

Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors



Abstract
1. Introduction
2. Results and Discussion
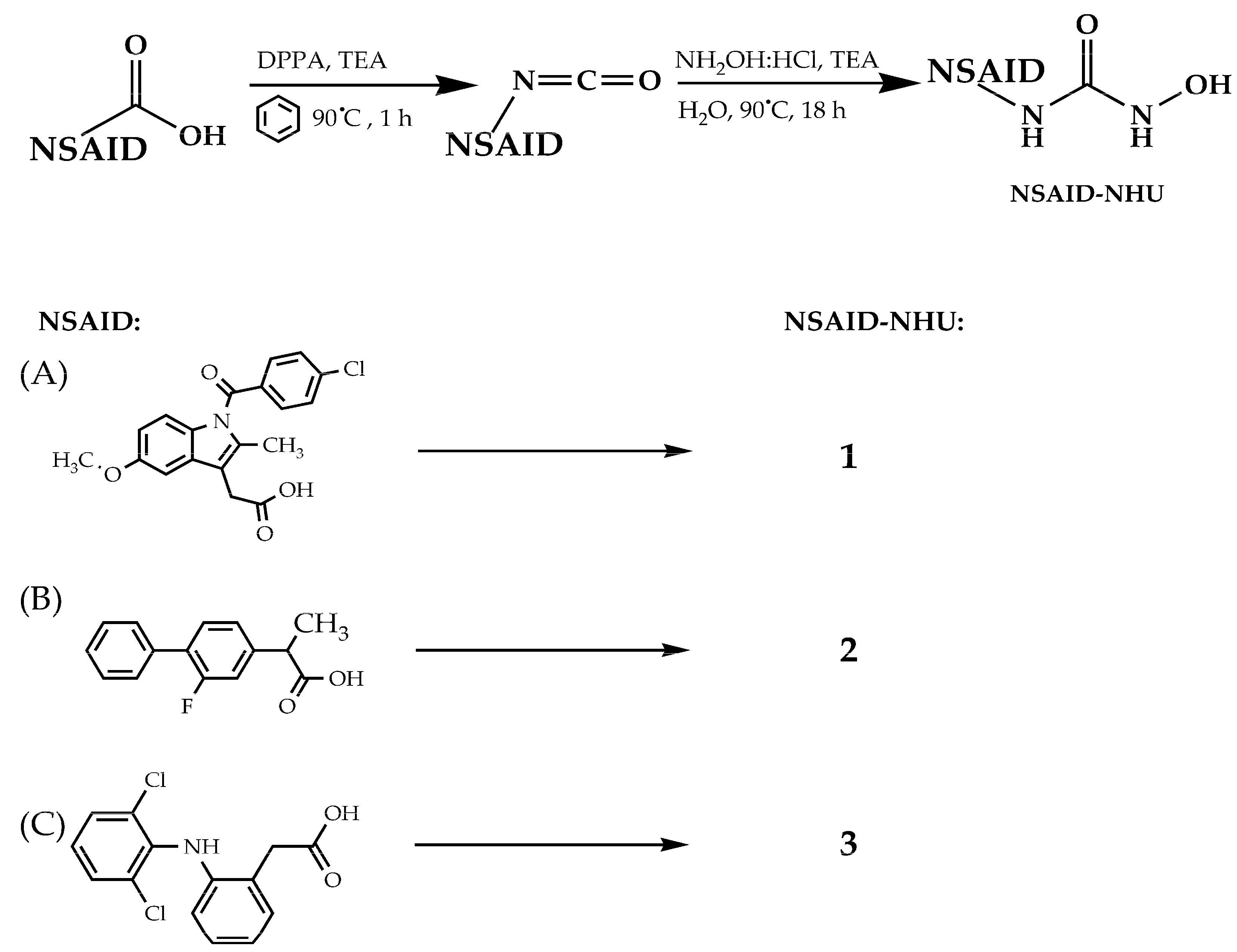
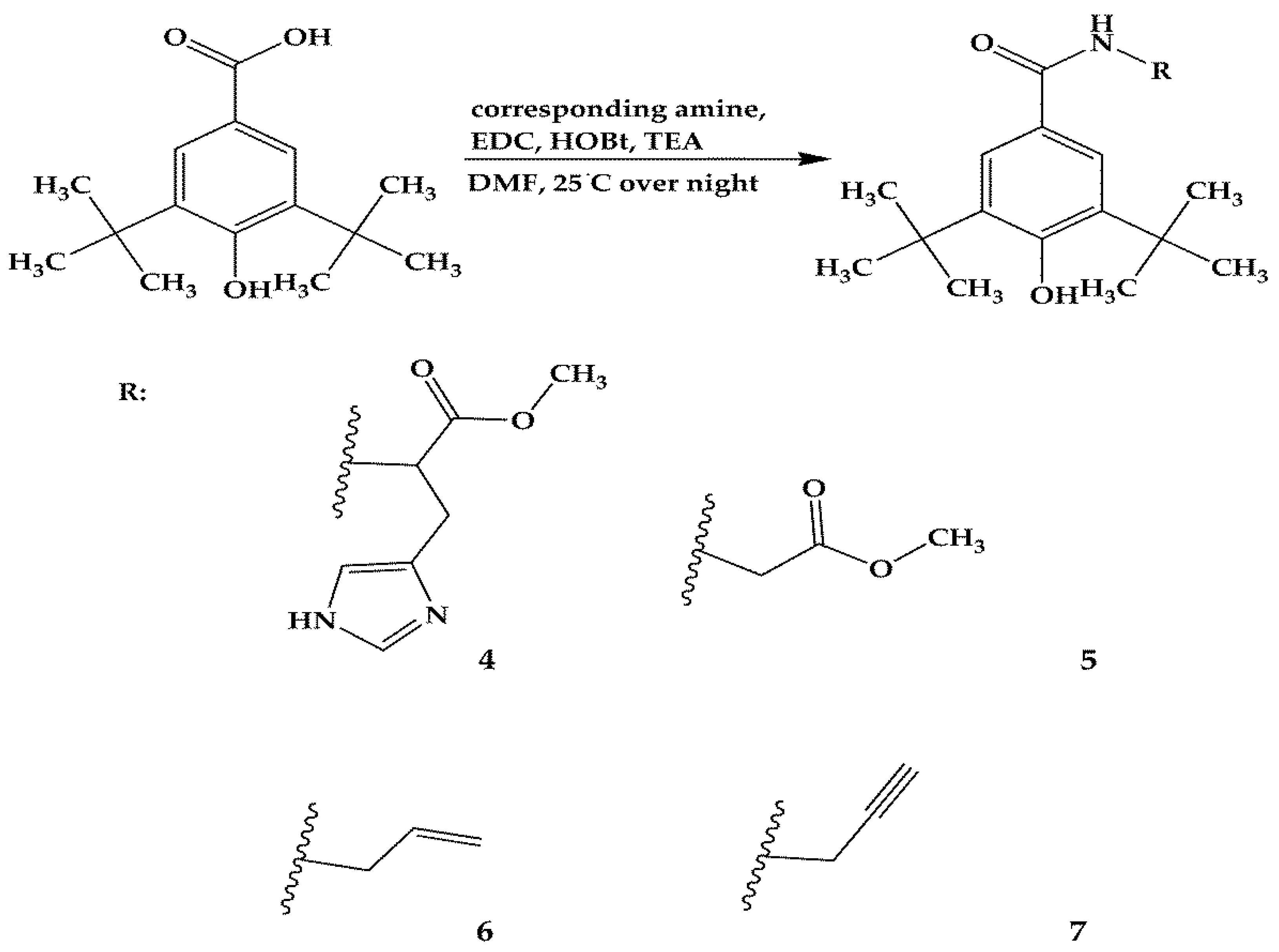
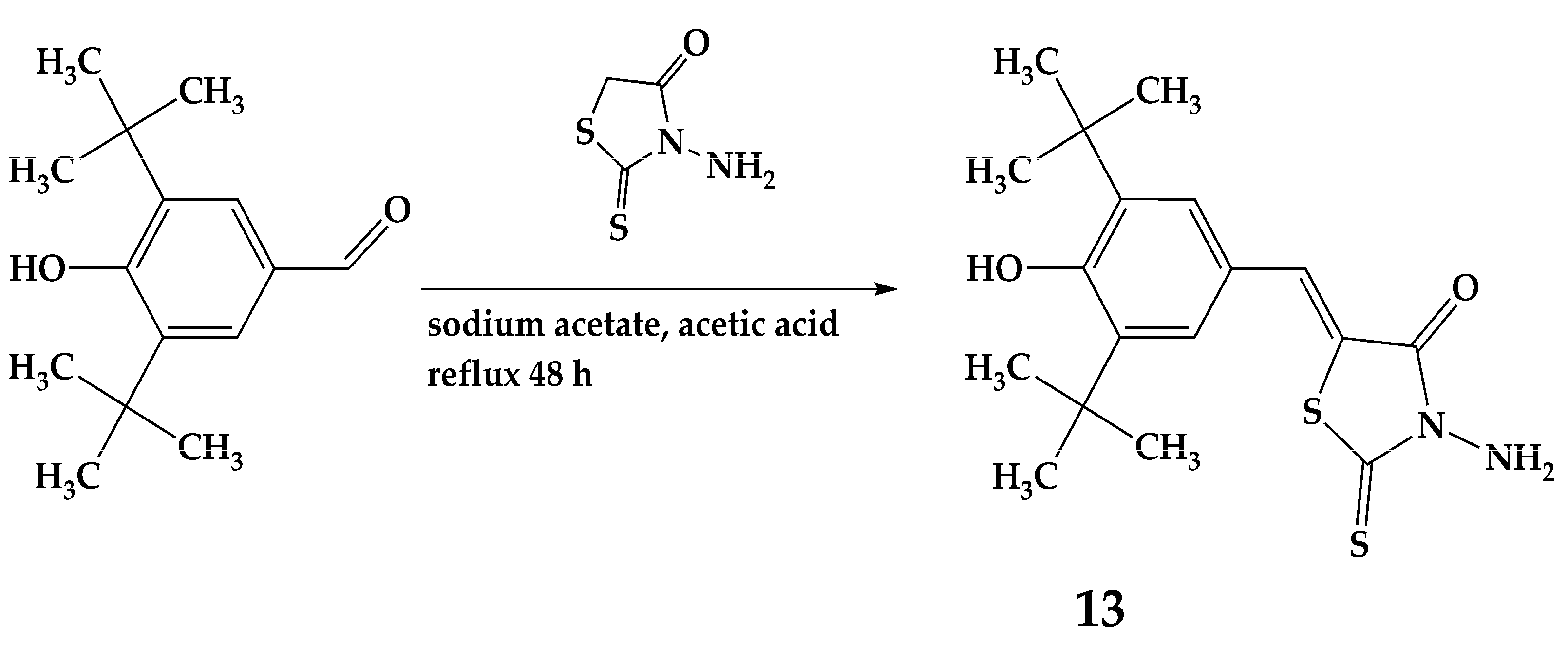
2.1. Design and Synthesis
2.2. In Vitro COX-1, COX-2 and 5-LOX Inhibition Assay
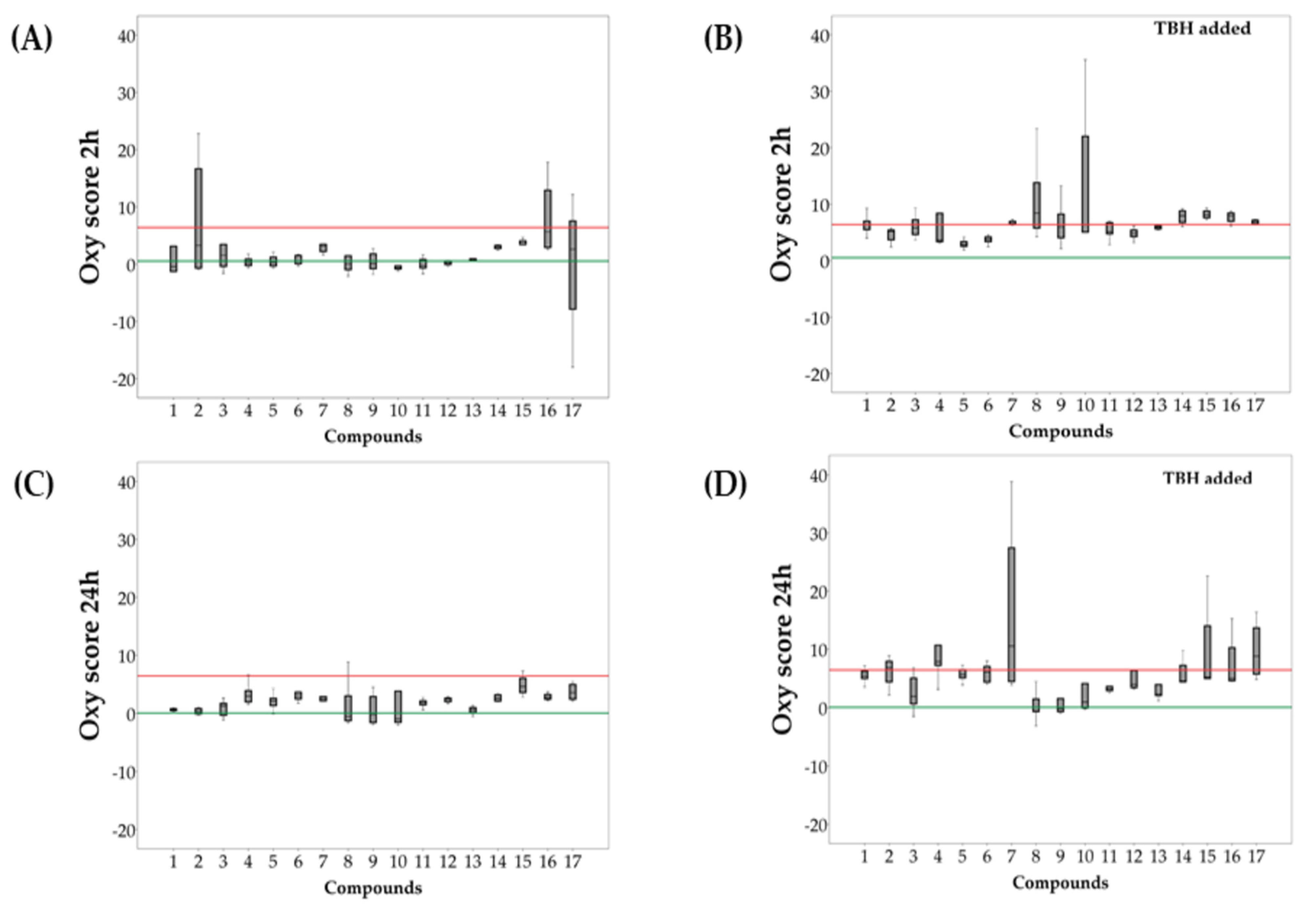
2.3. Evaluation of Redox Activity of Synthesized Compounds
3. Materials and Methods
3.1. General
3.2. Synthesis of Compounds 1–13
3.2.1. Synthesis of Compounds 1–3
3.2.2. Synthesis of Compounds 4–7
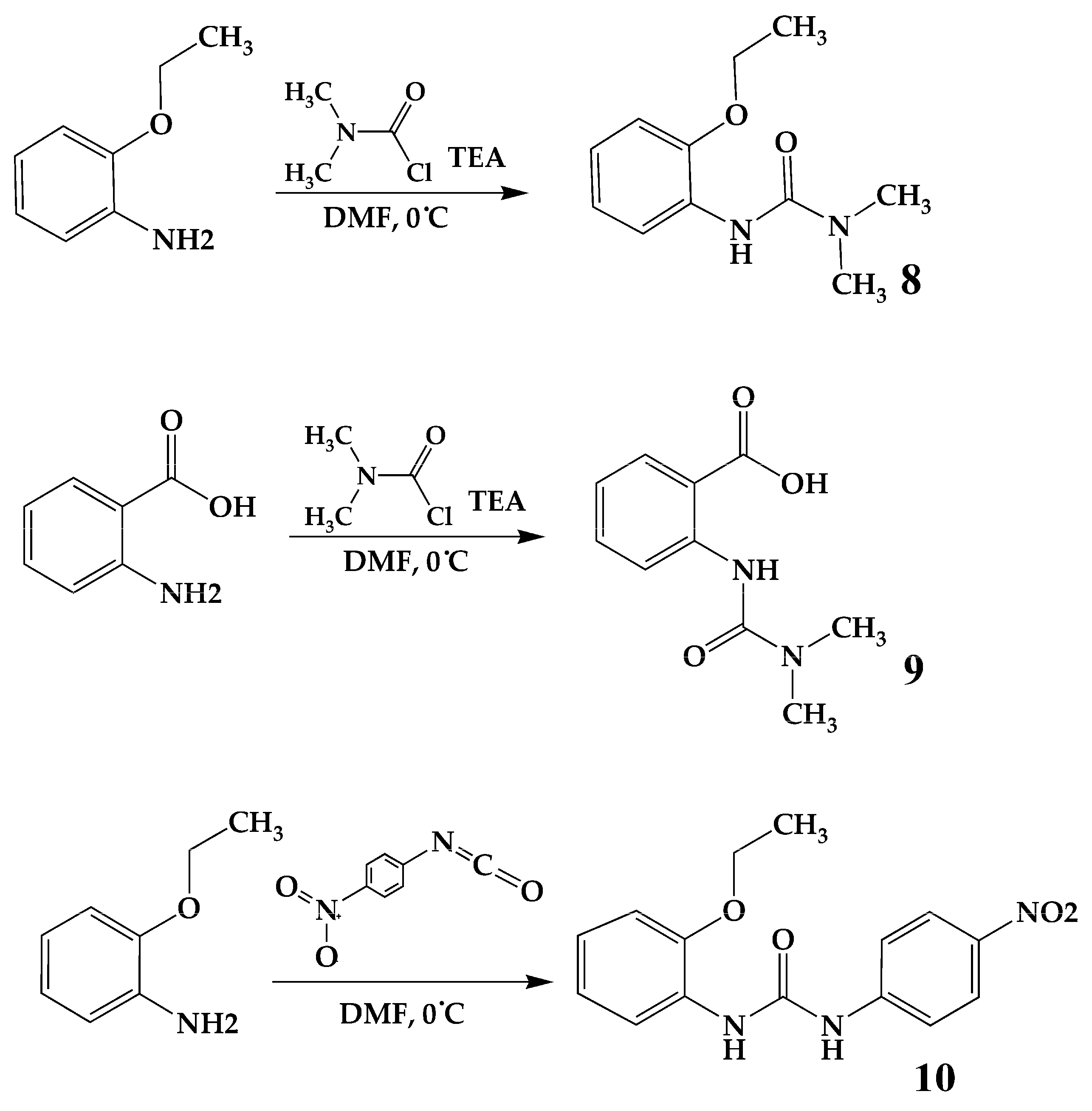
3.2.3. Synthesis of Compounds 8 and 9
3.2.4. Synthesis of Compound 10
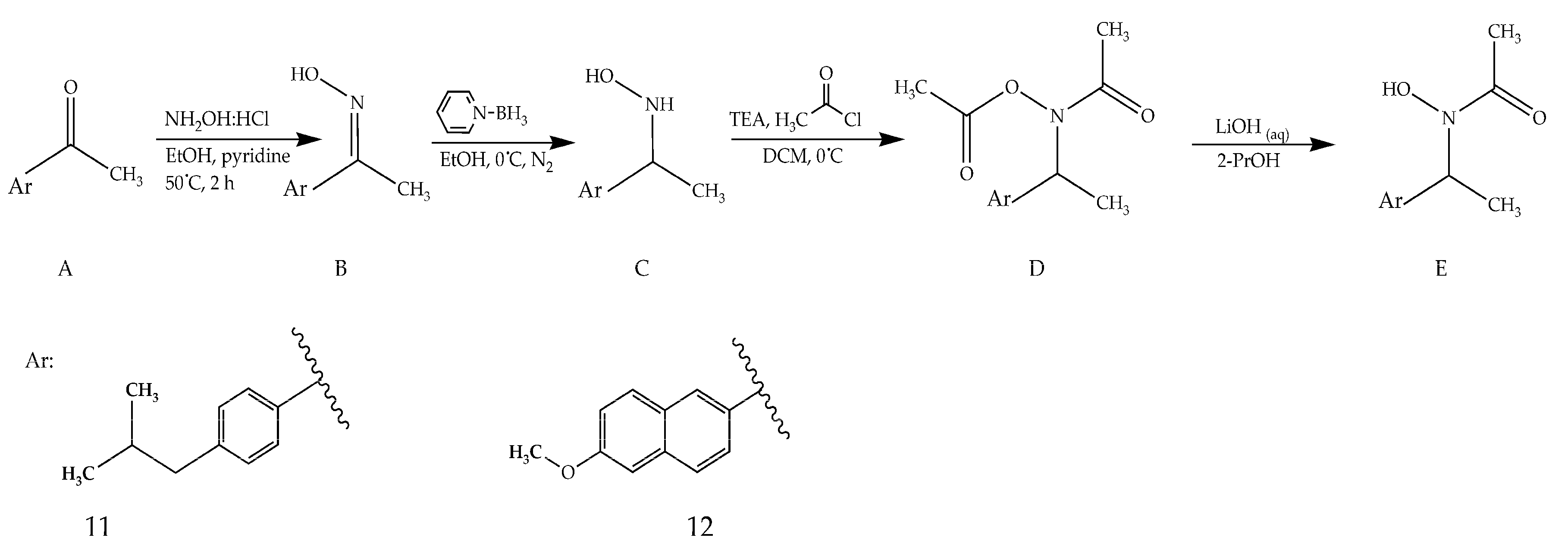
3.2.5. Synthesis of Compounds 11–12
3.2.6. Synthesis of Compound 13
3.3. In Vitro COX-1, COX-2 and 5-LOX Inhibition Assay
3.3.1. COX-1 and COX-2 Inhibition
3.3.2. 5-LOX Inhibition
3.4. Evaluation of Antioxidant Activity of Synthesized Compounds
3.4.1. Sample Collection
3.4.2. Biochemical Parameters
3.4.3. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, R.X.; Zhou, M.; Ma, H.L.; Qiao, Y.B.; Li, Q.S. The Role of Chronic Inflammation in Various Diseases and Anti-Inflammatory Therapies Containing Natural Products. ChemMedChem 2021, 16, 1576–1592. [Google Scholar] [CrossRef]
- P, J.J.; Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer Anti-Inflammatory Therapy through Dual COX-2/5-LOX Inhibitors: A Structure-Based Approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar] [CrossRef]
