
Rapid and Efficient Access to Novel Bio-Inspired 3-Dimensional Tricyclic SpiroLactams as Privileged Structures via Meyers’ Lactamization
 , , , and
, , , and
Abstract
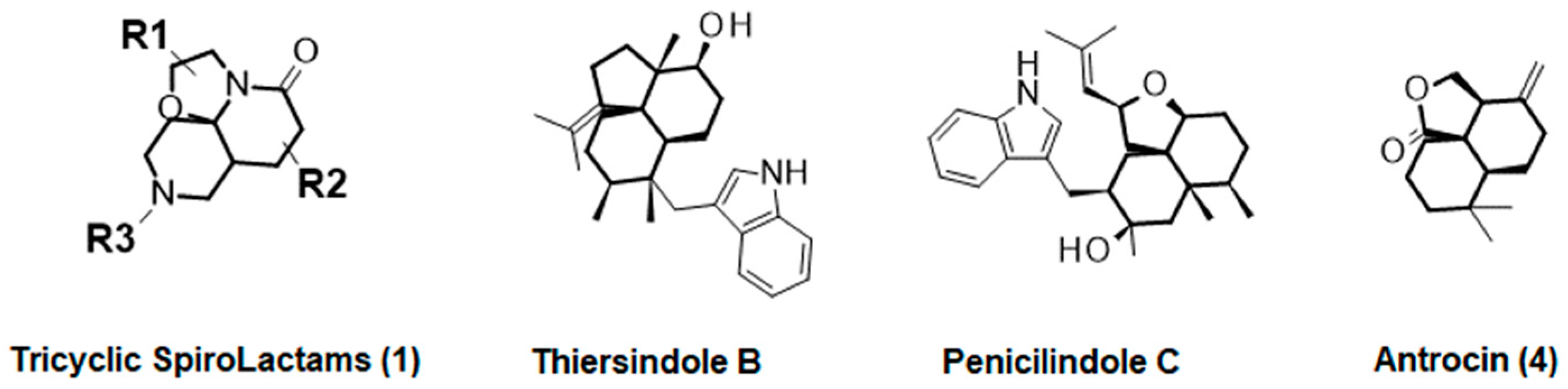
1. Introduction
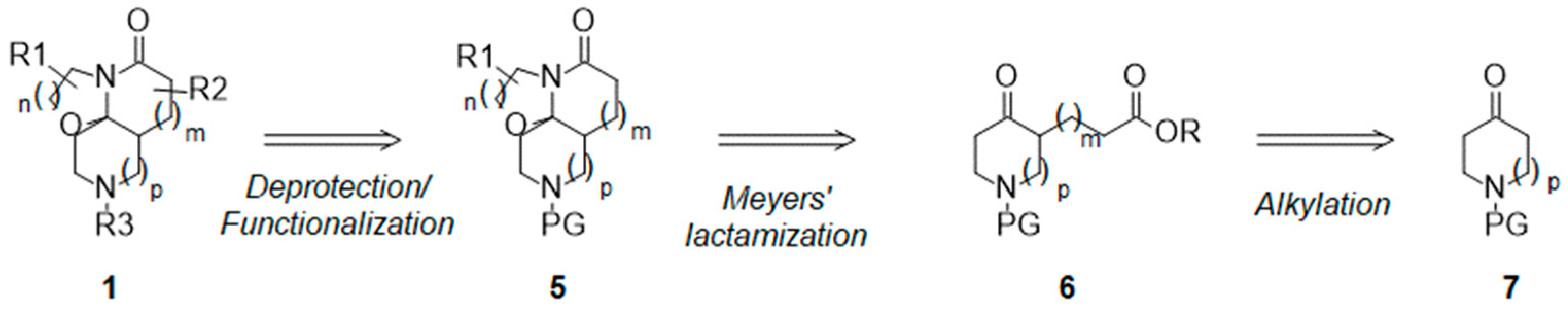
2. Results and Discussion
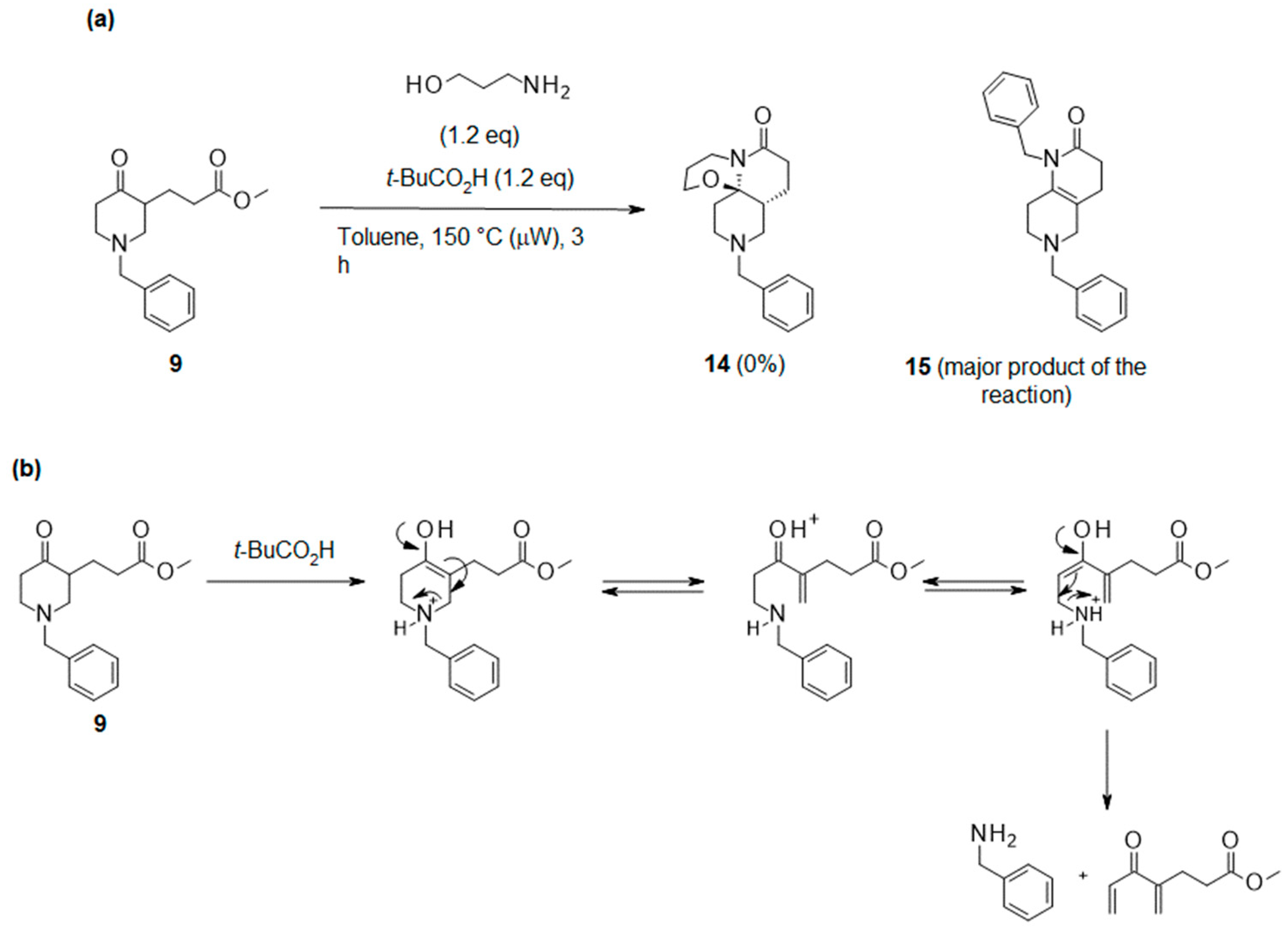
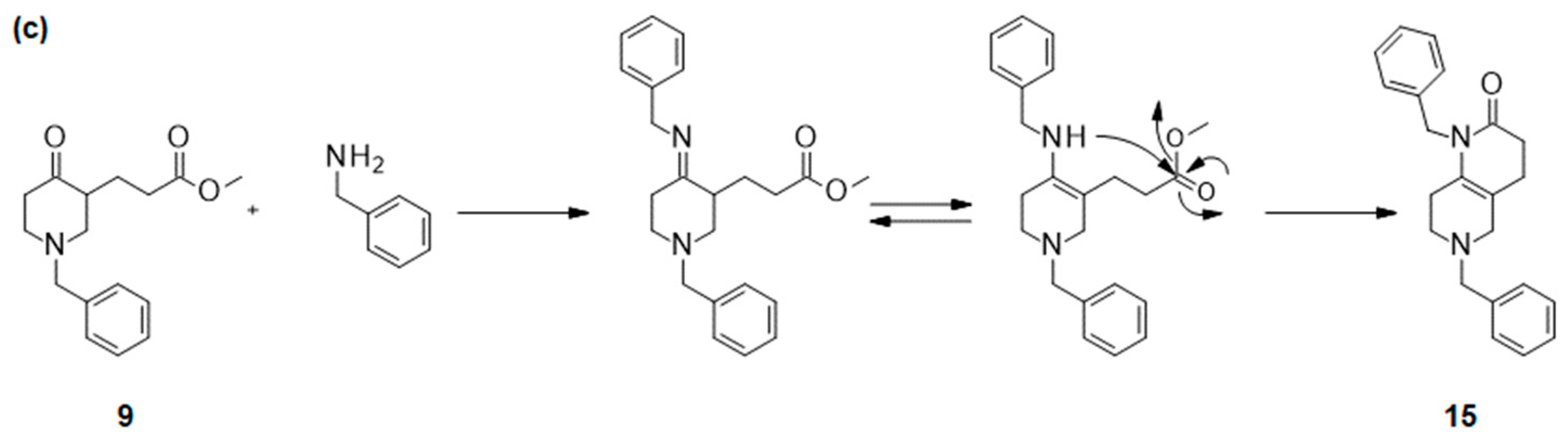
2.1. Optimization of Meyers’ Lactamization Reaction
2.2. Scope of Meyers’ Lactamization Reaction
2.2.1. Synthesis of Lactams with Octahydrooxazolo [2,3-j][1,6]naphthyridin-5-ones Core ([5.6.6] Ring System) from Benzyl-Keto-Ester
2.2.2. Synthesis of Lactams with Decahydro-[1,3]oxazino[2,3-j][1,6]naphthyridin-6-one core ([6,6,6] Ring System)
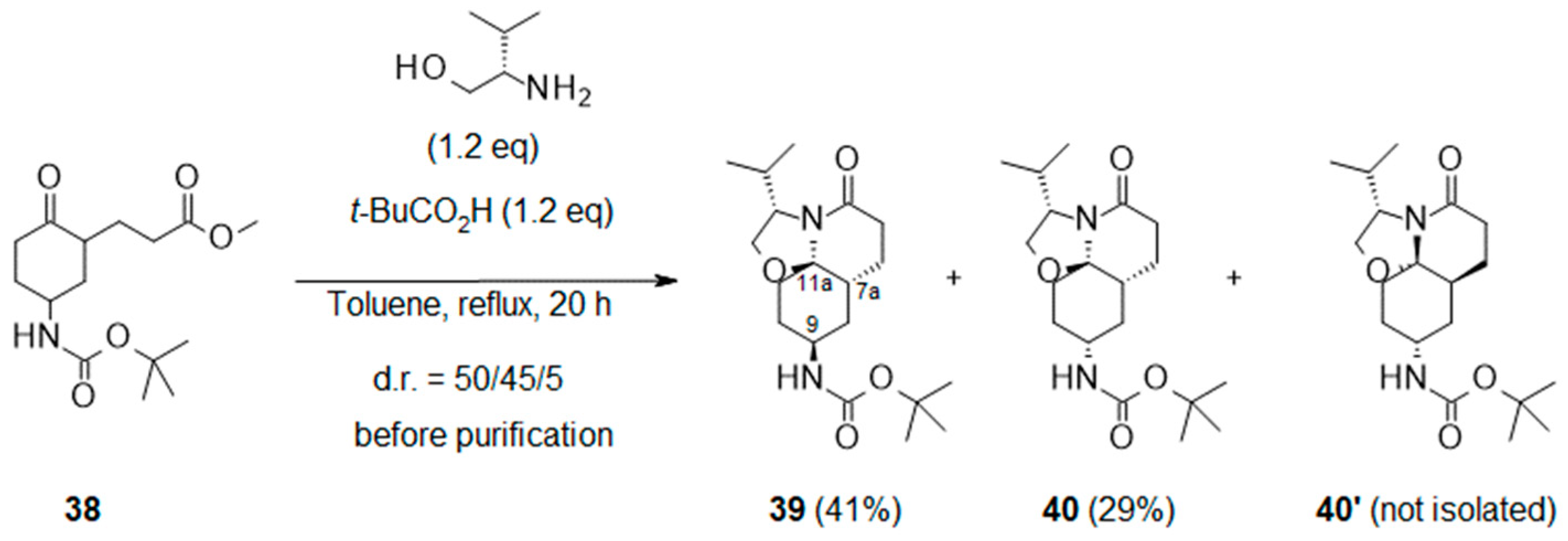
2.2.3. Application of Meyers’ Lactamization Reaction to Other Boc-Keto-Esters and Amino-Alcohols to Produce Tricyclic Spirolactams with Original Scaffolds
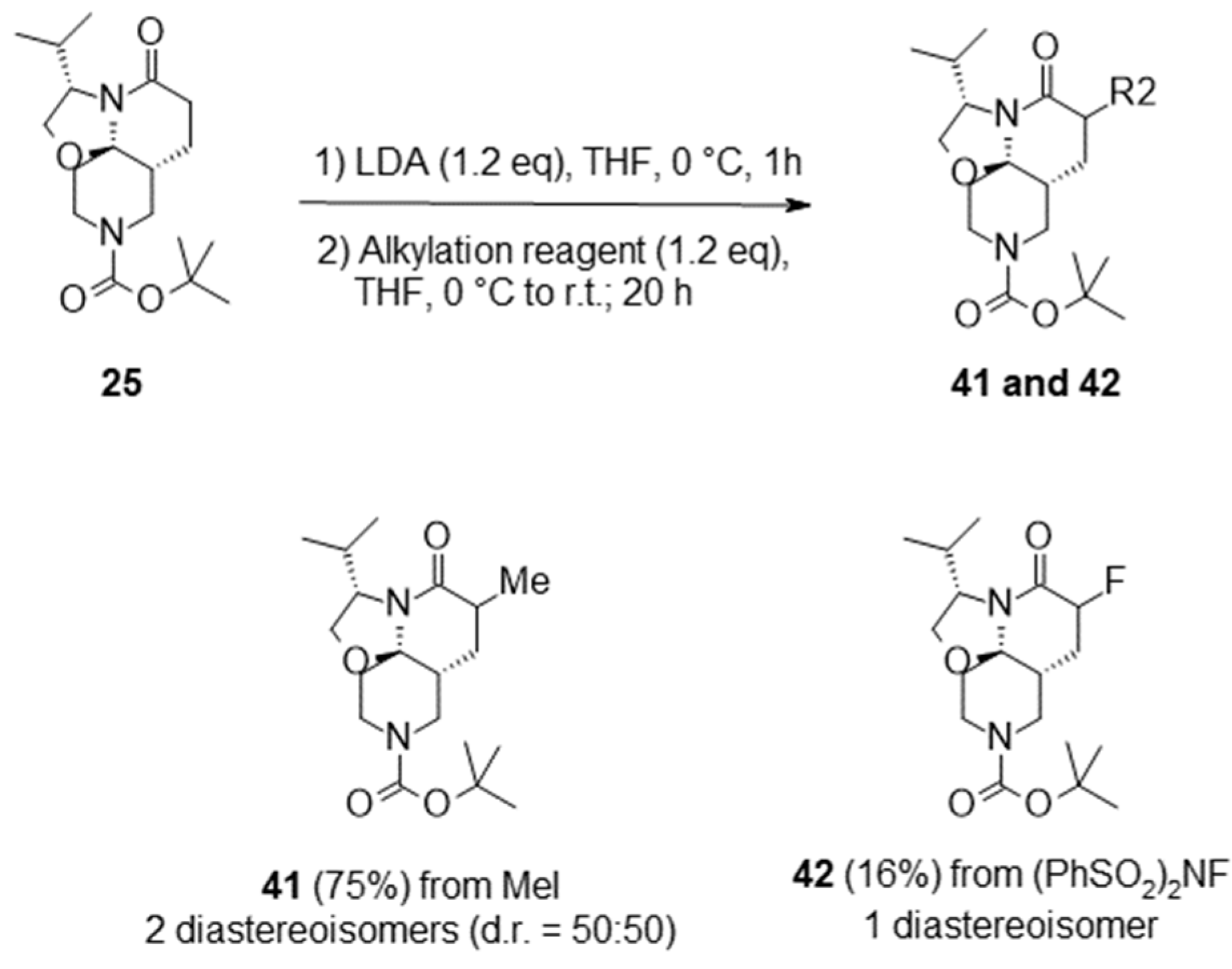
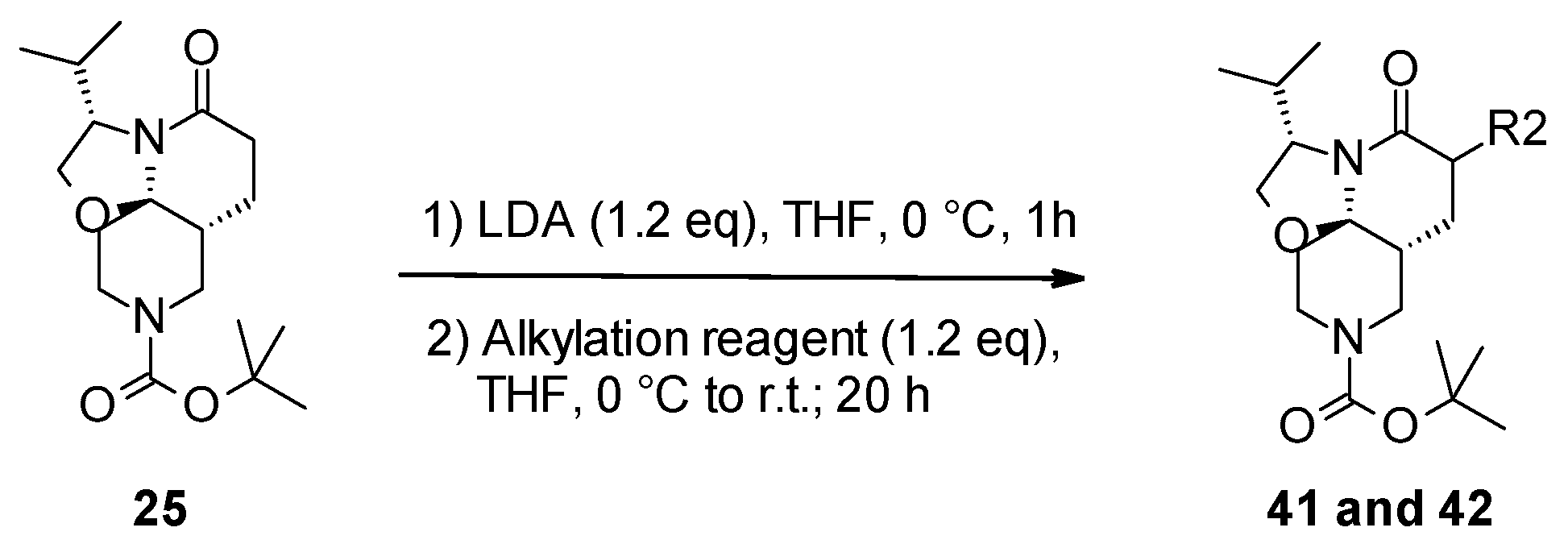
2.3. Functionalization of the Lactam Ring
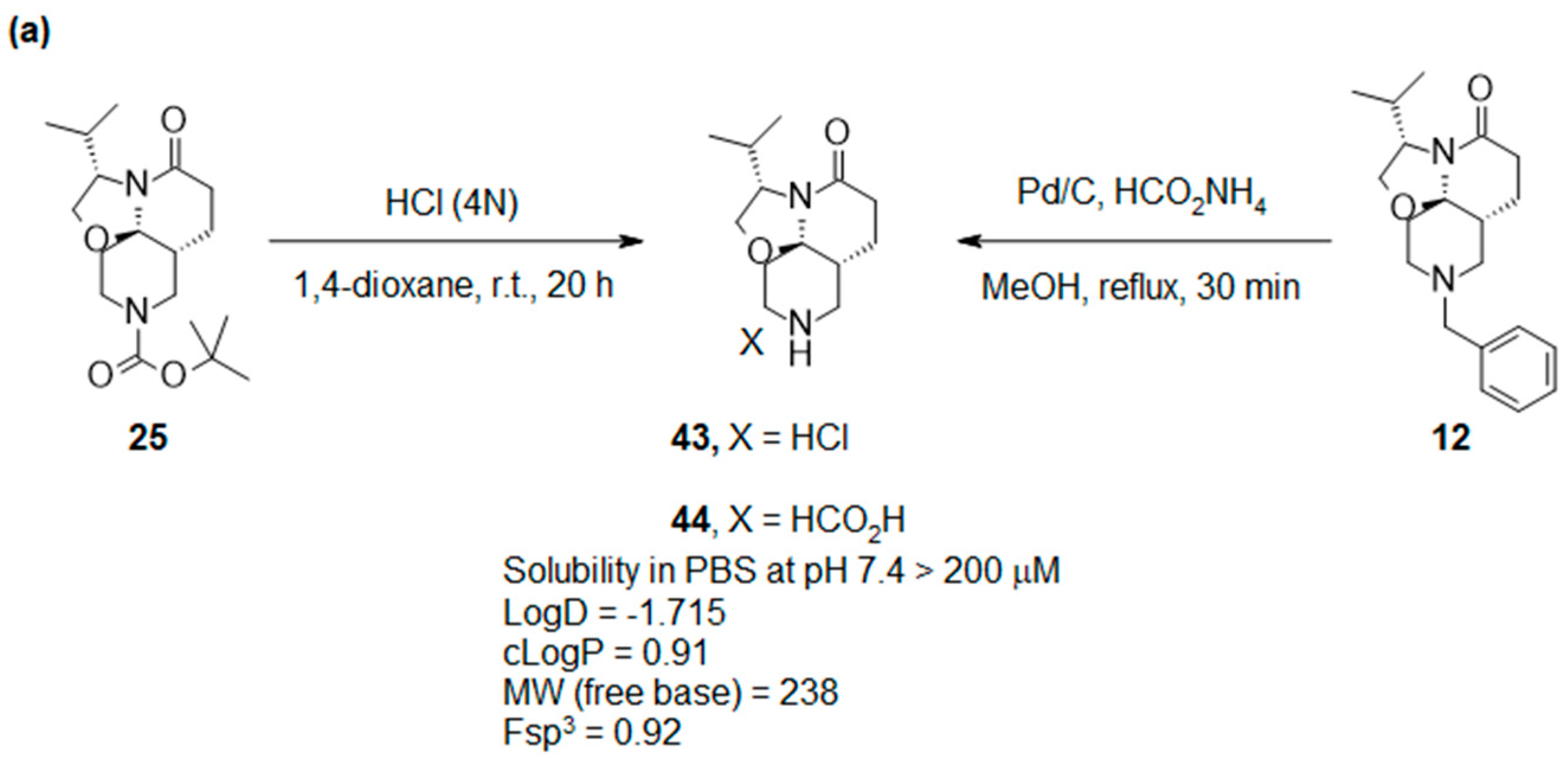
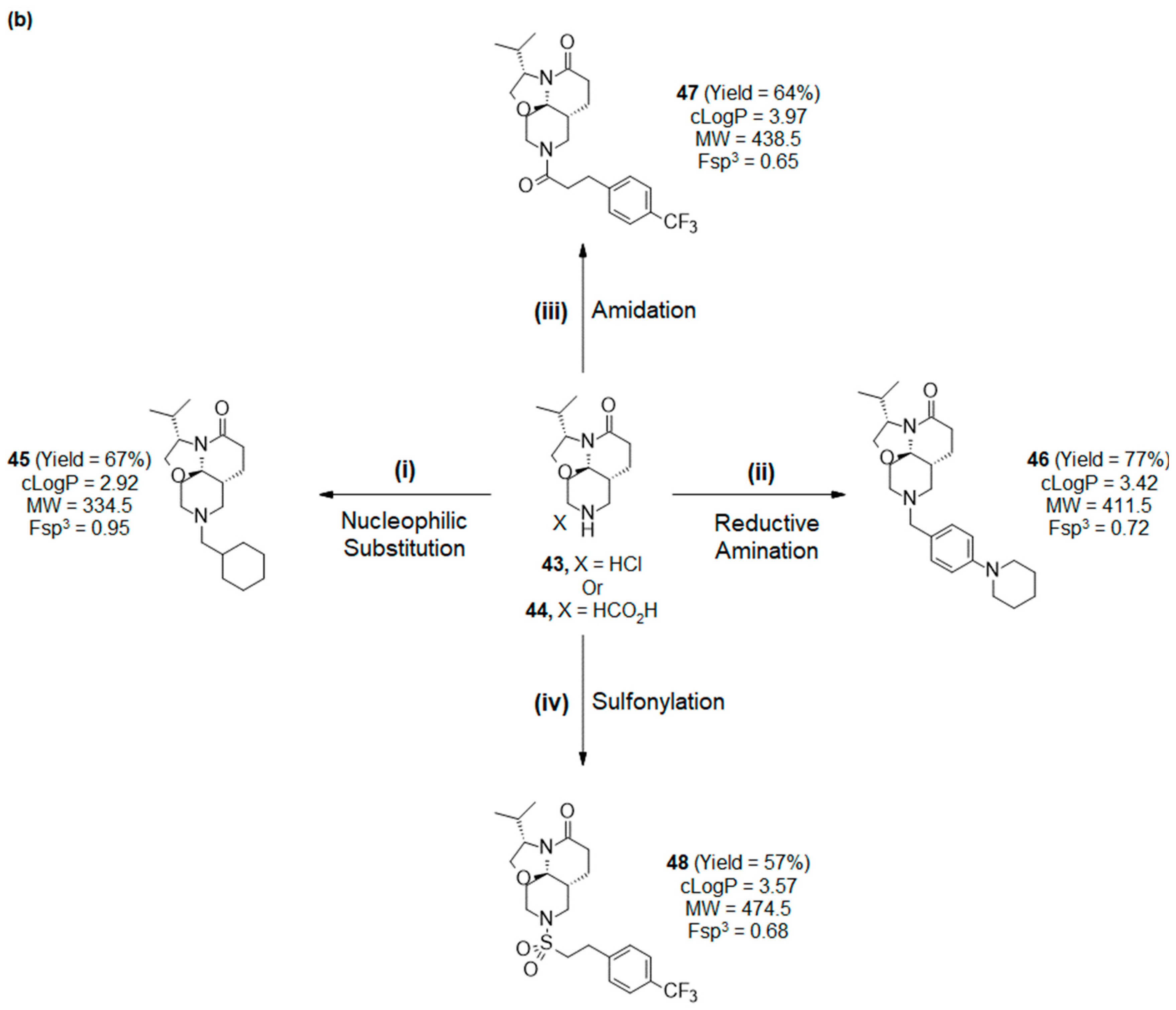
2.4. Synthesis and Drug-like Properties of Novel Tricyclic Spirolactams Obtained via R3 Position Functionalization
3. Materials and Methods
3.1. General Information
3.2. Chemistry
- 3-(1-benzyl-4-oxo-3-piperidyl)propanoic acid (8):
- Methyl 3-(1-benzyl-4-oxo-3-piperidyl)propanoate (9):
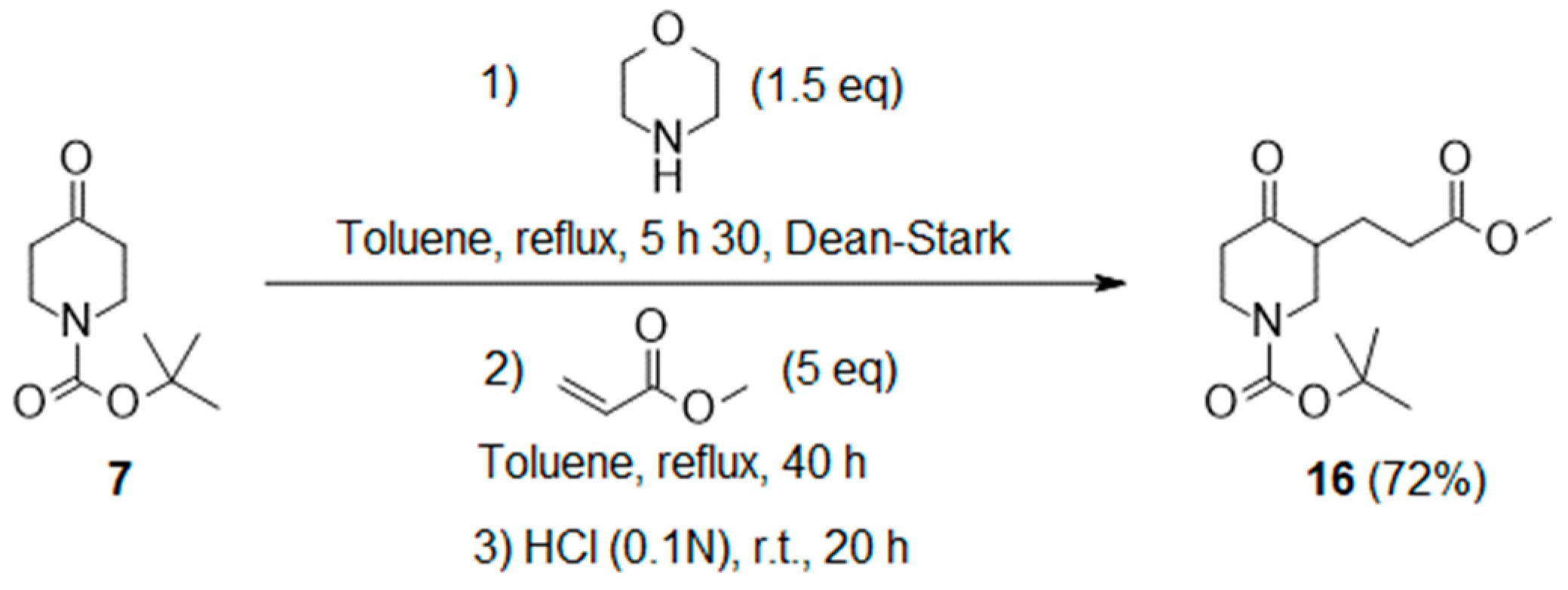
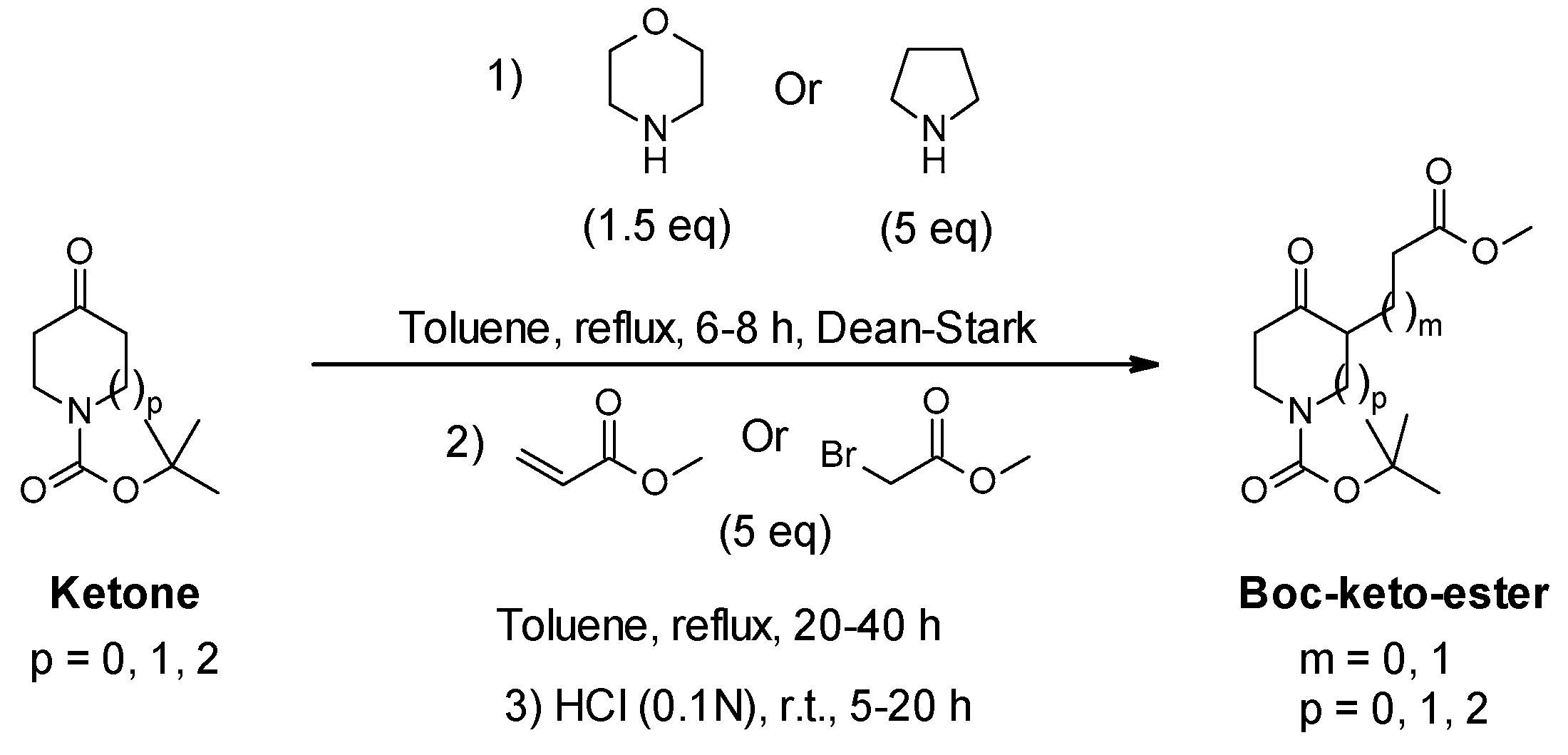
3.2.1. General Protocol 1: Synthesis of Boc-Keto-Esters via Stork Enamine Alkylation
- Tert-butyl 3-(3-methoxy-3-oxo-propyl)-4-oxo-piperidine-1-carboxylate (16):
- Tert-butyl 2-(3-methoxy-3-oxo-propyl)-3-oxo-piperidine-1-carboxylate (30):
- Tert-butyl 3-(3-methoxy-3-oxo-propyl)-4-oxo-pyrrolidine-1-carboxylate (32):
- Tert-butyl 3-(3-methoxy-3-oxo-propyl)-4-oxo-azepane-1-carboxylate (34):
- Tert-butyl 3-(2-methoxy-2-oxo-ethyl)-4-oxo-piperidine-1-carboxylate (36):
- Methyl 3-[5-(tert-butoxycarbonylamino)-2-oxo-cyclohexyl]propanoate (38):
3.2.2. General Protocol 2: Synthesis of Lactams via Meyers’ Lactamization of Keto-Esters with Amino-Alcohols
