
HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor-γ Activation in THP-1 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
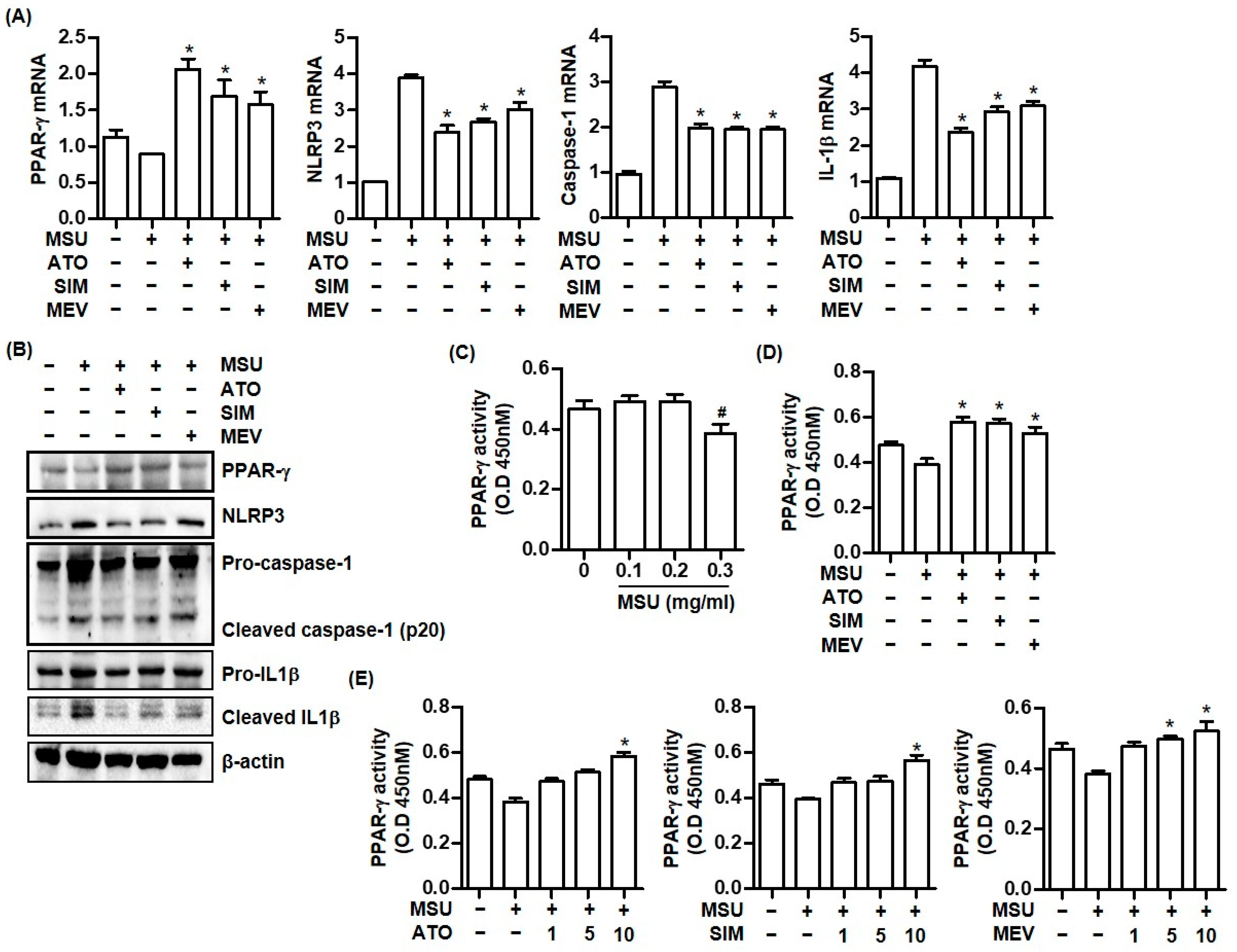
2.1. MSU Crystals Downregulate PPAR-γ Expression in THP-1 Cells
2.2. Statins Increase PPAR-γ Expression and Suppress the NLRP3 Inflammasome Activation Caused by MSU Crystal Stimulation
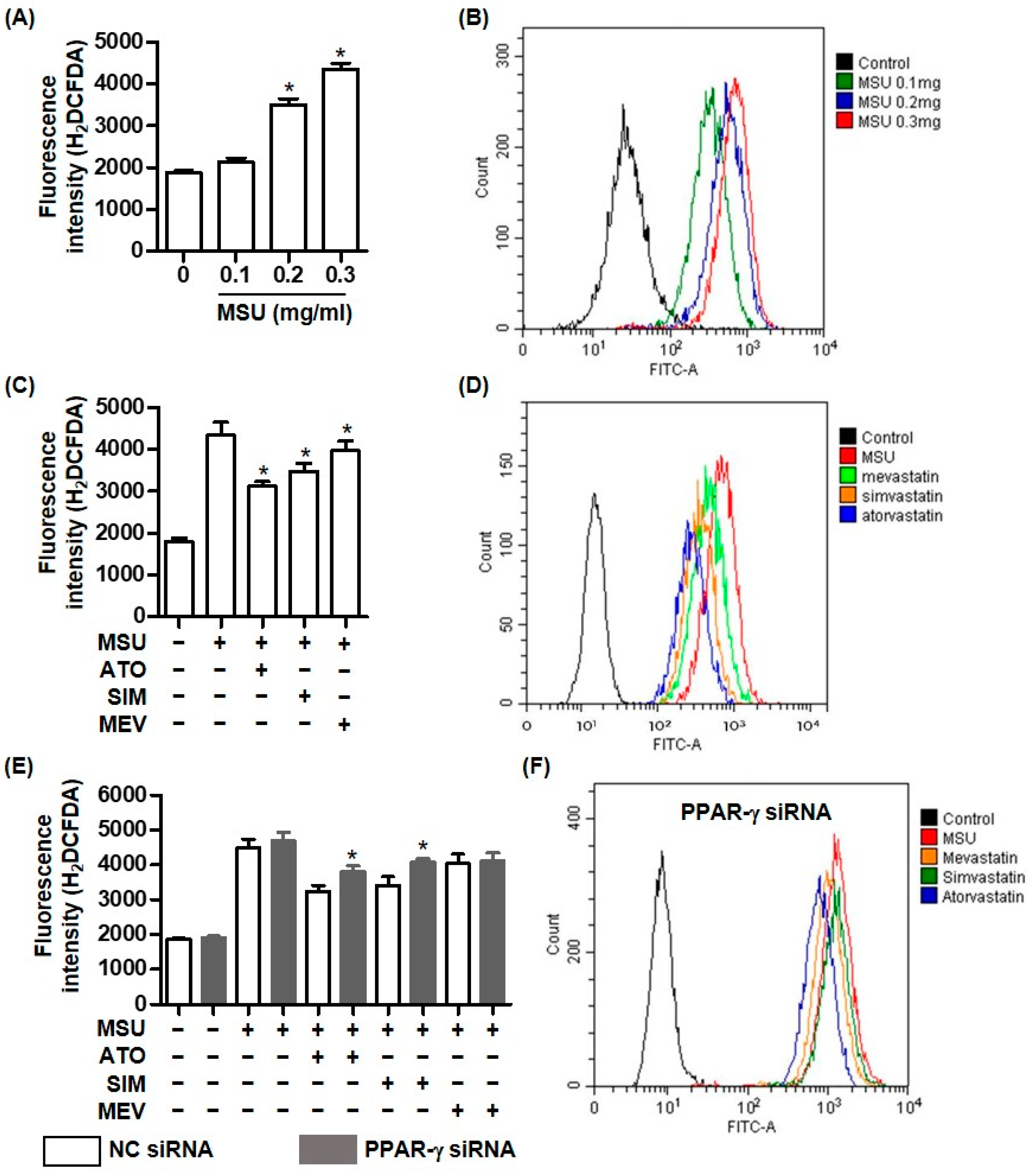
2.3. PPAR-γ Deficiency Attenuated the Inhibitory Effect of Statins on MSU Crystal-Mediated NLRP3 Inflammasome Activation
2.4. The Inhibitory Effect of Statins on Intracellular ROS Generation Depends on PPAR-γ in MSU Crystal-Mediated Inflammation
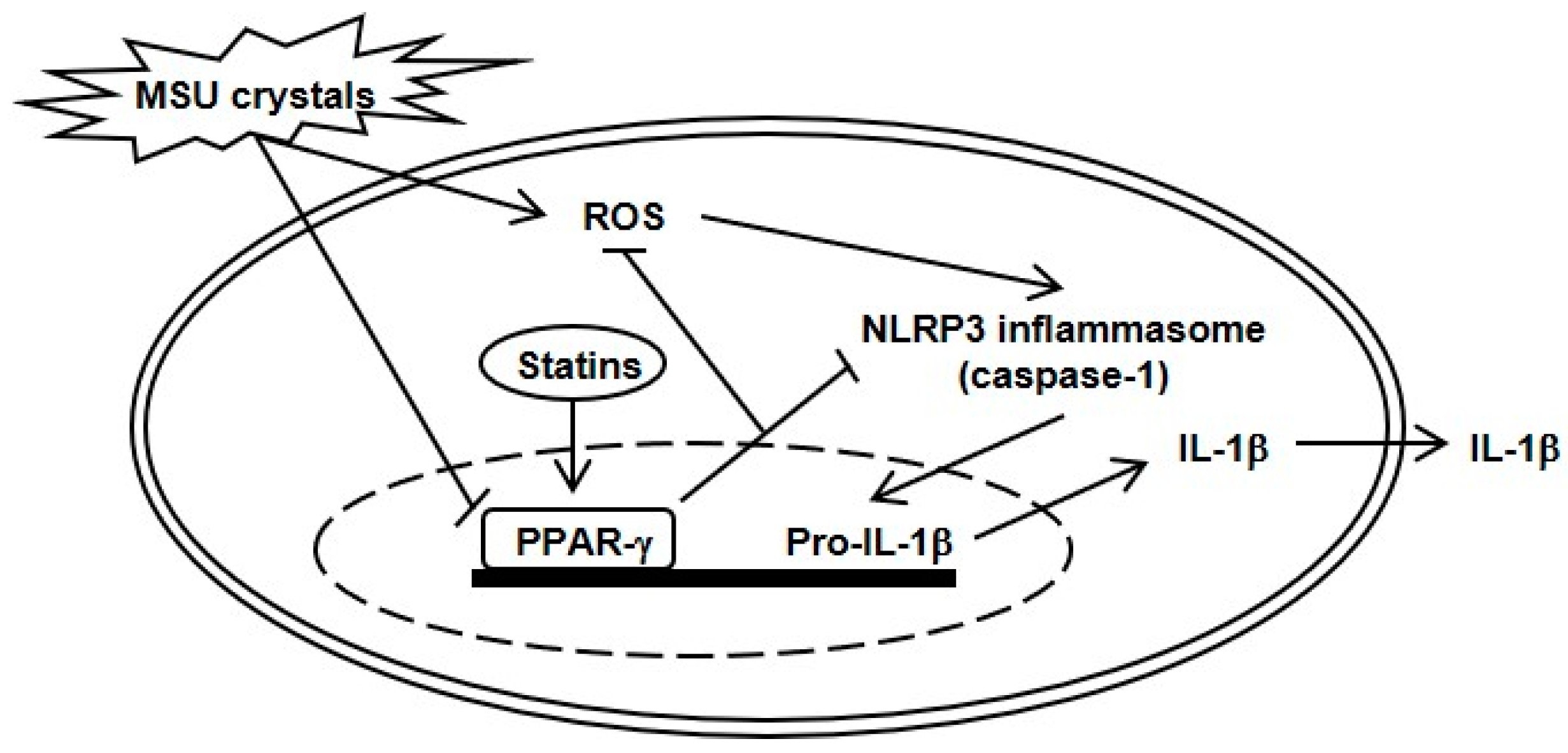
3. Discussion

4. Materials and Methods
4.1. Cell Culture
4.2. Measurement of Intracellular Reactive Oxygen Species (ROS)
4.3. Flow Cytometry Analysis of ROS
4.4. RNA Isolation and Quantitative Real Time-Polymerase Chain Reaction (RT-PCR)
4.5. Transfection of siRNA
4.6. Western Blotting
4.7. PPAR-γ Activity Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Wang, J.; Liu, H.; Kwong, J.S.; Chen, H.; Li, L.; Chung, S.C.; Shah, A.; Chen, Y.; et al. Diagnosis and treatment for hyperuricemia and gout: A systematic review of clinical practice guidelines and consensus statements. BMJ Open 2019, 9, e026677. [Google Scholar] [CrossRef]
- Keenan, R.T. The biology of urate. Semin. Arthritis Rheum. 2020, 50, S2–S10. [Google Scholar] [CrossRef] [PubMed]
- So, A.K.; Martinon, F. Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K. The mechanism of the NLRP3 inflammasome activation and pathogenic implication in the pathogenesis of gout. J. Rheum. Dis. 2022, 29, 140–153. [Google Scholar] [CrossRef]