- Pergola, C.; Werz, O. 5-Lipoxygenase Inhibitors: A Review of Recent Developments and Patents. Expert Opin. Ther. Pat. 2010, 20, 355–375. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.; Ravula, S.; Tatishchev, S.F.; Wang, H.L. Colorectal Carcinoma: Pathologic Aspects. J. Gastrointest. Oncol. 2012, 3, 153–173. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Rao, C.V. The Role of Inflammation in Colon Cancer. In Inflammation and Cancer. Advances in Experimental Medicine and Biology; Aggarwal, B., Sung, B., Gupta, S., Eds.; Springer: Basel, Switzerland, 2014; Volume 816, pp. 25–52. [Google Scholar] [CrossRef]
- Rao, C.V.; Janakiram, N.B.; Mohammed, A. Lipoxygenase and Cyclooxygenase Pathways and Colorectal Cancer Prevention. Curr. Color. Cancer Rep. 2012, 8, 316–324. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Sanidad, K.Z.; Shih, P.-A.; Zhao, X.; Zhang, G. Eicosanoid Signaling in Carcinogenesis of Colorectal Cancer. Cancer Metastasis Rev. 2018, 37, 257–267. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Misra, S.; Ghatak, S.; Patil, N.; Dandawate, P.; Ambike, V.; Adsule, S.; Unni, D.; Venkateswara Swamy, K.; Padhye, S. Novel Dual Cyclooxygenase and Lipoxygenase Inhibitors Targeting Hyaluronan–CD44v6 Pathway and Inducing Cytotoxicity in Colon Cancer Cells. Bioorg. Med. Chem. 2013, 21, 2551–2559. [Google Scholar] [CrossRef]
