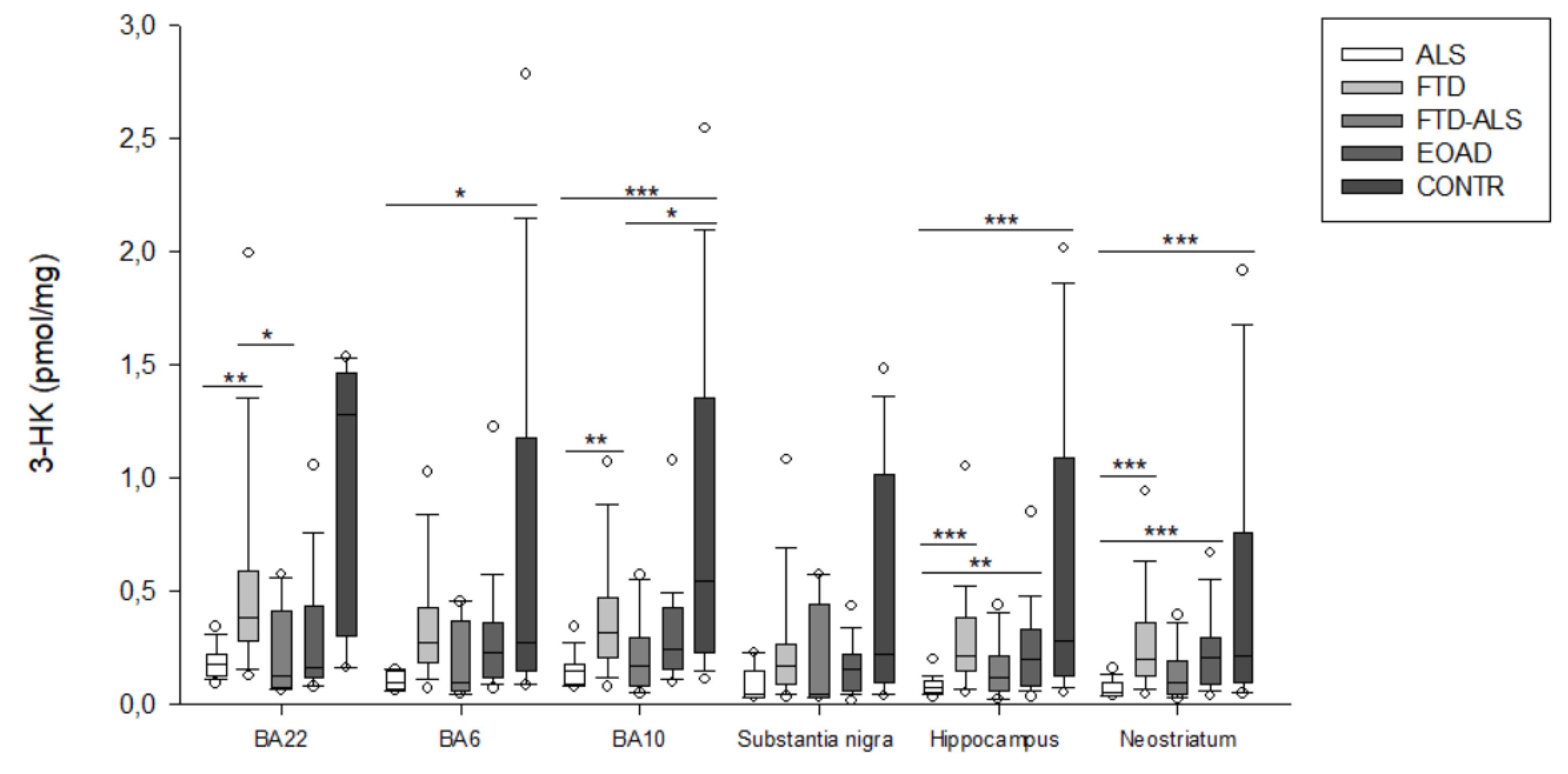
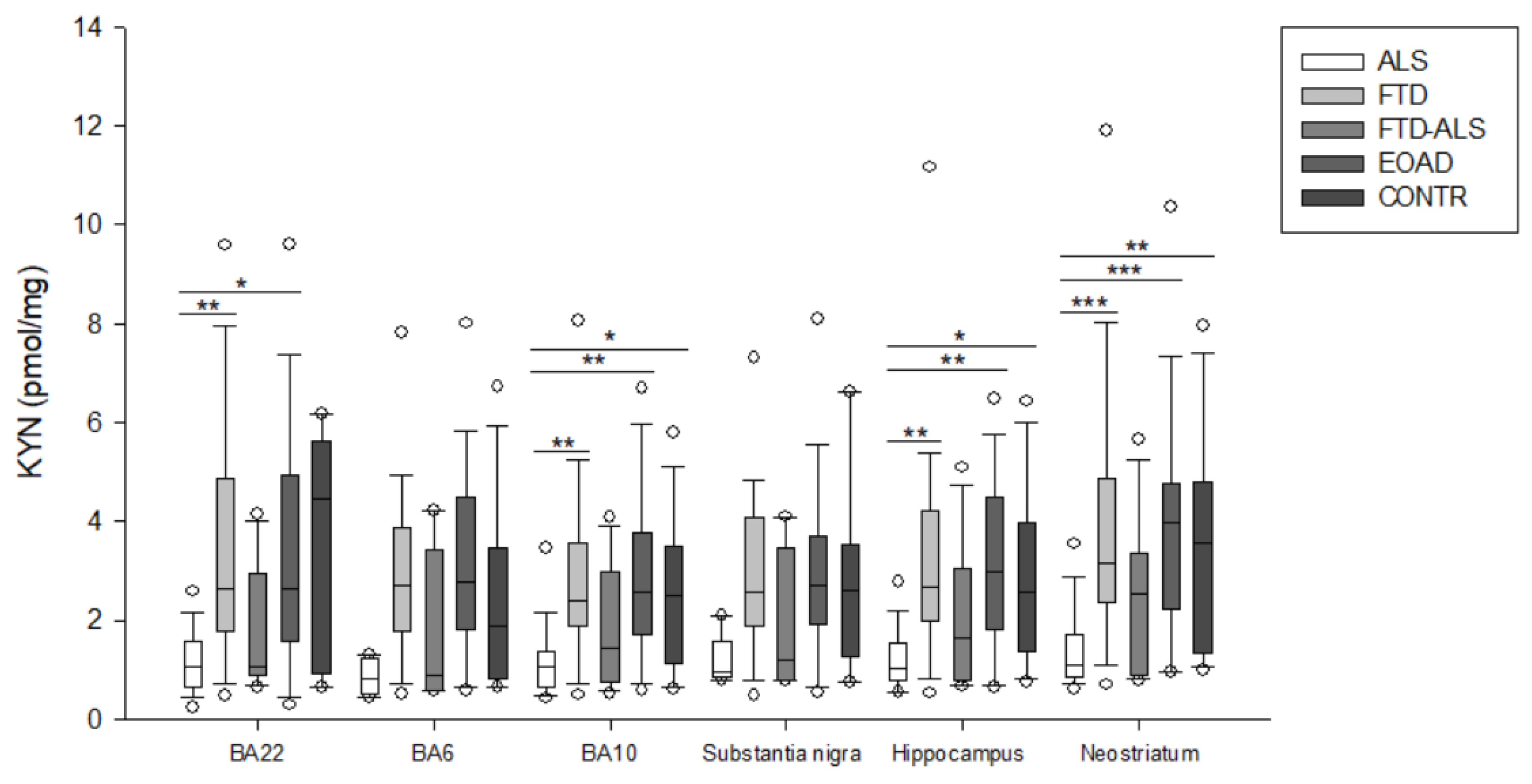
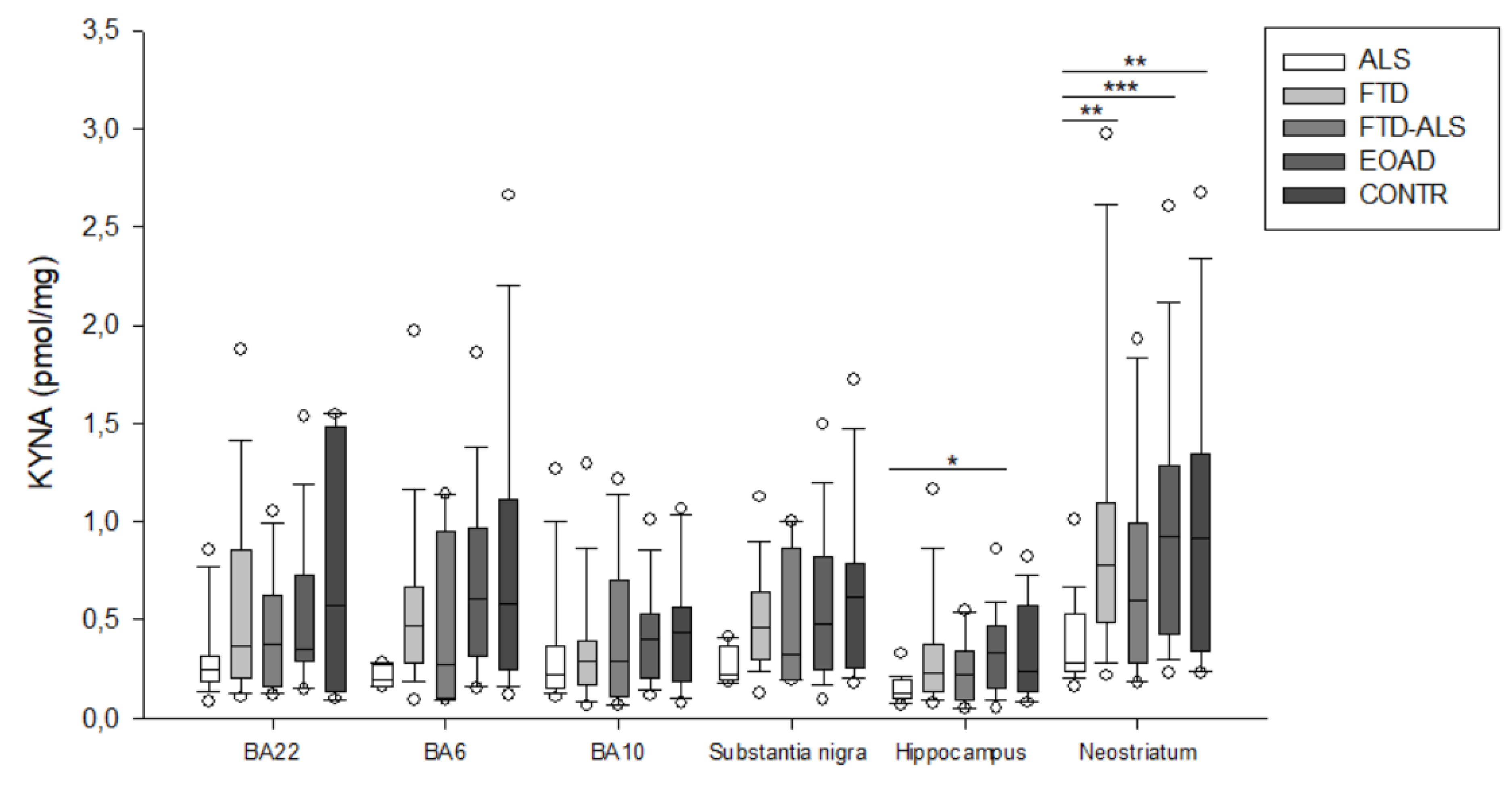
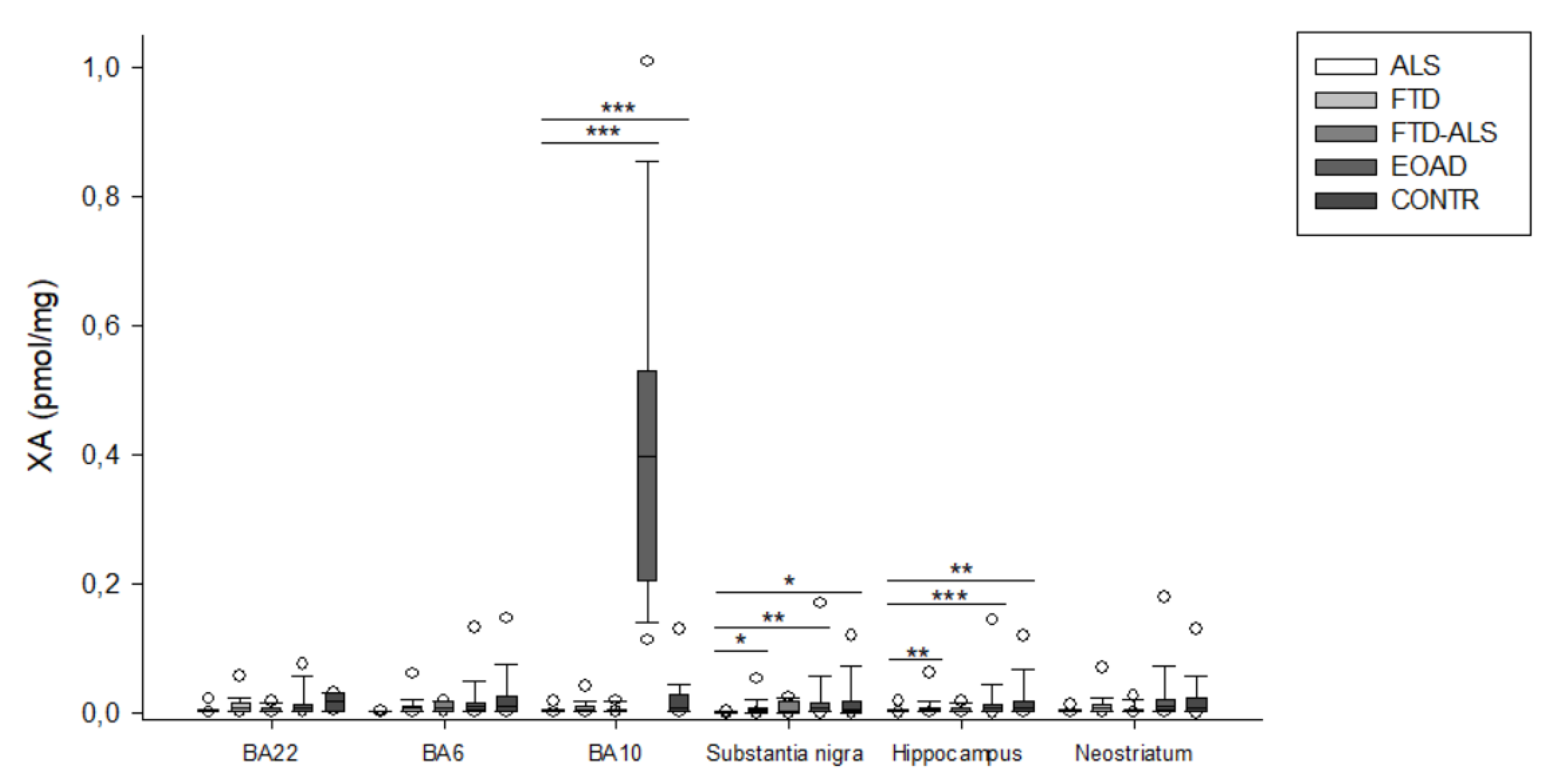
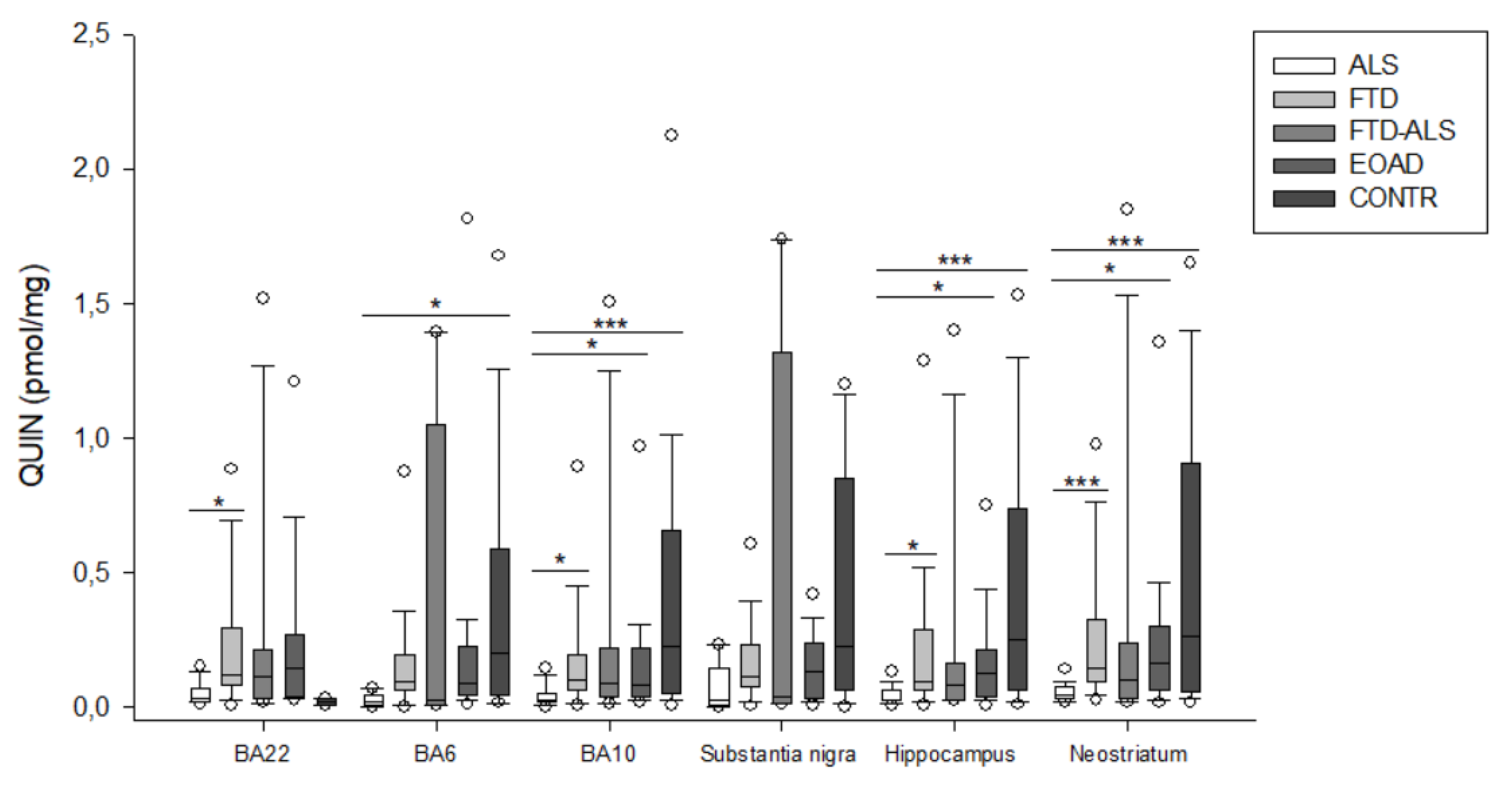
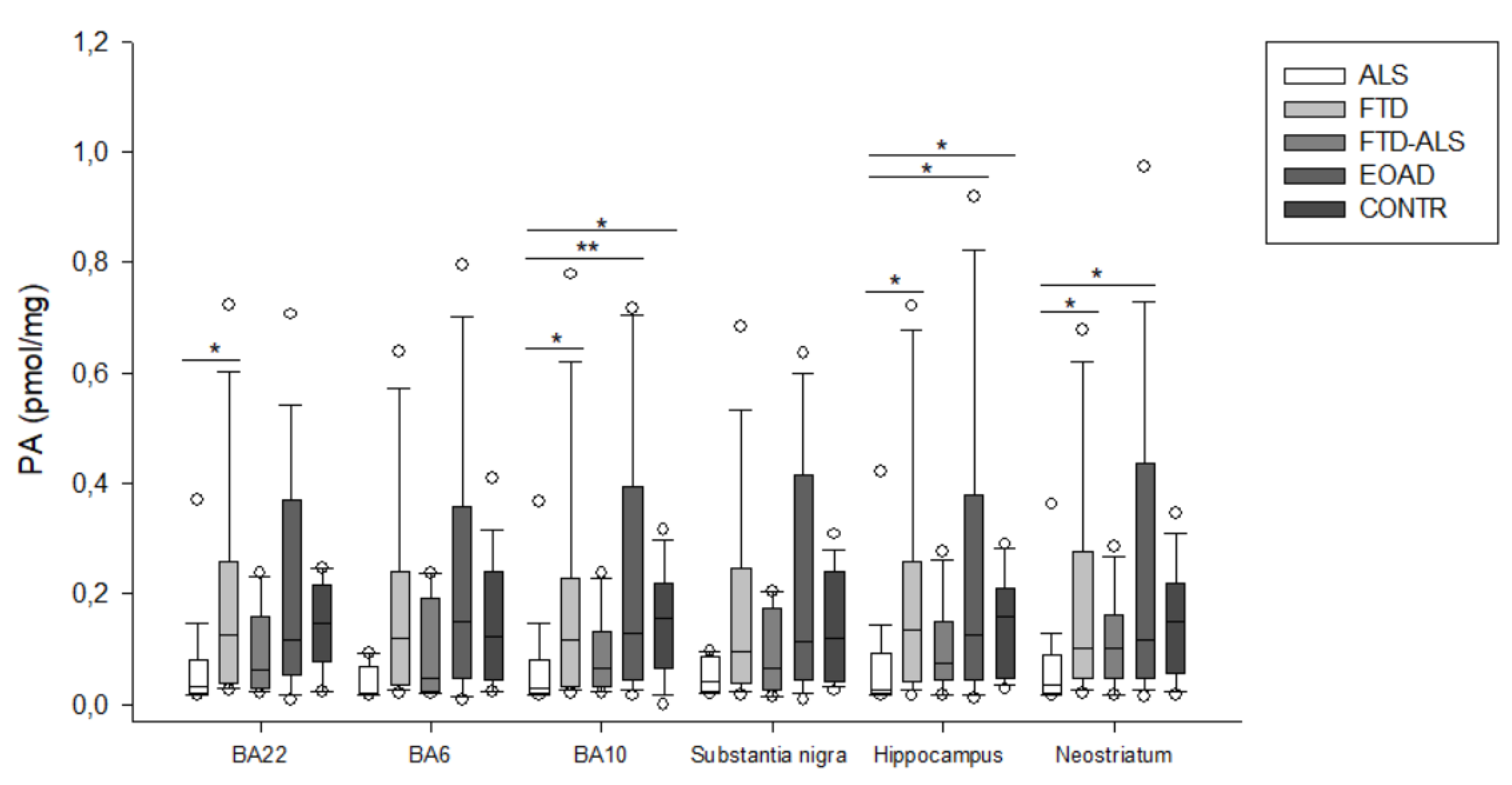
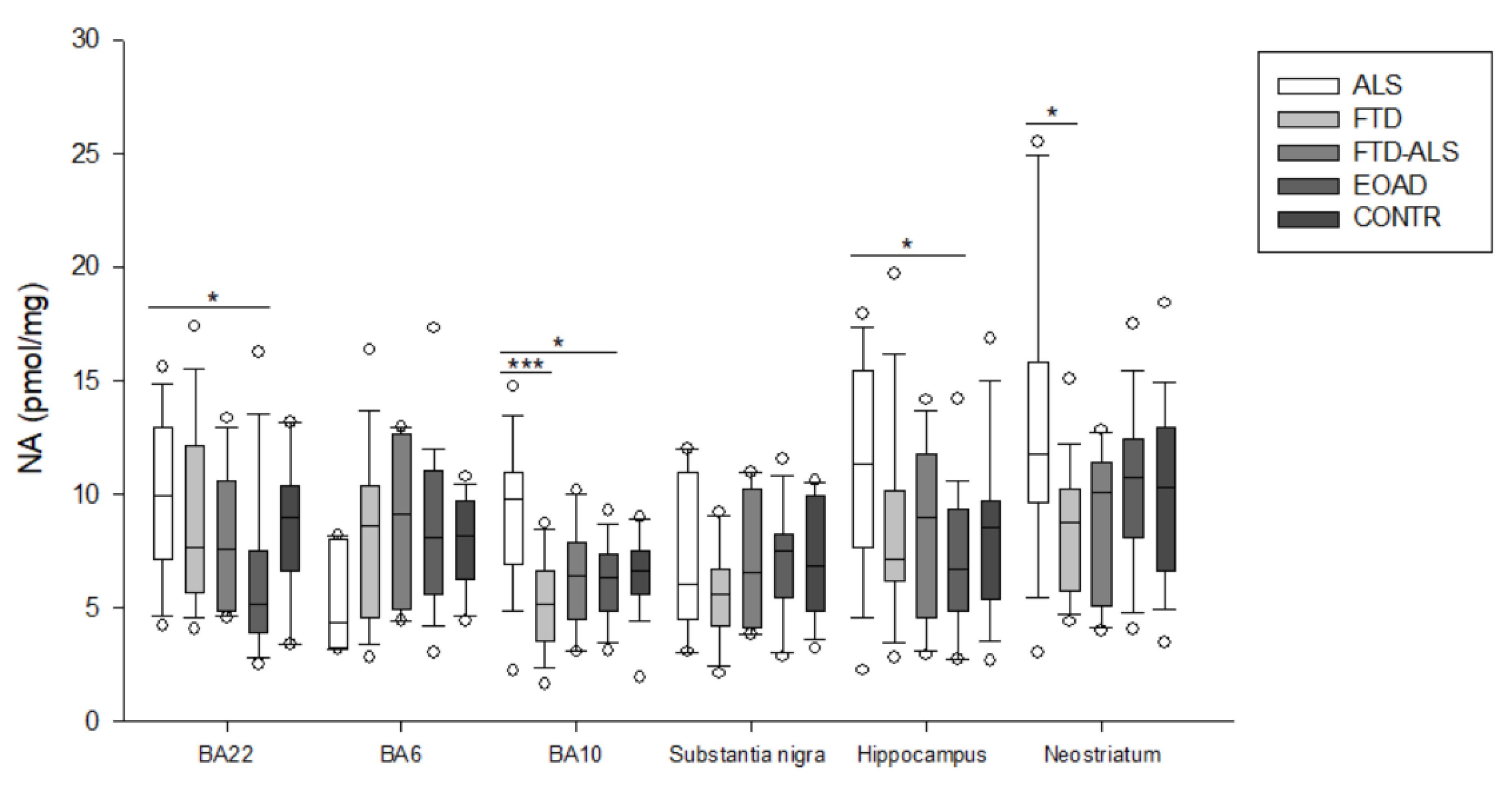
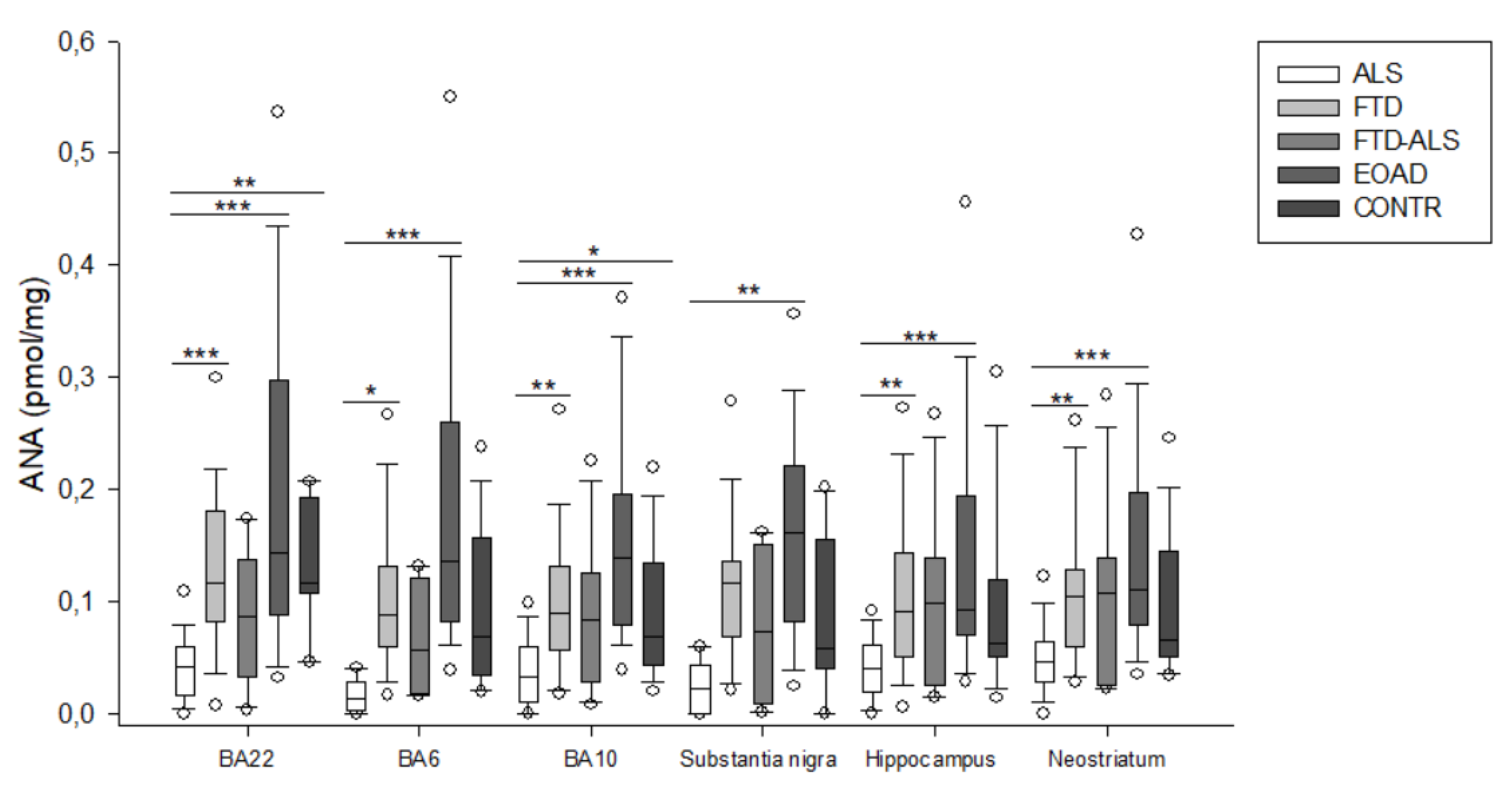
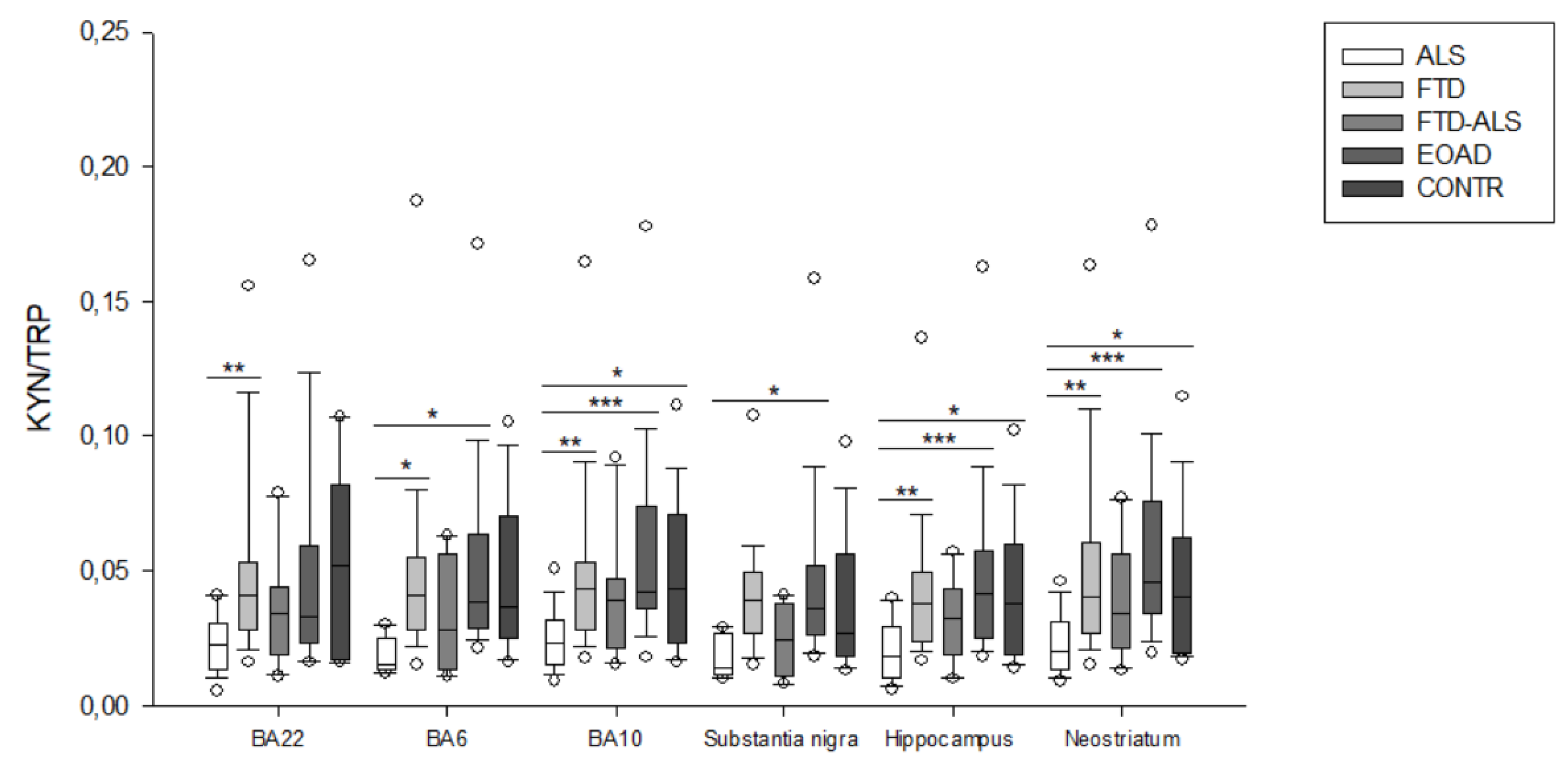
2.2. Kynurenine Pathway Findings
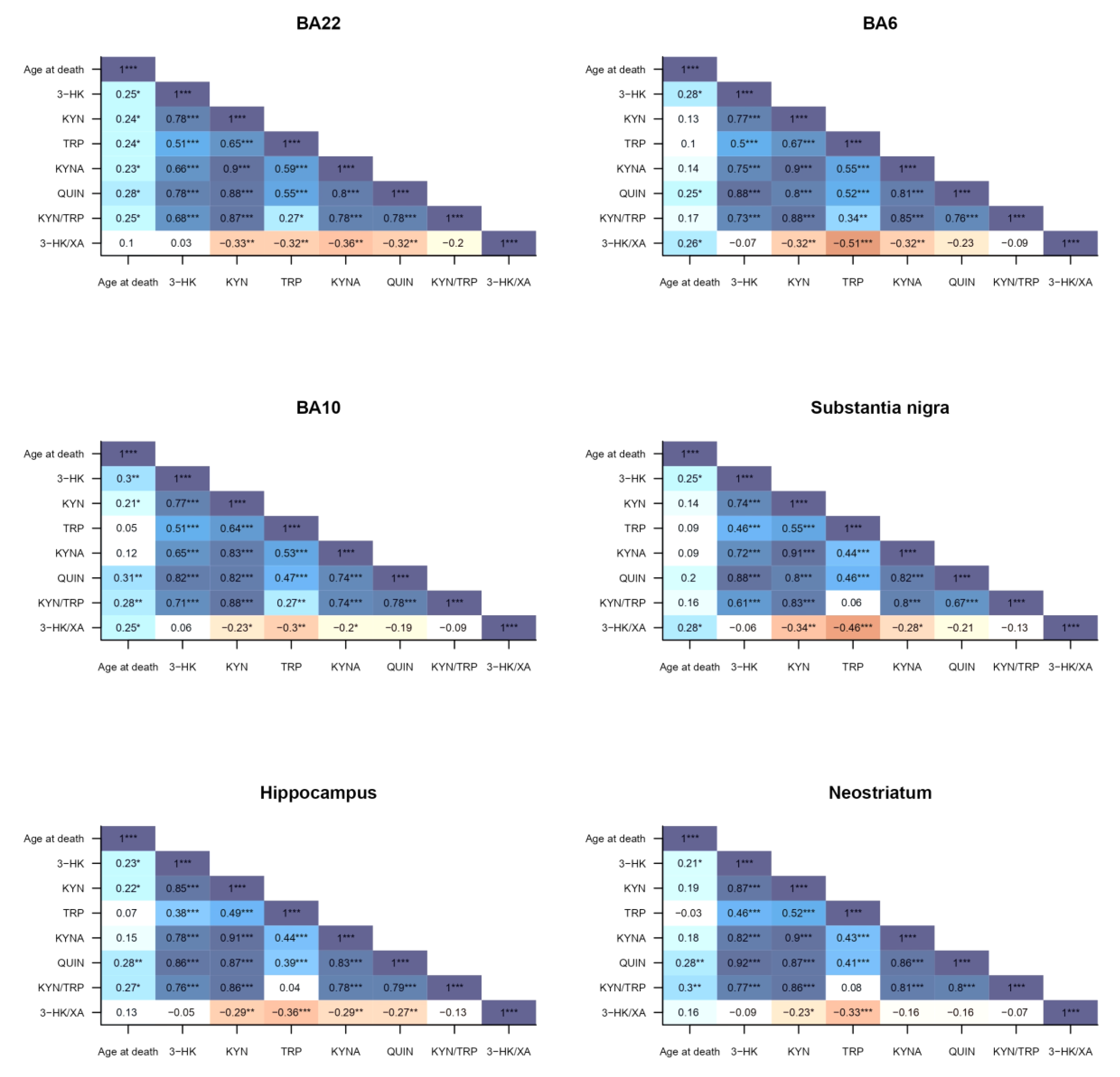
2.3. Correlations to Psychotropic Medication and Demographic Factors
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Brain Dissections
4.3. LC-MS/MS
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.097 (0.068–0.149) (n =5) c | 0.271 (0.187–0.432) (n = 23) | 0.100 (0.059–0.369) (n = 4) | 0.230 (0.121–0.363) (n = 20) | 0.277 (0.151–1.176) (n = 16) c | H = 12.073 p = 0.017 | H = 4.319 p > 0.05 |
| KYN (pmol/mg) | 0.826 (0.519–1.227) (n = 5) | 2.691 (1.768–3.882) (n = 23) | 0.874 (0.595–3.434) (n = 4) | 2.793 (1.814–4.488) (n = 20) | 1.901 (0.816–3.467) (n = 16) | H = 3.673 p = 0.042 | H = 5.819 p > 0.05 |
| TRP (pmol/mg) | 50.684 (36.098–55.231) (n =5) | 59.745 (38.969–89.684) (n = 23) | 41.376 (31.418–62.697) (n = 4) | 59.895 (37.411–75.573) (n = 20) | 47.572 (41.129–62.970) (n = 16) | H = 4.309 p > 0.05 | H = 2.060 p > 0.05 |
| KYNA (pmol/mg) | 0.197 (0.163–0.275) (n = 5) | 0.471 (0.279–0.670) (n = 23) | 0.272 (0.105–0.953) (n = 4) | 0.608 (0.313–0.967) (n = 20) | 0.584 (0.251–1.108) (n = 16) | H = 8.938 p > 0.05 | H = 5.736 p > 0.05 |
| XA (pmol/mg) | 0.002 (0.001–0.002) (n = 5) | 0.006 (0.002–0.011) (n = 23) | 0.006 (0.002–0.017) (n = 4) | 0.010 (0.004–0.015) (n = 20) | 0.010 (0.003–0.027) (n = 16) | H = 9.459 p > 0.05 | H = 3.934 p > 0.05 |
| QUIN (pmol/mg) | 0.019 (0.003–0.043) (n = 5) c | 0.090 (0.062–0.193) (n = 23) | 0.021 (0.006–1.050) (n = 4) | 0.087 (0.041–0.221) (n = 20) | 0.195 (0.040–0.587) (n = 16) c | H = 10.879 p = 0.028 | H = 6.176 p > 0.05 |
| PA (pmol/mg) | 0.019 (0.018–0.070) (n = 5) | 0.120 (0.036–0.240) (n = 23) | 0.049 (0.025–0.192) (n = 4) | 0.150 (0.046–0.357) (n = 20) | 0.123 (0.045–0.242) (n = 16) | H = 8.056 p > 0.05 | H = 6.280 p > 0.05 |
| NA (pmol/mg) | 4.322 (3.201–7.958) (n = 5) | 8.533 (4.509–10.374) (n = 23) | 9.128 (4.908–12.643) (n = 4) | 8.075 (5.557–11.014) (n = 20) | 8.109 (6.240–9.677) (n = 16) | H = 4.363 p > 0.05 | H = 1.077 p > 0.05 |
| ANA (pmol/mg) | 0.014 (0.003–0.029) (n = 5) a,bbb | 0.088 (0.060–0.131) (n = 23) a | 0.057 (0.018–0.122) (n = 4) | 0.136 (0.083–0.260) (n = 20) bbb | 0.069 (0.035–0.157) (n = 16) | H = 19.963 p < 0.001 | H = 6.396 p > 0.05 |
| KYN/TRP | 0.015 (0.014–0.025) (n = 5) a,b | 0.041 (0.028–0.055) (n = 23) a | 0.028 (0.014–0.056) (n = 4) | 0.039 (0.029–0.063) (n = 20) b | 0.0365 (0.025–0.070) (n = 16) | H = 10.961 p = 0.027 | H = 4.670 p > 0.05 |