- Alatshan, A.; Benkő, S. Nuclear Receptors as Multiple Regulators of NLRP3 Inflammasome Function. Front. Immunol. 2021, 12, 630569. [Google Scholar] [CrossRef]
- Janani, C.; Ranjitha Kumari, B.D. PPAR γ gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Miao, H.; Ou, J.; Ma, Y.; Guo, F.; Yang, Z.; Wiggins, M.; Liu, C.; Song, W.; Han, X.; Wang, M.; et al. Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep. 2014, 7, 223–235. [Google Scholar] [CrossRef]
- Kumar, N.; Gupta, G.; Anilkumar, K.; Fatima, N.; Karnati, R.; Reddy, G.V.; Giri, P.V.; Reddanna, P. 15-Lipoxygenase metabolites of α-linolenic acid, [13-(S)-HPOTrE and 13-(S)-HOTrE], mediate anti-inflammatory effects by inactivating NLRP3 inflammasome. Sci. Rep. 2016, 6, 31649. [Google Scholar] [CrossRef]
- Luo, H.; Fan, Z.; Xiang, D.; Jiang, Z.; Zhang, W.; Gao, L.; Feng, C. The protective effect of umbelliferone ameliorates myocardial injury following ischemia-reperfusion in the rat through suppression NLRP3 inflammasome and upregulating the PPAR-γ. Mol. Med. Rep. 2018, 17, 3404–3410. [Google Scholar] [CrossRef]
- Wang, X.; Li, R.; Wang, X.; Fu, Q.; Ma, S. Umbelliferone ameliorates cerebral ischemia-reperfusion injury via upregulating the PPAR γ expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 2015, 600, 182–187. [Google Scholar] [CrossRef]
- Yang, C.C.; Wu, C.H.; Lin, T.C.; Cheng, Y.N.; Chang, C.S.; Lee, K.T.; Tsai, P.J.; Tsai, Y.S. Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics 2021, 11, 2424–2441. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.I.; Muniyappa, R.; Francisco, R.; Sowers, J.R. Clinical review 145: Pleiotropic effects of statins: Lipid reduction and beyond. J. Clin. Endocrinol. Metab. 