- Fuchs-Tarlovsky, V. Role of Antioxidants in Cancer Therapy. Nutrition 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Stoia, M.; Oancea, S. Low-Molecular-Weight Synthetic Antioxidants: Classification, Pharmacological Profile, Effectiveness and Trends. Antioxidants 2022, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of Reactive Oxygen Species in Cell Signalling Pathways. Biochem. Soc. Trans. 2001, 29, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Patel, A.K.; Shah, N.; Choudhary, A.K.; Jha, U.K.; Yadav, U.C.; Gupta, P.K.; Pakuwal, U. Oxidative Stress and Antioxidants in Disease and Cancer: A Review. Asian Pac. J. Cancer Prev. 2014, 15, 4405–4409. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Eustress and Oxidative Distress: Introductory Remarks. In Oxidative Stress; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3–12. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxidative Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef]
- Chatterjee, S. Oxidative Stress, Inflammation, and Disease. In Oxidative Stress and Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 35–58. [Google Scholar] [CrossRef]
- Hwang, S.H.; Wecksler, A.T.; Wagner, K.; Hammock, B.D. Rationally Designed Multitarget Agents Against Inflammation and Pain. CMC 2013, 20, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Kolasa, T.; Brooks, C.D.W.; Rodriques, K.E.; Summers, J.B.; Dellaria, J.F.; Hulkower, K.I.; Bouska, J.; Bell, R.L.; Carter, G.W. Nonsteroidal Anti-Inflammatory Drugs as Scaffolds for the Design of 5-Lipoxygenase Inhibitors. J. Med. Chem. 1997, 40, 819–824. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Janusz, J.M.; Young, P.A.; Ridgeway, J.M.; Scherz, M.W.; Enzweiler, K.; Wu, L.I.; Gan, L.; Darolia, R.; Matthews, R.S.; Hennes, D.; et al. New Cyclooxygenase-2/5-Lipoxygenase Inhibitors. 