| 3-HK/XA | 82.000 (45.626–83.693) (n = 5) | 56.810 (37.367–87.727) (n = 23) | 34.670 (9.524–46.561) (n = 4) | 30.762 (15.469–57.833) (n = 20) | 45.744 (28.636–80.060) (n = 16) | H = 7.916 p > 0.05 | H = 5.308 p > 0.05 |
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.149 (0.089–0.181) (n =20) aa,ccc | 0.316 (0.211–0.470) (n = 23) aa | 0.174 (0.084–0.297) (n = 11) d | 0.244 (0.154–0.428) (n = 23) | 0.547 (0.227–1.353) (n = 19) ccc,d | H = 26.169 p < 0.001 | H = 4.368 p > 0.05 |
| KYN (pmol/mg) | 1.065 (0.657–1.381) (n = 20) aa,bb,c | 2.399 (1.894–3.574) (n = 23) aa | 1.442 (0.760–8.984) (n = 11) | 2.569 (1.714–3.774) (n = 23) bb | 2.500 (1.130–3.510) (n = 19) c | H = 19.692 p < 0.001 | H = 5.819 p > 0.05 |
| TRP (pmol/mg) | 43.599 (37.866–49.496) (n =20) | 58.552 (39.385–76.132) (n = 23) | 40.469 (33.934–60.961) (n = 11) | 50.185 (37.389–65.420) (n = 23) | 51.375 (42.655–60.816) (n = 19) | H = 7.567 p > 0.05 | H = 4.280 p > 0.05 |
| KYNA (pmol/mg) | 0.223 (0.152–0.368) (n = 20) | 0.285 (0.171–0.390) (n = 23) | 0.292 (0.105–0.701) (n = 11) | 0.398 (0.206–0.529) (n = 23) | 0.435 (0.190–0.564) (n = 19) | H = 4.791 p > 0.05 | H = 5.357 p > 0.05 |
| XA (pmol/mg) | 0.002 (0.001–0.003) (n = 20) bbb,ccc | 0.005 (0.003–0.009) (n = 23) | 0.004 (0.002–0.005) (n = 11) | 0.007 (0.003–0.013) (n = 23) bbb | 0.008 (0.003–0.027) (n = 19) ccc | H = 20.455 p < 0.001 | H = 5.165 p > 0.05 |
| QUIN (pmol/mg) | 0.025 (0.017–0.047) (n = 20) a,b,ccc | 0.101 (0.059–0.189) (n = 23) a | 0.088 (0.036–0.219) (n = 11) | 0.080 (0.034–0.214) (n = 23) b | 0.226 (0.050–0.653) (n = 19) ccc | H = 23.314 p < 0.001 | H = 6.176 p > 0.05 |
| PA (pmol/mg) | 0.031 (0.019–0.082) (n = 20) a,bb,c | 0.118 (0.033–0.227) (n = 23) a | 0.066 (0.033–0.132) (n = 11) | 0.128 (0.045–0.394) (n = 23) bb | 0.157 (0.066–0.220) (n = 19) c | H = 15.938 p = 0.003 | H = 6.429 p > 0.05 |
| NA (pmol/mg) | 9.736 (6.906–10.929) (n = 20) aaa,b | 5.147 (3.503–6.601) (n = 23) aaa | 6.383 (4.466–7.816) (n = 11) | 6.321 (4.858–7.336) (n = 23) b | 6.615 (5.533–7.477) (n = 19) | H = 22.059 p < 0.001 | H = 5.560 p > 0.05 |
| ANA (pmol/mg) | 0.033 (0.010–0.060) (n = 20) aa,bbb,c | 0.089 (0.057–0.132) (n = 23) aa | 0.084 (0.029–0.125) (n = 11) | 0.139 (0.080–0.196) (n = 23) bbb | 0.068 (0.043–0.134) (n = 19) c | H = 30.018 p < 0.001 | H = 6.636 p > 0.05 |
| KYN/TRP | 0.023 (0.015–0.032) (n = 20) aa,bbb,c | 0.043 (0.028–0.053) (n = 23) aa | 0.039 (0.021–0.047) (n = 11) | 0.042 (0.036–0.074) (n = 23) bbb | 0.043 (0.023–0.071) (n = 19) c | H = 20.528 p < 0.001 | H = 5.088 p > 0.05 |
| 3-HK/XA | 63.232 (44.304–78.804) (n = 20) | 68.138 (40.067–101.081) (n = 23) | 59.820 (23.694–65.210) (n = 11) | 38.242 (16.459–62.043) (n = 23) | 59.362 (33.222–102.185) (n = 19) | H = 8.826 p > 0.05 | H = 3.511 p > 0.05 |