2002, 87, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Yano, M.; Matsumura, T.; Senokuchi, T.; Ishii, N.; Murata, Y.; Taketa, K.; Motoshima, H.; Taguchi, T.; Sonoda, K.; Kukidome, D.; et al. Statins activate peroxisome proliferator-activated receptor γ through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ. Res. 2007, 100, 1442–1451. [Google Scholar] [CrossRef]
- Fukuda, K.; Matsumura, T.; Senokuchi, T.; Ishii, N.; Kinoshita, H.; Yamada, S.; Murakami, S.; Nakao, S.; Motoshima, H.; Kondo, T.; et al. Statins meditate anti-atherosclerotic action in smooth muscle cells by peroxisome proliferator-activated receptor-γ activation. Biochem. Biophys. Res. Commun. 2015, 457, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Grip, O.; Janciauskiene, S.; Lindgren, S. Atorvastatin activates PPAR-γ and attenuates the inflammatory response in human monocytes. Inflamm. Res. 2002, 51, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, X.; Lei, T.; Zhang, K.; Lai, B.; Zhang, Z.; Guan, Y.; Mao, G.; Xiao, L.; Wang, N. Statins Attenuate Activation of the NLRP3 Inflammasome by Oxidized LDL or TNFα in Vascular Endothelial Cells through a PXR-Dependent Mechanism. Mol. Pharmacol. 2017, 92, 256–264. [Google Scholar] [CrossRef]
- Kong, Y.; Feng, W.; Zhao, X.; Zhang, P.; Li, S.; Li, Z.; Lin, Y.; Liang, B.; Li, C.; Wang, W.; et al. Statins ameliorate cholesterol-induced inflammation and improve AQP2 expression by inhibiting NLRP3 activation in the kidney. Theranostics 2020, 10, 10415–10433. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Hu, S.; Zou, J.; Xiao, J.; Zhang, X.; Fu, C.; Feng, X.; Ye, Z. Peroxisome proliferator-activated receptor γ prevents the production of NOD-like receptor family, pyrin domain containing 3 inflammasome and interleukin 1β in HK-2 renal tubular epithelial cells stimulated by monosodium urate crystals. Mol. Med. Rep. 2015, 12, 6221–6226. [Google Scholar] [CrossRef]