- (3S,7aS,11aS)-9-benzyl-3-methyl-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (11) and (3S,7aS,11aS)-9-benzyl-3-methyl-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (11′):
- (3S,7aR,11aR)-9-benzyl-3-isopropyl-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (12):
- (3S,7aR,11aR)-9-benzyl-3-tert-butyl-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (13):
- Tert-butyl 6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3] oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (17):
- Tert-butyl (4R,8aR,12aR)-4-isopropyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (18):
- Tert-butyl (4S,8aR,12aR)-4-methyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (19) and tert-butyl (4S,8aS,12aS)-4-methyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (19′):
- Tert-butyl (3R,8aR,12aR)-3-methyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (20):
- Tert-butyl (3R,8aR,12aR)-3-ethyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (21) and Tert-butyl (3R,8aR,12aR)-3-ethyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (21′):
- Tert-butyl 3-isopropyl-6-oxo-3,4,7,8,8a,9,11,12-octahydro-2H-[1,3]oxazino[2,3-j][1,6]naphthyridine-10-carboxylate (22):
- Tert-butyl (8aR,12aR)-6-oxospiro[2,4,7,8,8a,9,11,12-octahydro-[1,3]oxazino[2,3-j][1,6]naphthyridine-3,1′-cyclopropane]-10-carboxylate (23):
- Tert-butyl (8aR,12aR)-6-oxospiro[2,4,7,8,8a,9,11,12-octahydro-[1,3]oxazino[2,3-j][1,6]naphthyridine-3,1′-cyclobutane]-10-carboxylate (24):
- Tert-butyl (3S,7aR,11aR)-3-isopropyl-5-oxo-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridine-9-carboxylate (25):
- Tert-butyl (3R,7aR,11aR)-3-[(1R)-1-benzyloxyethyl]-5-oxo-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridine-9-carboxylate (26):
- Tert-butyl (3R,7aR,11aR)-5-oxo-3-(trifluoromethyl)-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridine-9-carboxylate (27):
- Tert-butyl 9-oxo-17-oxa-4,10-diazatetracyclo[8.7.0.01,6.011,16]heptadeca-11(16),12,14-triene-4-carboxylate (28):
- Tert-butyl (1R,6R)-9-oxo-15-oxa-4,10-diazatricyclo[8.5.0.01,6]pentadecane-4-carboxylate (29):
- Tert-butyl (3S,7aS,11aR)-3-isopropyl-5-oxo-2,3,6,7,7a,9,10,11-octahydrooxazolo[2,3-e][1,5]naphthyridine-8-carboxylate (31):