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.045 (0.035–0.152) (n =5) | 0.171 (0.090–0.266) (n = 21) | 0.047 (0.033–0.445) (n = 4) | 0.154 (0.060–0.223) (n = 22) | 0.221 (0.101–1.018) (n = 15) | H = 9.109 p > 0.05 | H = 5.617 p > 0.05 |
| KYN (pmol/mg) | 0.960 (0.844–1.560) (n = 5) | 2.563 (1.885–4.069) (n = 21) | 1.208 (0.792–3.464) (n = 4) | 2.708 (1.848–3.738) (n = 22) | 2.601 (1.251–3.541) (n = 15) | H = 6.280 p > 0.05 | H = 3.461 p > 0.05 |
| TRP (pmol/mg) | 72.913 (54.697–77.153) (n = 5) | 66.919 (42.826–87.649) (n = 21) | 78.706 (42.791–108.929) (n = 4) | 62.102 (42.911–90.552) (n = 22) | 68.146 (57.883–95.652) (n = 15) | H = 1.812 p > 0.05 | H = 4.604 p > 0.05 |
| KYNA (pmol/mg) | 0.221 (0.198–0.365) (n = 5) | 0.463 (0.299–0.642) (n = 21) | 0.328 (0.197–0.860) (n = 4) | 0.479 (0.251–0.823) (n = 22) | 0.611 (0.256–0.783) (n = 15) | H = 5.113 p > 0.05 | H = 5.695 p > 0.05 |
| XA (pmol/mg) | 0.001 (0.000–0.002) (n = 5) | 0.005 (0.002–0.008) (n = 21) | 0.002 (0.000–0.019) (n = 4) | 0.008 (0.002–0.016) (n = 22) | 0.005 (0.001–0.017) (n = 15) | H = 7.521 p > 0.05 | H = 4.431 p > 0.05 |
| QUIN (pmol/mg) | 0.020 (0.003–0.0145) (n = 5) | 0.109 (0.073–0.227) (n = 21) | 0.033 (0.008–1.315) (n = 4) | 0.127 (0.028–0.239) (n = 22) | 0.220 (0.059–0.848) (n = 15) | H = 7.892 p > 0.05 | H = 5.485 p > 0.05 |
| PA (pmol/mg) | 0.041 (0.022–0.086) (n = 5) | 0.096 (0.038–0.245) (n = 21) | 0.067 (0.026–0.173) (n = 4) | 0.114 (0.046–0.415) (n = 22) | 0.120 (0.041–0.241) (n = 15) | H = 4.432 p > 0.05 | H = 6.649 p > 0.05 |
| NA (pmol/mg) | 6.018 (4.471–10.923) (n = 5) | 5.565 (4.155–6.690) (n = 21) | 6.495 (4.072–10.224) (n = 4) | 7.504 (5.426–8.194) (n = 22) | 6.843 (4.806–9.882) (n = 15) | H = 6.104 p > 0.05 | H = 1.071 p > 0.05 |
| ANA (pmol/mg) | 0.023 (0.000–0.043) (n = 5) bb | 0.116 (0.069–0.136) (n = 21) | 0.074 (0.009–0.151) (n = 4) | 0.162 (0.083–0.221) (n = 22) bb | 0.059 (0.040–0.156) (n = 15) | H = 16.271 p = 0.003 | H = 6.162 p > 0.05 |
| KYN/TRP | 0.014 (0.012–0.027) (n = 5) b | 0.039 (0.027–0.050) (n = 21) | 0.024 (0.011–0.038) (n = 4) | 0.036 (0.026–0.052) (n = 22) b | 0.027 (0.018–0.056) (n = 15) | H = 10.658 p = 0.031 | H = 5.325 p > 0.05 |
| 3-HK/XA | 106.530 (35.265–295.489) (n = 5) | 45.478 (16.334–81.452) (n = 21) | 36.504 (12.886–1107.970) (n = 4) | 21.733 (10.640–59.994) (n = 22) | 49.111 (26.341–174.329) (n = 15) | H = 8.443 p > 0.05 | H = 0.779 p > 0.05 |
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.078 (0.051–0.106) (n =19) aaa,bb,ccc | 0.218 (0.152–0.384) (n = 22) aaa | 0.121 (0.065–0.213) (n = 11) | 0.204 (0.087–0.336) (n = 22) bb | 0.280 (0.129–1.093) (n = 15) ccc | H = 27.456 p < 0.001 | H = 4.436 p > 0.05 |
| KYN (pmol/mg) | 1.041 (0.800–1.528) (n = 19) aa,bb,c | 2.691 (1.986–4.224) (n = 22) aa | 1.654 (0.772–3.037) (n = 11) | 2.991 (1.820–4.493) (n = 22) bb | 2.554 (1.375–3.992) (n = 15) c | H = 18.775 p < 0.001 | H = 5.527 p > 0.05 |