- Akahoshi, T.; Namai, R.; Murakami, Y.; Watanabe, M.; Matsui, T.; Nishimura, A.; Kitasato, H.; Kameya, T.; Kondo, H. Rapid induction of peroxisome proliferator-activated receptor γ expression in human monocytes by monosodium urate monohydrate crystals. Arthritis Rheum. 2003, 48, 231–239. [Google Scholar] [CrossRef]
- Wang, X.; Deng, J.; Xiong, C.; Chen, H.; Zhou, Q.; Xia, Y.; Shao, X.; Zou, H. Treatment with a PPAR-γ Agonist Protects Against Hyperuricemic Nephropathy in a Rat Model. Drug Des. Devel. Ther. 2020, 14, 2221–2233. [Google Scholar] [CrossRef]
- Niu, S.W.; Chang, K.T.; Lin, H.Y.; Kuo, I.C.; Chang, Y.H.; Chen, Y.H.; Hung, C.C.; Chiu, Y.W.; Hwang, S.J. Decreased incidence of gout in diabetic patients using pioglitazone. Rheumatology 2018, 57, 92–99. [Google Scholar] [CrossRef]
- Maalouf, N.M.; Poindexter, J.R.; Adams-Huet, B.; Moe, O.W.; Sakhaee, K. Increased production and reduced urinary buffering of acid in uric acid stone formers is ameliorated by pioglitazone. Kidney Int. 2019, 95, 1262–1268. [Google Scholar] [CrossRef]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the Innate Immune System Mediate the Resolution of Inflammation. PPAR. Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.P.; Liu, Y.H.; Chen, J.; Song, T.; You, Y.; Tang, Z.Y.; Li, Y.J.; Zhang, G.G. Rosiglitazone attenuates NF-κB-dependent ICAM-1 and TNF-α production caused by homocysteine via inhibiting ERK1/2/p38MAPK activation. Biochem. Biophys. Res. Commun. 2007, 360, 20–26. [Google Scholar] [CrossRef]