- Tert-butyl (1S,4S,9R)-4-isopropyl-6-oxo-2-oxa-5,11-diazatricyclo[7.3.0.01,5]dodecane-11-carboxylate (33):
- Tert-butyl (1R,4S,9R)-4-isopropyl-6-oxo-2-oxa-5,11-diazatricycl [7.5.0.01,5]tetradecane-11-carboxylate (35):
- Tert-butyl (1R,4S,8R)-4-isopropyl-6-oxo-2-oxa-5,10-diazatricyclo[6.4.0.01,5]dodecane-10-carboxylate (37):
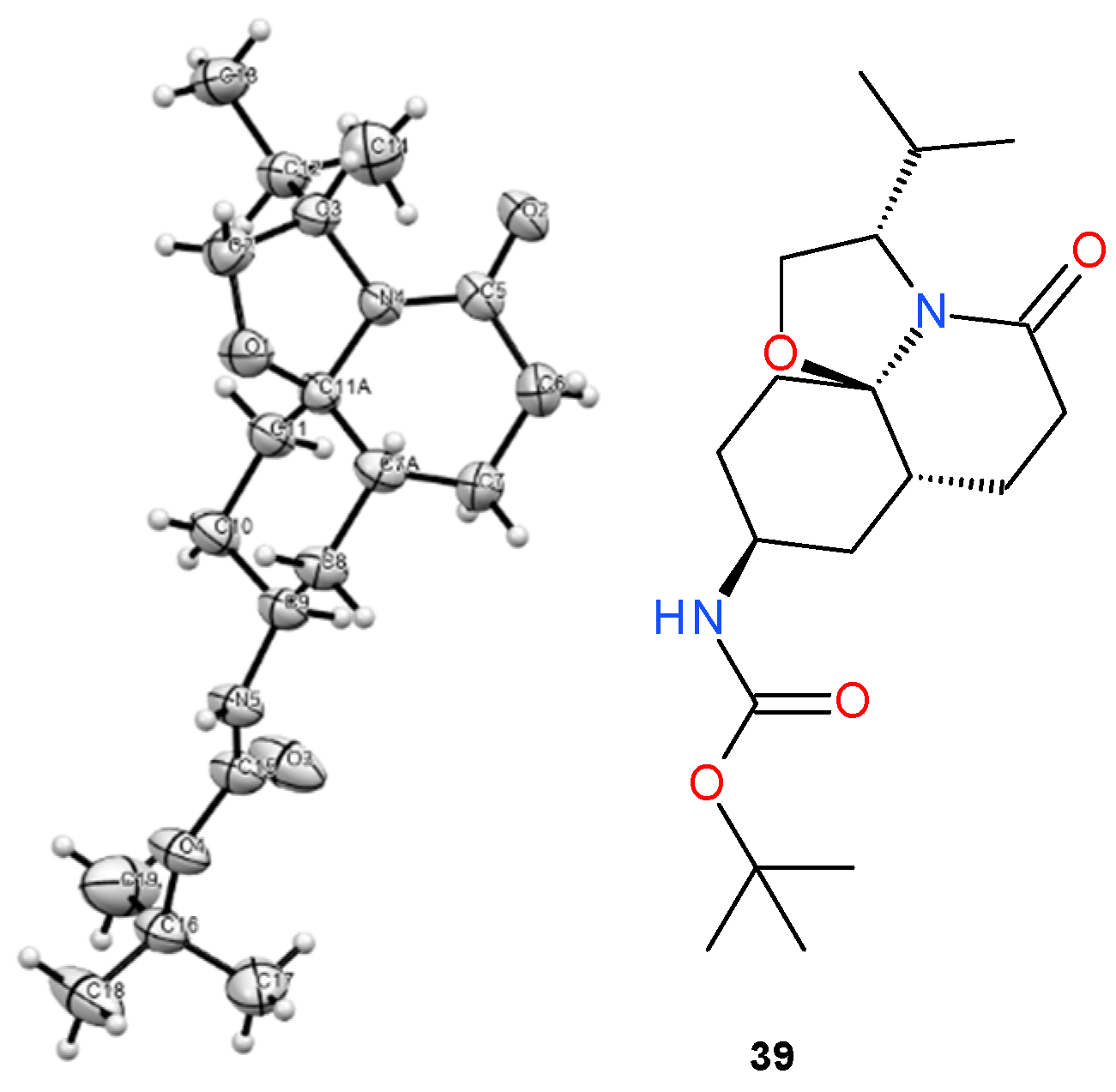
- Tert-butyl N-[(3S,7aR,9S,11aR)-3-isopropyl-5-oxo-3,6,7,7a,8,9,10,11-octahydro-2H-oxazolo[2,3-j]quinolin-9-yl]carbamate (39) and Tert-butyl N-[(3S,7aR,9R,11aR)-3-isopropyl-5-oxo-3,6,7,7a,8,9,10,11-octahydro-2H-oxazolo[2,3-j]quinolin-9-yl]carbamate (40):
3.2.3. General Protocol 3: Functionalization of Lactam Ring by Alkylation Reaction
- Tert-butyl (3S,7aR,11aR)-3-isopropyl-6-methyl-5-oxo-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridine-9-carboxylate (41):
- Tert-butyl (3S,7aR,11aR)-6-fluoro-3-isopropyl-5-oxo-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridine-9-carboxylate (42):
3.2.4. Deprotection of Lactams
- (3S,7aR,11aR)-3-isopropyl-3,6,7,7a,8,9,10,11-octahydro-2H-oxazolo[2,3-j][1,6]naphthyridin-5-one;hydrochloride (43):
3.2.5. R3 Functionalization of Lactams 43 and 44
- (3S,7aR,11aR)-9-(cyclohexylmethyl)-3-isopropyl-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (45):
- (3S,7aR,11aR)-3-isopropyl-9-[[4-(1-piperidyl)phenyl]methyl]-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (46):
- (3S,7aR,11aR)-3-isopropyl-9-[3-[4-(trifluoromethyl)phenyl]propanoyl]-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (47):
- (3S,7aR,11aR)-3-isopropyl-9-[2-[4-(trifluoromethyl)phenyl]ethylsulfonyl]-2,3,6,7,7a,8,10,11-octahydrooxazolo[2,3-j][1,6]naphthyridin-5-one (48):
3.3. X-ray Structural Determination
3.4. Determination of Compound 44 Solubility
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, R.W.; Currie, K.S.; Mitchell, S.A.; Darrow, J.W.; Pippin, D.A. Privileged Structures: Applications in Drug Discovery. Comb. Chem. High Throughput Screen. 2004, 7, 473–493. [Google Scholar] [CrossRef]
- Lovering, F.; Biccker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F. Escape from Flatland 2: Complexity and promiscuity. Med. Chem. Commun. 2013, 4, 515–519. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef]