1. 7-Tert-Butyl-2,3-Dihydro-3,3-Dimethylbenzofuran Derivatives as Gastrointestinal Safe Antiinflammatory and Analgesic Agents: Discovery and Variation of the 5-Keto Substituent. J. Med. Chem. 1998, 41, 1112–1123. [Google Scholar] [CrossRef]
- Inukai, S.; Agata, M.; Akiba, K.; Ohmura, T.; Horio, Y.; Ootake, Y.; Sawaki, S.; Goto, M. Aldose reductase inhibitor. EP0388967A1, 26 September 1990. [Google Scholar]
- Dobričić, V.; Marković, B.; Nikolic, K.; Savić, V.; Vladimirov, S.; Čudina, O. 17β-Carboxamide Steroids–in Vitro Prediction of Human Skin Permeability and Retention Using PAMPA Technique. Eur. J. Pharm. Sci. 2014, 52, 95–108. [Google Scholar] [CrossRef]
- Unangst, P.C.; Connor, D.T.; Cetenko, W.A.; Sorenson, R.J.; Kostlan, C.R.; Sircar, J.C.; Wright, C.D.; Schrier, D.J.; Dyer, R.D. Synthesis and Biological Evaluation of 5-[[3,5-Bis(1,1-Dimethylethyl)-4-Hydroxyphenyl]Methylene]Oxazoles, -Thiazoles, and -Imidazoles: Novel Dual 5-Lipoxygenase and Cyclooxygenase Inhibitors with Antiinflammatory Activity. J. Med. Chem. 1994, 37, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Chojkier, M. Treatment and Prevention of Hepatic Disorders. WO0121166A1, 29 March 2001. [Google Scholar]
- Prashanth, M.; Revanasiddappa, H. Synthesis and Antioxidant Activity of Novel Quinazolinones Functionalized with Urea/Thiourea/Thiazole Derivatives as 5-Lipoxygenase Inhibitors. LDDD 2014, 11, 712–720. [Google Scholar] [CrossRef]