- Dasu, M.R.; Park, S.; Devaraj, S.; Jialal, I. Pioglitazone inhibits Toll-like receptor expression and activity in human monocytes and db/db mice. Endocrinology 2009, 150, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.Q.; Feng, Z.C.; Zhang, X.L.; Hu, L.Q.; Wang, M.; Zhang, H.F.; Li, S.M. PPAR-γ Activation Exerts an Anti-inflammatory Effect by Suppressing the NLRP3 Inflammasome in Spinal Cord-Derived Neurons. Mediat. Inflamm. 2019, 2019, 6386729. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Schoonjans, K.; Gelman, L.; Kim, J.B.; Najib, J.; Martin, G.; Fruchart, J.C.; Briggs, M.; Spiegelman, B.M.; Auwerx, J. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: Implications for adipocyte differentiation and metabolism. Mol. Cell. Biol. 1999, 19, 5495–5503. [Google Scholar] [CrossRef]
- Moon, H.J.; Kim, S.E.; Yun, Y.P.; Hwang, Y.S.; Bang, J.B.; Park, J.H.; Kwon, I.K. Simvastatin inhibits osteoclast differentiation by scavenging reactive oxygen species. Exp. Mol. Med. 2011, 43, 605–612. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Zeiher, A.M.; Dimmeler, S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: A novel vasculoprotective function of statins. Circulation 2004, 110, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Jin, S.M.; Lee, M.K.; Kim, U.H.; Lee, Y.C. Peroxisome proliferator activated receptor-γ modulates reactive oxygen species generation and activation of nuclear factor-κB and hypoxia-inducible factor 1alpha in allergic airway disease of mice. J. Allergy Clin. Immunol. 2006, 118, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Hua, B.; Liu, Q.; Gao, S.; Li, W.; Li, H. Protective role of activating PPARγ in advanced glycation end products-induced impairment of coronary artery vasodilation via inhibiting p38 phosphorylation and reactive oxygen species production. Biomed. Pharm. 2022, 147, 112641. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef]