- Stratton, C.F.; Newman, D.J.; Tan, D.S. Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 2015, 25, 4802–4807. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef]
- Grabowski, K.; Baringhaus, K.-H.; Schneider, G. Scaffold diversity of natural products: Inspiration for combinatorial library design. Nat. Prod. Rep. 2008, 25, 892–904. [Google Scholar] [CrossRef]
- Lee, M.-L.; Schneider, G. Scaffold Architecture and Pharmacophoric Properties of Natural Products and Trade Drugs: Application in the Design of Natural Product-Based Combinatorial Libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef]
- Grigalunas, M.; Burhop, A.; Zinken, S.; Pahl, A.; Gally, J.-M.; Wild, N.; Mantel, Y.; Sievers, S.; Foley, D.J.; Scheel, R.; et al. Natural product fragment combination to performance-diverse pseudo-natural products. Nat. Commun. 2021, 12, 1883. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Flitsch, S.L.; Grigalunas, M.; Leeson, P.D.; Quinn, R.J.; Turner, N.J.; Waldmann, H. The Time and Place for Nature in Drug Discovery. JACS Au. 2022, 2, 2400–2416. [Google Scholar] [CrossRef]
- Thomas, G.L.; Johannes, C.W. Natural product-like synthetic libraries. Curr. Opin. Chem. Biol. 2011, 15, 516–522. [Google Scholar] [CrossRef]
- Wildermuth, R.; Speck, K.; Haut, F.-L.; Mayer, P.; Karge, B.; Brönstrup, M.; Magauer, T. A modular synthesis of tetracyclic meroterpenoid antibiotics. Nat. Commun. 2017, 8, 2083. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gloer, J.B.; Wicklow, D.T. Thiersindoles A–C: New Indole Diterpenoids from Penicillium thiersii . J. Nat. Prod. 2003, 66, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.-J.; Bai, M.; Zhou, X.-M.; Huang, G.-L.; Shao, T.-M.; Luo, Y.-P.; Niu, Z.-G.; Niu, Y.-Y.; Chen, G.-Y.; Han, C.-R. Penicilindoles A–C, Cytotoxic Indole Diterpenes from the Mangrove Derived Fungus Eupenicillium sp. HJ002. J. Nat. Prod. 2018, 81, 1045–1049. [Google Scholar] [CrossRef]
- Geethangili, M.; Tzeng, Y.-M. Review of Pharmacological Effects of Antrodia camphorata and Its Bioactive Compounds. Evid.-Based Complement. Altern. Med. 2009, 2011, nep108. [Google Scholar] [CrossRef]