| TRP (pmol/mg) | 70.418 (63.628–80.470) (n =19) | 77.954 (47.530–94.132) (n = 22) | 70.300 (40.110–94.641) (n = 11) | 67.003 (38.662–90.356) (n = 22) | 70.578 (61.559–90.414) (n = 15) | H = 1.444 p > 0.05 | H = 0.182 p > 0.05 |
| KYNA (pmol/mg) | 0.129 (0.102–0.193) (n = 19) b | 0.233 (0.137–0.376) (n = 22) | 0.225 (0.092–0.338) (n = 11) | 0.332 (0.155–0.467) (n = 22) b | 0.235 (0.136–0.575) (n = 15) | H = 12.793 p = 0.012 | H = 5.527 p > 0.05 |
| XA (pmol/mg) | 0.002 (0.001–0.003) (n = 19) a,bb,c | 0.005 (0.002–0.008) (n = 22) a | 0.003 (0.002–0.006) (n = 11) | 0.008 (0.002–0.122) (n = 22) bb | 0.007 (0.002–0.018) (n = 15) c | H = 16.989 p = 0.002 | H = 3.732 p > 0.05 |
| QUIN (pmol/mg) | 0.026 (0.021–0.058) (n = 19) a,b,ccc | 0.091 (0.061–0.289) (n = 22) a | 0.078 (0.025–0.158) (n = 11) | 0.126 (0.036–0.211) (n = 22) b | 0.251 (0.060–0.736) (n = 15) ccc | H = 18.680 p < 0.001 | H = 4.683 p > 0.05 |
| PA (pmol/mg) | 0.027 (0.019–0.092) (n = 19) a,b,c | 0.136 (0.040–0.258) (n = 22) a | 0.074 (0.044–0.150) (n = 11) | 0.125 (0.045–0.378) (n = 22) b | 0.160 (0.047–0.210) (n = 15) c | H = 14.362 p = 0.006 | H = 4.655 p > 0.05 |
| NA (pmol/mg) | 11.287 (7.588–15.388) (n = 19) b | 7.096 (6.143–10.102) (n = 22) | 8.932 (4.507–11.749) (n = 11) | 6.661 (3.330–9.263) (n = 22) b | 8.528 (5.334–9.702) (n = 15) | H = 10.292 p = 0.036 | H = 0.000 p > 0.05 |
| ANA (pmol/mg) | 0.041 (0.019–0.061) (n = 19) aa,bbb | 0.091 (0.051–0.144) (n = 22) aa | 0.098 (0.025–0.139) (n = 11) | 0.093 (0.070–0.195) (n = 22) bbb | 0.063 (0.051–0.120) (n = 15) | H = 19.546 p < 0.001 | H = 6.182 p = 0.045 |
| KYN/TRP | 0.018 (0.010–0.029) (n = 19) aa,bbb,c | 0.038 (0.024–0.050) (n = 22) aa | 0.032 (0.019–0.043) (n = 11) | 0.042 (0.025–0.057) (n = 22) bbb | 0.038 (0.019–0.060) (n = 15) c | H = 21.702 p < 0.001 | H = 3.915 p > 0.05 |
| 3-HK/XA | 56.835 (27.000–92.577) (n = 19) | 55.206 (34.042–87.430) (n = 22) | 38.211 (24.360–70.876) (n = 11) | 30.668 (11.502–67.397) (n = 22) | 61.354 (33.220–154.368) (n = 15) | H = 7.698 p > 0.05 | H = 0.295 p > 0.05 |
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.056 (0.042–0.095) (n = 19) aaa,bbb,ccc | 0.203 (0.130–0.363) (n = 24) aaa | 0.100 (0.047–0.192) (n = 11) | 0.207 (0.088–0.298) (n = 23) bbb | 0.218 (0.099–0.762) (n = 20) ccc | H = 28.811 p < 0.001 | H = 5.736 p > 0.05 |
| KYN (pmol/mg) | 1.108 (0.853–1.696) (n = 19) aaa,bbb,cc | 3.141 (2.364–4.862) (n = 24) aaa | 2.545 (0.900–3.353) (n = 11) | 3.989 (2.239–4.775) (n = 23) bbb | 3.564 (1.344–4.817) (n = 20) cc | H = 25.847 p < 0.001 | H = 6.615 p > 0.05 |
| TRP (pmol/mg) | 68.277 (48.502–82.532) (n = 19) | 77.566 (53.803–109.793) (n = 24) | 59.902 (42.723–73.137) (n = 11) | 67.920 (48.997–104.054) (n = 23) | 70.770 (61.050–84.172) (n = 20) | H = 6.231 p > 0.05 | H = 4.236 p > 0.05 |