- Hwang, S.H.; Wagner, K.M.; Morisseau, C.; Liu, J.-Y.; Dong, H.; Wecksler, A.T.; Hammock, B.D. Synthesis and Structure−Activity Relationship Studies of Urea-Containing Pyrazoles as Dual Inhibitors of Cyclooxygenase-2 and Soluble Epoxide Hydrolase. J. Med. Chem. 2011, 54, 3037–3050. [Google Scholar] [CrossRef]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Socha, J.; Kohout, V. Herbicide Means on the Basis of the Urea and Method of Preparation of the Active Substance. CS189171B1, 30 April 1979. [Google Scholar]
- Goldhamer, D.L.; Onyszkewycz, M.; Wilson, A. Base Dependence in the Selective Nucleophilic Attack of Anthranilic Acid on N, N-Dimethylcarbamoyl Chloride. Tetrahedron Lett. 1968, 9, 4077–4080. [Google Scholar] [CrossRef]
- Staiger, R.P.; Miller, E.B. Isatoic Anhydride. IV. Reactions with Various Nucleophiles. J. Org. Chem. 1959, 24, 1214–1219. [Google Scholar] [CrossRef]
- Summers, J.B.; Gunn, B.P.; Martin, J.G.; Martin, M.B.; Mazdiyasni, H.; Stewart, A.O.; Young, P.R.; Bouska, J.B.; Goetze, A.M. Structure-Activity Analysis of a Class of Orally Active Hydroxamic Acid Inhibitors of Leukotriene Biosynthesis. J. Med. Chem. 1988, 31, 1960–1964. [Google Scholar] [CrossRef]
- Yehye, W.A.; Rahman, N.A.; Ariffin, A.; Abd Hamid, S.B.; Alhadi, A.A.; Kadir, F.A.; Yaeghoobi, M. Understanding the Chemistry behind the Antioxidant Activities of Butylated Hydroxytoluene (BHT): A Review. Eur. J. Med. Chem. 2015, 101, 295–312. [Google Scholar] [CrossRef]