- Venkatachalam, A.; Tai, D.-F. Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction. Molecules 2020, 25, 831. [Google Scholar]
- Rao, Y.K.; Wu, A.T.H.; Geethangili, M.; Huang, M.-T.; Chao, W.-J.; Wu, C.-H.; Deng, W.-P.; Yeh, C.-T.; Tzeng, Y.-M. Identification of Antrocin from Antrodia camphorata as a Selective and Novel Class of Small Molecule Inhibitor of Akt/mTOR Signaling in Metastatic Breast Cancer MDA-MB-231 Cells. Chem. Res. Toxicol. 2011, 24, 238–245. [Google Scholar] [CrossRef]
- Wu, A.T.H.; Lawal, B.; Wei, L.; Wen, Y.-T.; Tzeng, D.T.W.; Lo, W.-C. Multiomics Identification of Potential Targets for Alzheimer Disease and Antrocin as a Therapeutic Candidate. Pharmaceutics 2021, 13, 1555. [Google Scholar] [CrossRef]
- Talele, T.T. Opportunities for Tapping into Three-Dimensional Chemical Space through a Quaternary Carbon. J. Med. Chem. 2020, 63, 13291–13315. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Lenci, E.; Trabocchi, A.; Medina-Franco, J.L. Exploring the chemical space and the bioactivity profile of lactams: A chemoinformatic study. RSC Adv. 2019, 9, 27105–27116. [Google Scholar] [CrossRef]
- Willand, N.; Beghyn, T.; Nowogrocki, G.; Gesquiere, J.-C.; Deprez, B. Synthesis and structural studies of a novel scaffold for drug discovery: A 4,5-dihydro-3H-spiro[1,5-benzoxazepine-2,40-piperidine]. Tetrahedron Lett. 2004, 45, 1051–1054. [Google Scholar] [CrossRef]
- Beghyn, T.; Deprez-Poulain, R.; Willand, N.; Folleas, B.; Deprez, B. Natural Compounds: Leads or Ideas? Bioinspired Molecules for Drug Discovery. Chem. Biol. Drug Des. 2008, 72, 3–15. [Google Scholar] [PubMed]
- Tran, N.C.; Dhondt, H.; Flipo, M.; Deprez, B.; Willand, N. Synthesis of functionalized 2-isoxazolines as three-dimensional fragments for fragment-based drug discovery. Tetrahedron Lett. 2015, 56, 4119–4123. [Google Scholar] [CrossRef]
- Prevet, H.; Flipo, M.; Roussel, P.; Deprez, B.; Willand, N. Microwave-assisted synthesis of functionalized spirohydantoins as 3-D privileged fragments for scouting the chemical space. Tetrahedron Lett. 2016, 57, 2888–2894. [Google Scholar] [CrossRef]