| KYNA (pmol/mg) | 0.280 (0.236–0.533) (n = 19) aa,bbb,cc | 0.781 (0.488–1.096) (n = 24) aa | 0.600 (0.282–0.988) (n= 11) | 0.926 (0.424–1.282) (n = 23) bbb | 0.916 (0.339–1.342) (n = 20) cc | H = 19.868 p < 0.001 | H = 5.885 p > 0.05 |
| XA (pmol/mg) | 0.002 (0.001–0.003) (n = 19) aa,bbb,cc | 0.007 (0.002–0.013) (n = 24) aa | 0.003 (0.002–0.005) (n = 11) | 0.010 (0.004–0.020) (n = 23) bbb | 0.007 (0.002–0.022) (n = 20) cc | H = 21.450 p < 0.001 | H = 5.078 p > 0.05 |
| QUIN (pmol/mg) | 0.042 (0.029–0.072) (n = 19) aaa,b,ccc | 0.142 (0.094–0.321) (n = 24) aaa | 0.096 (0.030–0.237) (n = 11) | 0.161 (0.059–0.301) (n = 23) b | 0.262 (0.056–0.905) (n = 20) ccc | H = 22.167 p < 0.001 | H = 6.176 p > 0.05 |
| PA (pmol/mg) | 0.037 (0.021–0.090) (n = 19) a,b | 0.102 (0.049–0.276) (n = 24) a | 0.102 (0.048–0.163) (n = 11) | 0.117 (0.047–0.435) (n = 23) b | 0.149 (0.056–0.220) (n = 20) | H = 13.247 p = 0.010 | H = 6.265 p > 0.05 |
| NA (pmol/mg) | 11.759 (9.617–15.757) (n = 19) a | 8.743 (5.695–10.223) (n = 24) a | 10.043 (5.028–11.347) (n = 11) | 10.708 (8.047–12.392) (n = 23) | 10.264 (6.596–12.884) (n = 20) | H = 10.685 p = 0.030 | H = 3.665 p > 0.05 |
| ANA (pmol/mg) | 0.046 (0.029–0.065) (n = 19) aa,bbb | 0.105 (0.060–0.129) (n = 24) aa | 0.107 (0.026–0.139) (n = 11) | 0.111 (0.080–0.197) (n = 23) bbb | 0.066 (0.051–0.146) (n = 20) | H = 18.811 p < 0.001 | H = 7.005 p > 0.05 |
| KYN/TRP | 0.020 (0.013–0.031) (n = 19) aa,bbb,c | 0.041 (0.027–0.061) (n = 24) aa | 0.034 (0.021–0.056) (n = 11) | 0.046 (0.034–0.076) (n = 23) bbb | 0.041 (0.020–0.063) (n = 20) c | H = 19.857 p < 0.001 | H = 5.621 p > 0.05 |
| 3-HK/XA | 36.663 (23.324–64.789) (n = 19) | 37.106 (20.606–75.004) (n = 24) | 35.959 (15.867–45.849) (n = 11) | 22.208 (11.574–38.688) (n = 23) | 37.123 (25.112–57.850) (n = 20) | H = 9.064 p > 0.05 | H = 2.473 p > 0.05 |
| QC Low | QC Medium | QC High | LOQ | ||
|---|---|---|---|---|---|
| 3-HK | Average | 37.2 | 147 | 487 | 1.6 |
| (µmol/L) | CV | 5.8 | 4.4 | 3.7 | |
| KYN | Average | 1.79 | 4.6 | 17.3 | 0.0036 |
| (nmol/L) | CV | 4.7 | 4.1 | 3.0 | |
| TRP | Average | 52.5 | 109 | 271 | 0.17 |
| (µmol/L) | CV | 2.1 | 2.3 | 2.4 | |
| KYNA | Average | 37.5 | 132 | 612 | 0.74 |
| (nmol/L) | CV | 4.6 | 3.0 | 2.6 | |
| XA | Average | 17.2 | 76.7 | 378 | 0.55 |
| (nmol/L) | CV | 6.1 | 3.1 | 3.5 | |
| QUIN | Average | 342 | 1492 | 7779 | 2.5 |
| (nmol/L) | CV | 3.8 | 4.0 | 2.8 | |
| PA | Average | 29.2 | 157 | 718 | 1.9 |
| (nmol/L) | CV | 5.6 | 3.6 | 3.2 | |
| NA | Average | 11.8 | 59.3 | 365 | 1.7 |
| (nmol/L) | CV | 14.3 | 6.7 | 4.9 | |
| ANA | Average | 51.7 | 253 | 1565 | 5.5 |
| (nmol/L) | CV | 13.6 | 6.3 | 3.6 |
References