- Wang, X.; Wu, L.; Aouffen, M.; Mateescu, M.-A.; Nadeau, R.; Wang, R. Novel Cardiac Protective Effects of Urea: From Shark to Rat: Cardioprotective Effects of Urea and Its Derivatives. Br. J. Pharmacol. 1999, 128, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Erel, O. A Novel Automated Direct Measurement Method for Total Antioxidant Capacity Using a New Generation, More Stable ABTS Radical Cation. Clin. Biochem. 2004, 37, 277–285. [Google Scholar] [CrossRef]
- Kotur-Stevuljevic, J.; Bogavac-Stanojevic, N.; Jelic-Ivanovic, Z.; Stefanovic, A.; Gojkovic, T.; Joksic, J.; Sopic, M.; Gulan, B.; Janac, J.; Milosevic, S. Oxidative Stress and Paraoxonase 1 Status in Acute Ischemic Stroke Patients. Atherosclerosis 2015, 241, 192–198. [Google Scholar] [CrossRef]
- Erel, O. A New Automated Colorimetric Method for Measuring Total Oxidant Status. Clin. Biochem. 2005, 38, 1103–1111. [Google Scholar] [CrossRef]
- Alamdari, D.H.; Paletas, K.; Pegiou, T.; Sarigianni, M.; Befani, C.; Koliakos, G. A Novel Assay for the Evaluation of the Prooxidant–Antioxidant Balance, before and after Antioxidant Vitamin Administration in Type II Diabetes Patients. Clin. Biochem. 2007, 40, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COX-2 IC50 (µM) | 5-LOX IC50 (µM) | COX-1 IC50 (µM) |
|---|---|---|---|
| 1 | 18.28 ± 2.17 | 5.71 ± 0.15 | >100 |
| 2 | 6.72 ± 0.79 | 1.62 ± 0.67 | >100 |
| 3 | 5.26 ± 0.34 | 1.73 ± 0.64 | >100 |
| 4 | >100 | 8.10 ± 2.12 | Not tested |
| 5 | 10.64 ± 0.80 | 9.30 ± 1.87 | >100 |
| 6 | 6.89 ± 0.83 | 53.84 ± 11.87 | >100 |
| 7 | >100 | 12.55 ± 3.36 | Not tested |
| 8 | >100 | 14.52 ± 3.62 | Not tested |
| 9 | >100 | >100 | Not tested |
| 10 | >100 | 13.01 ± 2.67 | Not tested |