- Meyers, A.I.; Brengel, G.P. Chiral bicyclic lactams: Useful precursors and templates for asymmetric syntheses. Chem. Commun. 1997, 1, 1–8. [Google Scholar] [CrossRef]
- Groaming, M.D.; Meyers, A.I. Chiral Non-Racemic Bicyclic Lactams. Auxiliary-Based Asymmetric Reactions. Tetrahedron 2000, 56, 9843–9873. [Google Scholar] [CrossRef]
- Amat, M.; Llor, N.; Griera, R.; Pérez, M.; Bosch, J. Enantioselective Synthesis of Alkaloids from Phenylglycinol-Derived Lactams. Nat. Prod. Commun. 2011, 6, 515–526. [Google Scholar] [CrossRef]
- Pinto, A.; Piccichè, M.; Griera, R.; Molins, E.; Bosch, J.; Amat, M. Studies on the Synthesis of Phlegmarine-Type Lycopodium Alkaloids: Enantioselective Synthesis of (−)-Cermizine B, (+)-Serratezomine E, and (+)-Luciduline. J. Org. Chem. 2018, 83, 8364–8375. [Google Scholar] [CrossRef]
- Piccichè, M.; Pinto, A.; Griera, R.; Bosch, J.; Amat, M.J. Total Synthesis of (−)-Cylindricine H. J. Org. Chem. 2022, 24, 5356–5360. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, S.; Jida, M.; Courtin, J.; Laconde, G.; Willand, N.; Deprez, B.; Deprez-Poulain, R. Water-based conditions for the microscale parallel synthesis of bicyclic lactams. Tetrahedron Lett. 2013, 54, 562–567. [Google Scholar] [CrossRef]
- Deprez-Poulain, R.; Willand, N.; Boutillon, C.; Nowogrocki, G.; Azaroual, N.; Deprez, B. A simple reaction to produce small structurally complex and diverse molecules. Tetrahedron Lett. 2004, 45, 5287–5290. [Google Scholar] [CrossRef]
- Penhoat, M.; Levacher, v.; Dupas, G. Novel Extension of Meyers’ Methodology: Stereoselective Construction of Axially Chiral 7,5-Fused Bicyclic Lactams. J. Org. Chem. 2003, 68, 9517–9520. [Google Scholar] [CrossRef] [PubMed]
- Ennis, M.D.; Hoffman, R.L.; Ghazal, N.B.; Old, D.W.; Mooney, P.A. Asymmetric Synthesis of Cis-Fused Bicyclic Pyrrolidines and Pyrrolidinones via Chiral Polycyclic Lactams. J. Org. Chem. 1996, 61, 5813–5817. [Google Scholar] [CrossRef]
- Jida, M.; Deprez-Poulain, R.; Malaquin, S.; Roussel, P.; Agbossou-Niedercorn, F.; Deprez, B.; Laconde, G. Solvent-free microwave-assisted Meyers’ lactamization. Green Chem. 2010, 12, 961–964. [Google Scholar] [CrossRef]