- Vieira, R.T.; Caixeta, L.; Machado, S.; Silva, A.C.; Nardi, A.E.; Arias-Carrion, O.; Carta, M.G. Epidemiology of early-onset dementia: A review of the literature. Clin. Pract. Epidemiol. Ment. Health 2013, 9, 88–95. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Engelborghs, S.; Saerens, J.; Goeman, J.; Mariën, P.; Maertens, K.; Nagels, G.; Martin, J.-J.; Pickut, B.A. The Middelheim Frontality Score: A behavioural assessment scale that discriminates frontotemporal dementia from Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2005, 20, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.-J.; Boon, P.; Cras, P.; De Deyn, P.-P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Grossman, M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int. Rev. Psychiatry 2013, 25, 145–158. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134 Pt 9, 2456–2477. [Google Scholar] [CrossRef]
- Salameh, J.S.; Brown, R.H., Jr.; Berry, J.D. Amyotrophic Lateral Sclerosis: Review. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J. The WFN Research Group On ALS/MND A revision of the El Escorial criteria—2015. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C.; Murphy, J.; Langmore, S.; Kramer, J.H.; Olney, R.K.; Miller, B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 2003, 60, 1094–1097. [Google Scholar] [CrossRef]
- Tsuji, H.; Arai, T.; Kametani, F.; Nonaka, T.; Yamashita, M.; Suzukake, M.; Hosokawa, M.; Yoshida, M.; Hatsuta, H.; Takao, M.; et al. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 2012, 135 Pt 11, 3380–3391. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Philtjens, S.; Heeman, B.; Engelborghs, S.; Vandenbulcke, M.; De Baets, G.; Bäumer, V.; et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 2015, 85, 2116–2125. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Strong, M.J.; Donison, N.S.; Volkening, K. Alterations in Tau Metabolism in ALS and ALS-FTSD. Front. Neurol. 2020, 11, 598907. [Google Scholar] [CrossRef]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of Frontotemporal Lobar Degeneration-Tau (FTLD-Tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef]
- Tsai, R.M.; Boxer, A.L. Therapy and clinical trials in frontotemporal dementia: Past, present, and future. J. Neurochem. 2016, 138, 211–221. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H.L.M. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2002, 2, CD001447. [Google Scholar] [CrossRef]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [Google Scholar] [CrossRef]
- Shefner, J.; Heiman-Patterson, T.; Pioro, E.P.; Wiedau-Pazos, M.; Liu, S.; Zhang, J.; Agnese, W.; Apple, S. Long-term edaravone efficacy in amyotrophic lateral sclerosis: Post-hoc analyses of Study 19 (MCI186-19). Muscle Nerve 2020, 61, 218–221. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/newsevents/press-announcements/fda-approves-drug-treat-als (accessed on 15 August 2022).