- Paumelle, R.; Staels, B. Peroxisome proliferator-activated receptors mediate pleiotropic actions of statins. Circ. Res. 2007, 100, 1394–1395. [Google Scholar] [CrossRef]
- Shou, X.; Zhou, R.; Zhu, L.; Ren, A.; Wang, L.; Wang, Y.; Zhou, J.; Liu, X.; Wang, B.; Emodin, A. Chinese Herbal Medicine, Inhibits Reoxygenation-Induced Injury in Cultured Human Aortic Endothelial Cells by Regulating the Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) and Endothelial Nitric Oxide Synthase (eNOS) Signaling Pathway. Med. Sci. Monit. 2018, 24, 643–651. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-K.; Choe, J.-Y.; Kim, J.-W.; Park, K.-Y. HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor-γ Activation in THP-1 Cells. Pharmaceuticals 2023, 16, 522. https://doi.org/10.3390/ph16040522
Kim S-K, Choe J-Y, Kim J-W, Park K-Y. HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor-γ Activation in THP-1 Cells. Pharmaceuticals. 2023; 16(4):522. https://doi.org/10.3390/ph16040522
Chicago/Turabian StyleKim, Seong-Kyu, Jung-Yoon Choe, Ji-Won Kim, and Ki-Yeun Park. 2023. "HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor-γ Activation in THP-1 Cells" Pharmaceuticals 16, no. 4: 522. https://doi.org/10.3390/ph16040522
APA StyleKim, S.-K., Choe, J.-Y., Kim, J.-W., & Park, K.-Y. (2023). HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor-γ Activation in THP-1 Cells. Pharmaceuticals, 16(4), 522. https://doi.org/10.3390/ph16040522