- Postikova, S.; Sabbah, M.; Wightman, D.; Nguyen, I.T.; Sanselme, M.; Besson, T.; Brière, J.-F.; Oudeyer, S.; Levacher, V. Developments in Meyers’ Lactamization Methodology: En Route to Bi(hetero)aryl Structures with Defined Axial Chirality. J. Org. Chem. 2013, 78, 8191–8197. [Google Scholar] [CrossRef] [PubMed]
- Membrat, R.; Vasseur, A.; Moraleda, D.; Michaud-Chevallier, S.; Martinez, A.; Giordano, L.; Nuel, D. Platinum–(phosphinito–phosphinous acid) complexes as bi-talented catalysts for oxidative fragmentation of piperidinols: An entry to primary amines. RSC Adv. 2019, 9, 37825–37829. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Baty, J.D.; Jones, G.; Moore, C. Alkylation of 4-Piperidones; Intermediates in the Synthesis of Reduced 2-Pyrindin-6-ones. J. Chem. Soc. C 1969, 11, 1520–1528. [Google Scholar] [CrossRef]
- Schwehm, C.; Li, J.; Song, H.; Hu, X.; Kellam, B.; Stocks, M. Synthesis of New DPP-4 Inhibitors Based on a Novel Tricyclic Scaffold. ACS Med. Chem. Lett. 2015, 6, 324–328. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Idzik, T.J.; Myk, Z.M.; Peruzynska, M.; Maciejewska, G.; Drozdzik, M.; Sosnicki, J.G. Arylation of enelactams using TIPSOTf: Reaction scope and mechanistic insight. Org. Chem. Front. 2021, 8, 708–720. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, J.G.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dam, S.; Tangara, S.; Hamela, C.; Hattabi, T.; Faїon, L.; Carre, P.; Antoine, R.; Herledan, A.; Leroux, F.; Piveteau, C.; et al. Tricyclic SpiroLactams Kill Mycobacteria in vitro and in vivo by Inhibiting Type II NADH Dehydrogenases. J. Med. Chem. 2022, 65, 16651–16664. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
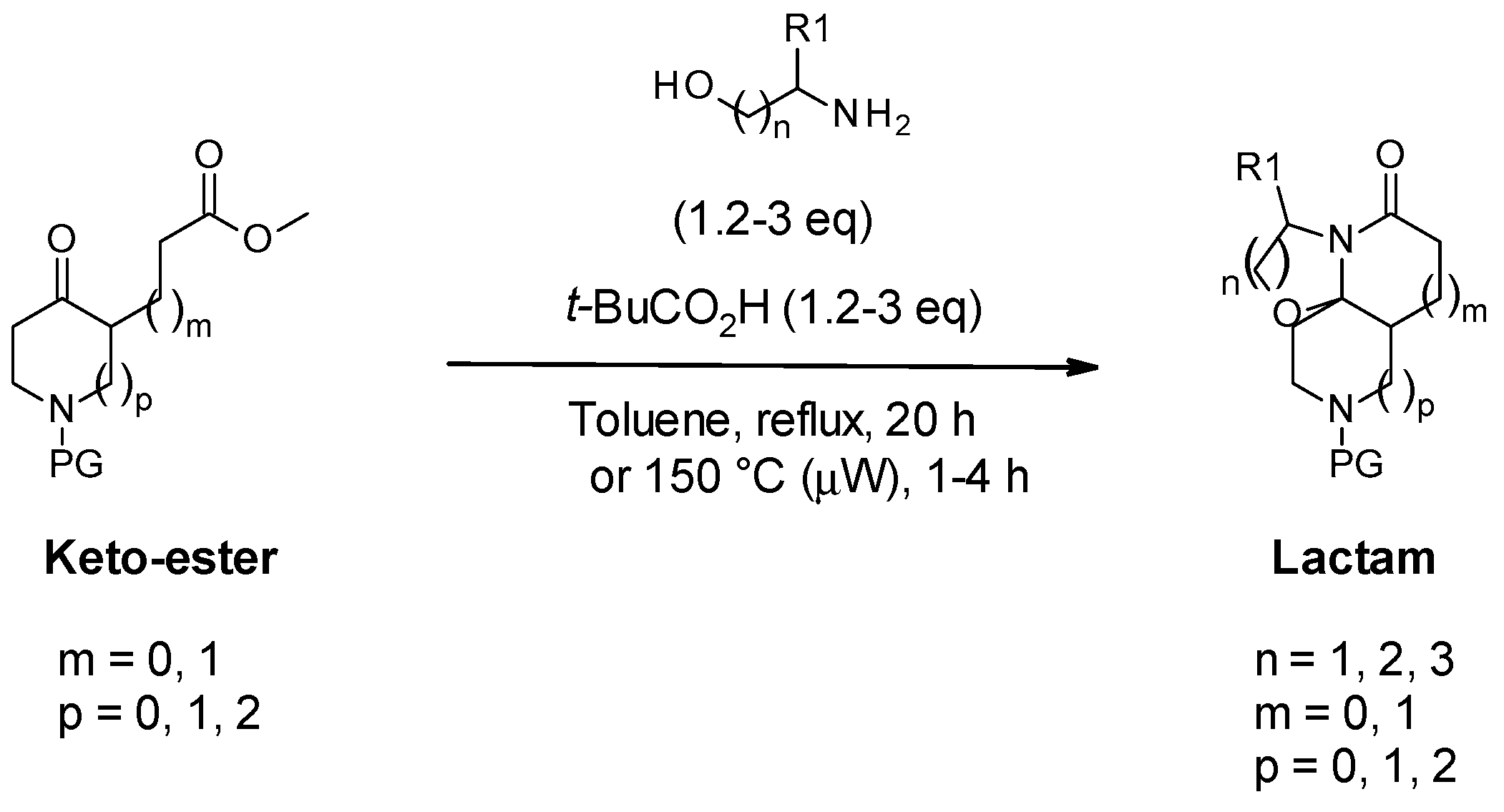{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Substrate a | Conditions | Conversion b |
| 1 | 8 | Toluene, reflux, 20 h | 66% |
| 2 | 8 | Toluene, 150 °C (µW), 2 h | 76% |
| 3 | 9 | Pivalic acid (1.2 eq), Toluene, 150 °C (µW), 1 h | 100% |
 | ||||
|---|---|---|---|---|
| Entry | Amino-Alcohol | Product | Isolated Compound (Yield) a | d.r. b |
| 1 |  |  | 11 + 11′ (46%) | 70:30 |
| 2 |  |  | 12 (75%) | 100:0 |
| 3 |  |  | 13 (40%) | 100:0 |
 | ||||
|---|---|---|---|---|