- Available online: https://www.radicava.com/patient/ (accessed on 15 August 2022).
- Smith, R.; Pioro, E.; Myers, K.; Sirdofsky, M.; Goslin, K.; Meekins, G.; Yu, H.; Wymer, J.; Cudkowicz, M.; Macklin, E.A.; et al. Enhanced Bulbar Function in Amyotrophic Lateral Sclerosis: The Nuedexta Treatment Trial. Neurotherapeutics 2017, 14, 762–772. [Google Scholar] [CrossRef]
- Oskarsson, B.; Maragakis, N.; Bedlack, R.S.; Goyal, N.; Meyer, J.A.; Genge, A.; Bodkin, C.; Maiser, S.; Staff, N.; Zinman, L.; et al. MN-166 (ibudilast) in amyotrophic lateral sclerosis in a Phase IIb/III study: COMBAT-ALS study design. Neurodegener. Dis. Manag. 2021, 11, 431–443. [Google Scholar] [CrossRef]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211030365. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of subjects in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Available online: https://alsnewstoday.com/amx0035/ (accessed on 17 August 2022).
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Meininger, V.; Brew, B.J. Implications for the Kynurenine Pathway and Quinolinic Acid in Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 166–176. [Google Scholar] [CrossRef]
- Chen, Y.; Stankovic, R.; Cullen, K.M.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The Kynurenine Pathway and Inflammation in Amyotrophic Lateral Sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef]
- Dantzer, R.; O’connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef]
- Aaldijk, E.; Vermeiren, Y. The role of serotonin within the microbiota-gut-brain axis in the development of Alzheimer’s disease: A narrative review. Ageing Res. Rev. 2022, 75, 101556. [Google Scholar] [CrossRef]
- Janssens, J.; Vermeiren, Y.; van Faassen, M.; van der Ley, C.; Kema, I.P.; De Deyn, P.P. Monoaminergic and Kynurenergic Characterization of Frontotemporal Dementia and Amyotrophic Lateral Sclerosis in Cerebrospinal Fluid and Serum. Neurochem. Res. 2020, 45, 1191–1201. [Google Scholar] [CrossRef]
- Sorgdrager, F.J.H.; Van Vermeiren, Y.; Faassen, M.; Ley, C.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef]
- Iłzecka, J.; Kocki, T.; Stelmasiak, Z.; Turski, W.A. Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine metabolism in Alzheimer’s disease. J. Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef]
- Woodling, N.S.; Colas, D.; Wang, Q.; Minhas, P.; Panchal, M.; Liang, X.; Mhatre, S.D.; Brown, H.; Ko, N.; Zagol-Ikapitte, I.; et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer’s disease model mice. Brain 2016, 139 Pt 7, 2063–2081. [Google Scholar] [CrossRef]
- Breda, C.; Sathyasaikumar, K.V.; Idrissi, S.S.; Notarangelo, F.M.; Estranero, J.G.; Moore, G.G.L.; Green, E.W.; Kyriacou, C.P.; Schwarcz, R.; Giorgini, F. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc. Natl. Acad. Sci. USA 2016, 113, 5435–5440. [Google Scholar] [CrossRef]
- van der Goot, A.T.; Zhu, W.; Vazquez-Manrique, R.P.; Seinstra, R.I.; Dettmer, K.; Michels, H.; Farina, F.; Krijnen, J.; Melki, R.; Buijsman, R.C.; et al. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. USA 2012, 109, 14912–14917. [Google Scholar] [CrossRef]
- Sorgdrager, F.; Van Der Ley, C.P.; Van Faassen, M.; Calus, E.; Nollen, E.A.; Kema, I.P.; Van Dam, D.; De Deyn, P.P. The Effect of Tryptophan 2,3-Dioxygenase Inhibition on Kynurenine Metabolism and Cognitive Function in the APP23 Mouse Model of Alzheimer’s Disease. Int. J. Tryptophan Res. 2020, 13, 1178646920972657. [Google Scholar] [CrossRef]
- Meltzer, C.C.; Smith, G.; DeKosky, S.; Pollock, B.G.; Mathis, C.A.; Moore, R.Y.; Kupfer, D.J.; Iii, C.F.R. Serotonin in Aging, Late-Life Depression, and Alzheimer’s Disease: The Emerging Role of Functional Imaging. Neuropsychopharmacology 1998, 18, 407–430. [Google Scholar] [CrossRef]
- Myint, A.; Kim, Y. Cytokine–serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Strac, D.Š.; Pivac, N.; Mück-Šeler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Skorobogatov, K.; De Picker, L.; Verkerk, R.; Coppens, V.; Leboyer, M.; Müller, N.; Morrens, M. Brain Versus Blood: A Systematic Review on the Concordance Between Peripheral and Central Kynurenine Pathway Measures in Psychiatric Disorders. Front. Immunol. 2021, 12, 716980. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef]
- Schwieler, L.; Trepci, A.; Krzyzanowski, S.; Hermansson, S.; Granqvist, M.; Piehl, F.; Venckunas, T.; Brazaitis, M.; Kamandulis, S.; Lindqvist, D.; et al. A novel, robust method for quantification of multiple kynurenine pathway metabolites in the cerebrospinal fluid. Bioanalysis 2020, 12, 379–392. [Google Scholar] [CrossRef]
- Sparkman, N.L.; Johnson, R.W. Neuroinflammation Associated with Aging Sensitizes the Brain to the Effects of Infection or Stress. Neuroimmunomodulation 2008, 15, 323–330. [Google Scholar] [CrossRef]
- Meier, T.B.; Drevets, W.C.; Teague, T.K.; Wurfel, B.E.; Mueller, S.C.; Bodurka, J.; Dantzer, R.; Savitz, J. Kynurenic acid is reduced in females and oral contraceptive users: Implications for depression. Brain Behav. Immun. 2018, 67, 59–64. [Google Scholar] [CrossRef]
- Reininghaus, E.Z.; Dalkner, N.; Riedrich, K.; Fuchs, D.; Gostner, J.M.; Reininghaus, B. Sex Specific Changes in Tryptophan Breakdown Over a 6 Week Treatment Period. Front. Psychiatry 2019, 10, 74. [Google Scholar] [CrossRef]
- Schröcksnadel, K.; Widner, B.; Neurauter, G.; Fuchs, D.; Schröcksnadel, H.; Bergant, A. Tryptophan Degradation During and After Gestation. Adv. Exp. Med. Biol. 2003, 527, 77–83. [Google Scholar] [CrossRef]
- Widner, B.; Werner, E.R.; Schennach, H.; Wachter, H.; Fuchs, D. Simultaneous Measurement of Serum Tryptophan and Kynurenine by HPLC. Clin. Chem. 1997, 43, 2424–2426. [Google Scholar] [CrossRef]
- Moursi, G.E.; Abdel-Daim, M.H.; Kelada, N.L.; Abdel-Tawab, G.A.; Girgis, L.H. The influence of sex, age, synthetic oestrogens, progestogens and oral contraceptives on the excretion of urinary tryptophan metabolites. Bull. World Health Organ. 1970, 43, 651–661. [Google Scholar]
- Rose, D.P.; Braidman, I.P. Excretion of tryptophan metabolites as affected by pregnancy, contraceptive steroids, and steroid hormones. Am. J. Clin. Nutr. 1971, 24, 673–683. [Google Scholar] [CrossRef]
- Rose, D.P.; Adams, P.W. Oral contraceptives and tryptophan metabolism: Effects of oestrogen in low dose combined with a progestagen and of a low-dose progestagen (megestrol acetate) given alone. J. Clin. Pathol. 1972, 25, 252–258. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.R.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Vermeiren, Y.; Janssens, J.; Van Dam, D.; De Deyn, P.P. Serotonergic Dysfunction in Amyotrophic Lateral Sclerosis and Parkinson’s Disease: Similar Mechanisms, Dissimilar Outcomes. Front. Neurosci. 2018, 12, 185. [Google Scholar] [CrossRef]
- Brazill, J.M.; Li, C.; Zhu, Y.; Zhai, R.G. NMNAT: It’s an NAD+ synthase… It’s a chaperone… It’s a neuroprotector. Curr. Opin. Genet. Dev. 2017, 44, 156–162. [Google Scholar] [CrossRef]
- Tan, V.X.; Guillemin, G.J. Kynurenine Pathway Metabolites as Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurosci. 2019, 13, 1013. [Google Scholar] [CrossRef]
- Ferrari, M.D.; Odink, J.; Tapparelli, C.; Van Kempen, G.M.; Pennings, E.J.; Bruyn, G.W. Serotonin metabolism in migraine. Neurology 1989, 39, 1239–1242. [Google Scholar] [CrossRef]
- Myint, A.; Halaris, A. Imbalances in Kynurenines as Potential Biomarkers in the Diagnosis and Treatment of Psychiatric Disorders. Front. Psychiatry 2022, 13, 913303. [Google Scholar] [CrossRef]
- Oxenkrug, G. Serotonin—Kynurenine Hypothesis of Depression: Historical Overview and Recent Developments. Curr. Drug Targets 2013, 14, 514–521. [Google Scholar] [CrossRef]