| 11 | 36.18 ± 3.08 | 1.04 ± 0.22 | >100 |
| 12 | 83.42 ± 4.37 | 1.29 ± 0.10 | >100 |
| 13 | >100 | 13.58 ± 3.54 | Not tested |
| Celecoxib | 0.07 ± 0.01 | Not tested | Not tested |
| Zileuton | Not tested | 0.36 ± 0.10 | Not tested |
| 2 h Incubation | 24 h Incubation | |||||
|---|---|---|---|---|---|---|
| TBH added | p | TBH added | p | |||
| 1 | −0.43 (−1.27–3.13) | 6.44 (5.51–7.02) | 0.023 | 0.71 (0.51–0.80) | 5.46 (4.96–6.20) | <0.001 |
| 2 | 3.28 (−0.68–16.66) | 5.20 (3.70–5.42) | 0.597 | 0.33 (−0.07–0.85) | 6.79 (4.37–7.90) | <0.001 |
| 3 | 1.58 (−0.38–3.45) | 5.81 (4.64–7.25) | 0.023 | 1.31 (−0.31–1.75) | 1.93 (0.65–5.02) # | 0.226 |
| 4 | 0.24 (−0.24–0.89) | 3.68 (3.36–8.40) | 0.001 | 3.00 (1.99–3.93) ** | 7.86 (7.20–10.68) | 0.016 |
| 5 | 0.29 (−0.30–1.23) | 2.83 (2.55–3.39) | <0.001 | 2.14 (1.37–2.62) ** | 5.64 (5.07–6.47) | <0.001 |
| 6 | 0.52 (0.08–1.52) | 3.77 (3.37–4.20) | 0.002 | 3.09 (2.50–3.67) | 6.07 (4.34–7.02) | 0.016 |
| 7 | 3.12 (2.21–3.47) | 6.62 (6.42–6.98) | 0.021 | 2.55 (2.18–2.92) | 10.54 (4.45–27.42) | 0.021 |
| 8 | 0.03 (−0.96–1.46) | 8.43 (5.76–13.81) | 0.010 | −0.52 (−1.24–3.03) | −0.55 (−0.67–1.48) ## | 0.762 |
| 9 | 0.09 (−0.79–1.80) | 5.97 (4.06–8.25) | 0.003 | −0.18 (−1.55–2.91) | −0.19 (−0.68–1.56) | 0.450 |
| 10 | −0.64 (−0.84−0.30) | 6.34 (5.07–22.02) | <0.001 | −0.97 (−1.53–3.85) | 0.97 (−0.04–4.08) | 0.199 |
| 11 | −0.22 (−0.65–0.82) | 5.15 (4.76–6.74) | <0.001 | 1.83 (1.49–2.18) * | 3.18 (2.92–3.60) ## | <0.001 |
| 12 | 0.24 (−0.06–0.58) | 4.79 (4.25–5.43) | <0.001 | 2.49 (2.03–2.66) ** | 3.97 (3.44–6.33) | <0.001 |