| Entry | Amino-Alcohol | Product | Isolated Compound (Yield) d | d.r. e |
| 1 |  |  | 17 a (79%) | |
| 2 |  |  | 18 # (18%) | 90:10 |
| 3 |  |  | 19 + 19′ b (86%) | 50:50 |
| 4 |  |  | 20 c (63%) | 60:40 |
| 5 |  |  | 21 + 21′ b (89%) | 70:30 |
| 6 |  |  | 22 c (92%) | 96:4 |
| 7 |  |  | 23 a (88%) | |
| 8 |  |  | 24 a (98%) | |
 | |||||
|---|---|---|---|---|---|
| Entry | Boc-Keto-Ester a | Amino-Alcohol | Product | Isolated Compound (Yield) c | d.r. d |
| 1 |  |  |  | 25 (84%) * 25 (88%) | 90:10 |
| 2 |  |  |  | 26 (50%) | 94:6 |
| 3 |  |  |  | 27 (59%) | 100:0 |
| 4 |  |  |  | 28 b (98%) | |
| 5 |  |  |  | 29 b (81%) | |
| 6 |  |  |  | 31 (49%) | 100:0 |
| 7 |  |  |  | 33 (78%) | 100:0 |
| 8 |  |  |  | 35 (89%) | 90:10 |
| 9 |  |  |  | 37 (90%) | 100:0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangara, S.; Faïon, L.; Piveteau, C.; Capet, F.; Godelier, R.; Michel, M.; Flipo, M.; Deprez, B.; Willand, N.; Villemagne, B. Rapid and Efficient Access to Novel Bio-Inspired 3-Dimensional Tricyclic SpiroLactams as Privileged Structures via Meyers’ Lactamization. Pharmaceuticals 2023, 16, 413. https://doi.org/10.3390/ph16030413
Tangara S, Faïon L, Piveteau C, Capet F, Godelier R, Michel M, Flipo M, Deprez B, Willand N, Villemagne B. Rapid and Efficient Access to Novel Bio-Inspired 3-Dimensional Tricyclic SpiroLactams as Privileged Structures via Meyers’ Lactamization. Pharmaceuticals. 2023; 16(3):413. https://doi.org/10.3390/ph16030413
Chicago/Turabian StyleTangara, Salia, Léo Faïon, Catherine Piveteau, Frédéric Capet, Romain Godelier, Marion Michel, Marion Flipo, Benoit Deprez, Nicolas Willand, and Baptiste Villemagne. 2023. "Rapid and Efficient Access to Novel Bio-Inspired 3-Dimensional Tricyclic SpiroLactams as Privileged Structures via Meyers’ Lactamization" Pharmaceuticals 16, no. 3: 413. https://doi.org/10.3390/ph16030413
APA StyleTangara, S., Faïon, L., Piveteau, C., Capet, F., Godelier, R., Michel, M., Flipo, M., Deprez, B., Willand, N., & Villemagne, B. (2023). Rapid and Efficient Access to Novel Bio-Inspired 3-Dimensional Tricyclic SpiroLactams as Privileged Structures via Meyers’ Lactamization. Pharmaceuticals, 16(3), 413. https://doi.org/10.3390/ph16030413