- Friedman, M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int. J. Tryptophan Res. 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vermeiren, Y.; Janssens, J.; Aerts, T.; Martin, J.-J.; Sieben, A.; Van Dam, D.; De Deyn, P.P. Brain Serotonergic and Noradrenergic Deficiencies in Behavioral Variant Frontotemporal Dementia Compared to Early-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 1079–1096. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L.; et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.A.; Lee, V.M.Y. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef]
- Vermeiren, Y.; Van Dam, D.; Aerts, T.; Engelborghs, S.; De Deyn, P.P. Brain Region-Specific Monoaminergic Correlates of Neuropsychiatric Symptoms in Alzheimer’s Disease. J. Alzheimers Dis. 2014, 41, 819–833. [Google Scholar] [CrossRef]
- de Jong, W.H.; Smit, R.; Bakker, S.J.; de Vries, E.G.; Kema, I.P. Plasma tryptophan, kynurenine and 3-hydroxykynurenine measurement using automated on-line solid-phase extraction HPLC–tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 603–609. [Google Scholar] [CrossRef]
- Meinitzer, A.; Tomaschitz, A.; Pilz, S.; Truber, M.; Zechner, G.; Gaksch, M.; Prietl, B.; Treiber, G.; Schwarz, M.; Baranyi, A. Development of a liquid chromatography–mass spectrometry method for the determination of the neurotoxic quinolinic acid in human serum. Clin. Chim. Acta 2014, 436, 268–272. [Google Scholar] [CrossRef]
- Schröcksnadel, K.; Wirleitner, B.; Winkler, C.; Fuchs, D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta 2006, 364, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann. N. Y. Acad. Sci. 2010, 1199, 1–14. [Google Scholar] [CrossRef]
- Ciorba, M.A. Kynurenine pathway metabolites: Relevant to vitamin B-6 deficiency and beyond. Am. J. Clin. Nutr. 2013, 98, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Ulvik, A.; Theofylaktopoulou, D.; Midttun, Ø.; Nygård, O.; Eussen, S.J.; Ueland, P.M. Substrate product ratios of enzymes in the kynurenine pathway measured in plasma as indicators of functional vitamin B-6 status. Am. J. Clin. Nutr. 2013, 98, 934–940. [Google Scholar] [CrossRef] [PubMed]










| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | |
|---|---|---|---|---|---|---|
| Age at death (years) | 63.5 (54.2–67.5) c | 67.6 (60.9–73.4) | 71.0 (64.7–76.1) | 67.0 (59.5–72.2) | 69.0 (62.8–78.0) c | H = 10.777 p = 0.029 |
| Sex (Male/Female) | 6/14 | 14/10 | 6/5 | 14/9 | 12/8 | Fisher’s Exact Test: 5.488 p > 0.05 |
| Psychotropic medication (No/Yes/Missing) | 0/5/15 | 1/17/6 | 2/2/7 | 2/20/1 | 7/7/6 | Fisher’s Exact Test: 13.348 p = 0.004 |
| Antidepressants (No/Yes/Missing) | 1/4/15 | 9/9/6 | 4/0/7 | 10/12/1 ddd | 13/1/6 ddd | Fisher’s Exact Test: 14.839 p = 0.003 |
| Antipsychotics (No/Yes/Missing) | 5/0/15 | 10/8/6 | 3/1/7 | 10/12/1 ddd | 13/1/6 ddd | Fisher’s Exact Test: 11.927 p = 0.012 |
| Antiepileptics (No/Yes/Missing) | 4/1/15 | 15/3/6 | 4/0/7 | 21/1/1 | 14/0/6 | Fisher’s Exact Test: 4.362 p > 0.05 |
| Anxiolytics & hypnotics (No/Yes/Missing) | 5/0/15 | 11/7/6 | 4/0/7 | 16/6/1 | 10/4/6 | Fisher’s Exact Test: 3.783 p > 0.05 |
| Analgesics (No/Yes/Missing) | 3/2/15 | 12/6/6 | 4/0/7 | 17/5/1 | 10/4/6 | Fisher’s Exact Test: 2.339 p > 0.05 |
| Anticholinergic medication (No/Yes/Missing) | 5/0/15 | 15/3/6 | 4/0/7 | 16/6/1 | 14/0/6 | Fisher’s Exact Test: 5.501 p > 0.05 |
| Antidementia medication (No/Yes/Missing) | 5/0/15 | 14/4/6 | 4/0/7 | 13/9/1 | 14/0/6 | Fisher’s Exact Test: 10.085 p = 0.023 |
| CNS-acting medication (No/Yes/Missing) | 0/5/15 aaa,bbb,ccc | 18/0/6 aaa | 3/1/7 | 22/0/1 bbb | 14/0/6 ccc | Fisher’s Exact Test: 28.277 p < 0.001 |
| Hormones (No/Yes/Missing) | 5/0/15 | 17/1/6 | 4/0/7 | 21/1/1 | 13/1/6 | Fisher’s Exact Test: 1.486 p > 0.05 |
| Corticosteroids (No/Yes/Missing) | 5/0/15 | 17/1/6 | 4/0/7 | 22/0/1 | 13/1/6 | Fisher’s Exact Test: 3.200 p > 0.05 |
| Adrenergic medication (No/Yes/Missing) | 4/1/15 | 18/0/6 | 4/0/7 | 22/0/1 | 11/3/6 | Fisher’s Exact Test: 8.047 p = 0.029 |
| Antiemetics (No/Yes/Missing) | 4/1/15 | 18/0/6 | 4/0/7 | 21/1/1 | 13/1/6 | Fisher’s Exact Test: 4.408 p > 0.05 |
| Antiparkinson medication (No/Yes/Missing) | 5/0/15 | 16/2/6 | 4/0/7 | 21/1/1 | 14/0/6 | Fisher’s Exact Test: 2.484 p > 0.05 |
| ALS (n = 20) | FTD (n = 24) | FTD–ALS (n = 11) | EOAD (n = 23) | CONTR (n = 20) | Kruskal–Wallis | Kruskal–Wallis (Psychotropic Medication Free) | |
|---|---|---|---|---|---|---|---|
| 3-HK (pmol/mg) | 0.179 (0.130–0.240) (n = 20) aa | 0.382 (0.284–0.646) (n = 23) aa,d | 0.129 (0.072–0.413) (n = 11) d | 0.243 (0.114–0.457) (n = 14) | 0.315 (0.167–1.466) (n = 7) | H = 21.832 p < 0.001 | H = 4.250 p > 0.05 |
| KYN (pmol/mg) | 1.119 (0.660–1.655) (n = 20) aa,b | 2.808 (1.781–4.865) (n = 23) aa | 1.069 (0.887–2.957) (n = 11) | 2.680 (1.775–5.164) (n = 14) b | 4.467 (0.670–5.621) (n = 7) | H = 18.205 p = 0.001 | H = 3.583 p > 0.05 |
| TRP (pmol/mg) | 49.790 (43.674–61.748) (n =20) | 63.595 (44.317–80.268) (n = 23) | 52.714 (31.859–76.818) (n = 11) | 64.399 (43.471–80.521) (n = 14) | 57.480 (35.792–68.562) (n = 7) | H = 6.340 p > 0.05 | H = 0.500 p > 0.05 |
| KYNA (pmol/mg) | 0.247 (0.186–0.405) (n = 20) | 0.405 (0.200–0.859) (n = 23) | 0.365 (0.157–0.622) (n = 11) | 0.376 (0.301–0.723) (n = 14) | 0.575 (0.134–1.480) (n = 7) | H = 6.902 p > 0.05 | H = 2.056 p > 0.05 |
| XA (pmol/mg) | 0.003 (0.002–0.004) (n = 20) | 0.007 (0.003–0.014) (n = 23) | 0.003 (0.002–0.006) (n = 11) | 0.008 (0.003–0.013) (n = 14) | 0.009 (0.003–0.022) (n = 7) | H = 18.207 p = 0.001 | H = 5.389 p > 0.05 |
| QUIN (pmol/mg) | 0.031 (0.020–0.076) (n = 20) aa | 0.115 (0.079–0.289) (n = 23) aa | 0.109 (0.032–0.210) (n = 11) | 0.144 (0.046–0.291) (n = 14) | 0.281 (0.037–0.636) (n = 7) | H = 18.865 p < 0.001 | H = 4.722 p > 0.05 |
| PA (pmol/mg) | 0.036 (0.024–0.096) (n = 20) aa | 0.126 (0.039–0.243) (n = 23) aa | 0.063 (0.030–0.205) (n = 11) | 0.122 (0.068–0.374) (n = 14) | 0.145 (0.038–0.217) (n = 7) | H = 14.833 p = 0.005 | H = 5.139 p > 0.05 |
| NA (pmol/mg) | 9.879 (7.085–12.940) (n = 20) b | 8.178 (5.613–12.078) (n = 23) | 7.564 (4.855–10.562) (n = 11) | 5.642 (3.810–7.730) (n = 14) b | 8.964 (4.119–10.348) (n = 7) | H = 10.965 p = 0.030 | H = 4.806 p > 0.05 |
| ANA (pmol/mg) | 0.043 (0.016–0.065) (n = 20) aaa,bbb,cc | 0.115 (0.082–0.181) (n = 23) aaa | 0.087 (0.033–0.137) (n = 11) | 0.171 (0.084–0.305) (n = 14) bbb | 0.144 (0.107–0.193) (n = 7) cc | H = 27.681 p < 0.001 | H = 6.500 p > 0.05 |
| KYN/TRP | 0.023 (0.013–0.031) (n = 20) aa | 0.041 (0.028–0.053) (n = 23) aa | 0.034 (0.019–0.044) (n = 11) | 0.033 (0.023–0.059) (n = 14) | 0.052 (0.017–0.082) (n = 7) | H = 14.879 p = 0.005 | H = 3.583 p > 0.05 |
| 3-HK/XA | 64.377 (47.660–91.572) (n = 20) | 62.705 (43.536–92.817) (n = 23) | 64.260 (37.645–83.200) (n = 11) | 36.403 (21.814–63.653) (n = 14) | 64.857 (41.607–77.034) (n = 7) | H = 7.918 p > 0.05 | H = 2.333 p > 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heylen, A.; Vermeiren, Y.; Kema, I.P.; van Faassen, M.; van der Ley, C.; Van Dam, D.; De Deyn, P.P. Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD. Pharmaceuticals 2023, 16, 615. https://doi.org/10.3390/ph16040615
Heylen A, Vermeiren Y, Kema IP, van Faassen M, van der Ley C, Van Dam D, De Deyn PP. Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD. Pharmaceuticals. 2023; 16(4):615. https://doi.org/10.3390/ph16040615
Chicago/Turabian StyleHeylen, Annelies, Yannick Vermeiren, Ido P. Kema, Martijn van Faassen, Claude van der Ley, Debby Van Dam, and Peter P. De Deyn. 2023. "Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD" Pharmaceuticals 16, no. 4: 615. https://doi.org/10.3390/ph16040615
APA StyleHeylen, A., Vermeiren, Y., Kema, I. P., van Faassen, M., van der Ley, C., Van Dam, D., & De Deyn, P. P. (2023). Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD. Pharmaceuticals, 16(4), 615. https://doi.org/10.3390/ph16040615