| 13 | 0.75 (0.56–0.94) | 5.89 (5.63–6.14) | <0.001 | 0.64 (0.19–1.01) | 2.35 (2.10–3.92) # | <0.001 |
| BHT | 3.08 (2.73–3.26) | 7.98 (6.77–8.81) | 0.021 | 2.62 (2.14–3.22) | 4.51 (4.38–7.22) | 0.021 |
| Celecoxib | 5.66 (2.92–12.90) | 7.94 (6.97–8.45) | 0.564 | 2.77 (2.46–3.27) | 4.95 (4.57–10.25) | 0.021 |
| Zileuton | 2.60 (−7.86–7.54) | 6.87 (6.52–7.18) | 0.248 | 3.61 (2.47–4.93) | 8.80 (5.71–13.66) | 0.043 |
| Urea | 3.48 (3.44–4.08) | 8.11 (7.61–8.79) | 0.021 | 4.60 (3.62–6.02) | 5.19 (4.96–14.00) | 0.149 |
| Trolox | 0.54 (−0.66–2.43) | / | / | 0.07 (−0.59–0.59) * | / | / |
| TBH | 6.40 (4.66–11.59) | / | / | 6.43 (4.73–12.00) | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bošković, J.; Dobričić, V.; Mihajlović, M.; Kotur-Stevuljević, J.; Čudina, O. Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals 2023, 16, 549. https://doi.org/10.3390/ph16040549
Bošković J, Dobričić V, Mihajlović M, Kotur-Stevuljević J, Čudina O. Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals. 2023; 16(4):549. https://doi.org/10.3390/ph16040549
Chicago/Turabian StyleBošković, Jelena, Vladimir Dobričić, Marija Mihajlović, Jelena Kotur-Stevuljević, and Olivera Čudina. 2023. "Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors" Pharmaceuticals 16, no. 4: 549. https://doi.org/10.3390/ph16040549
APA StyleBošković, J., Dobričić, V., Mihajlović, M., Kotur-Stevuljević, J., & Čudina, O. (2023). Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals, 16(4), 549. https://doi.org/10.3390/ph16040549