



Discovery of Ureido-Based Apcin Analogues as Cdc20-specific Inhibitors against Cancer


Abstract
1. Introduction
2. Results and Discussion
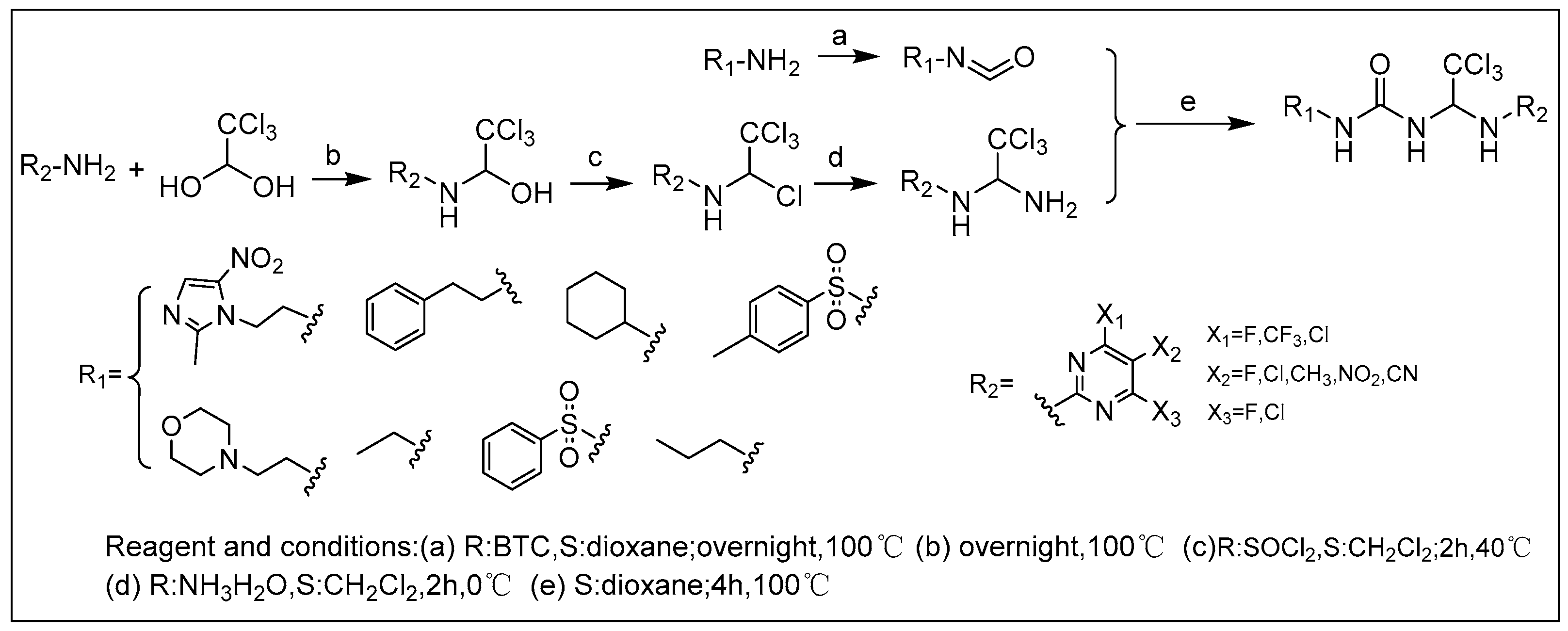
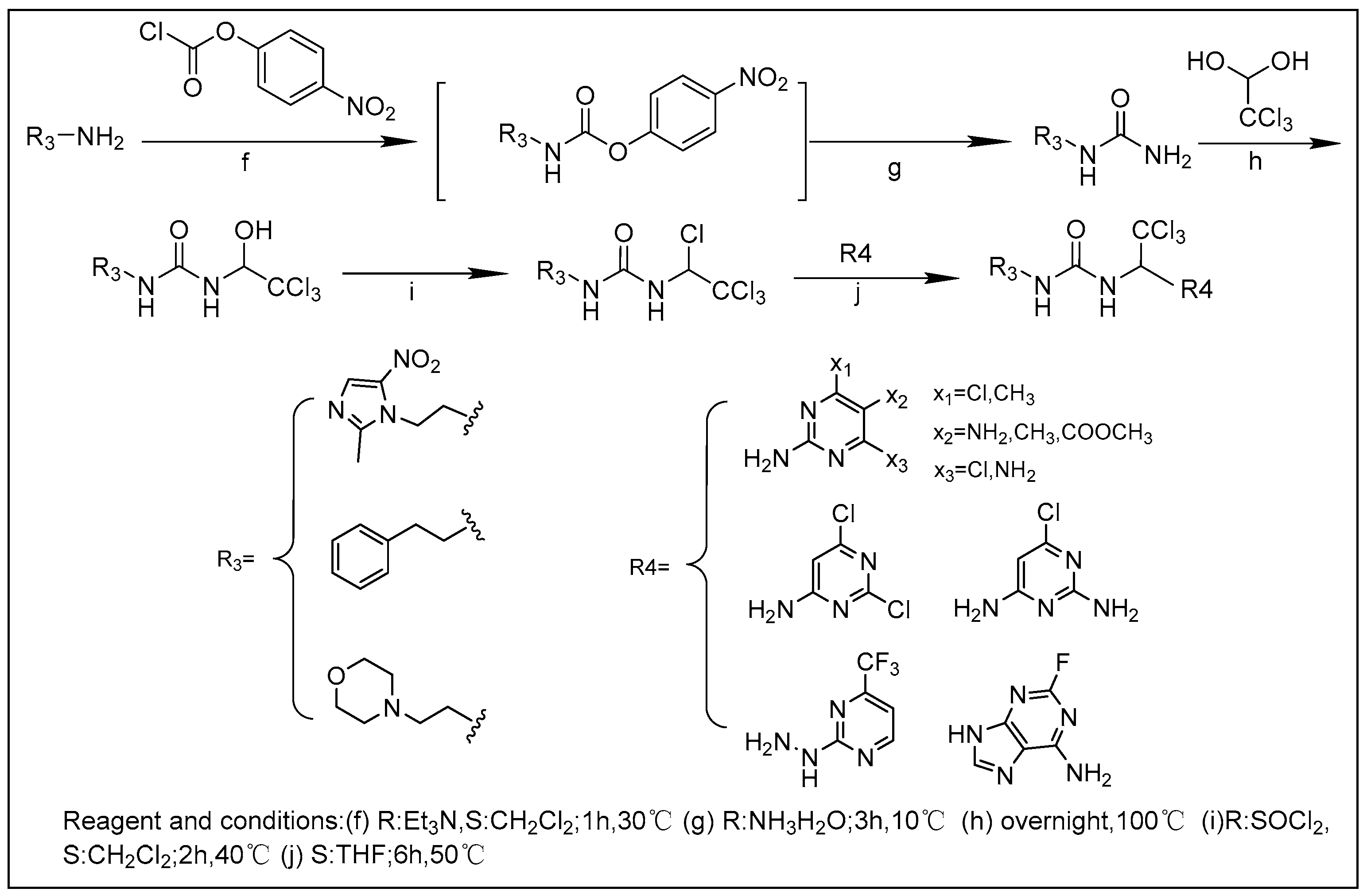
2.1. Chemistry
2.2. In Vitro Antiproliferative Assay and Prediction of Lipid Permeability and Toxicity in Silico
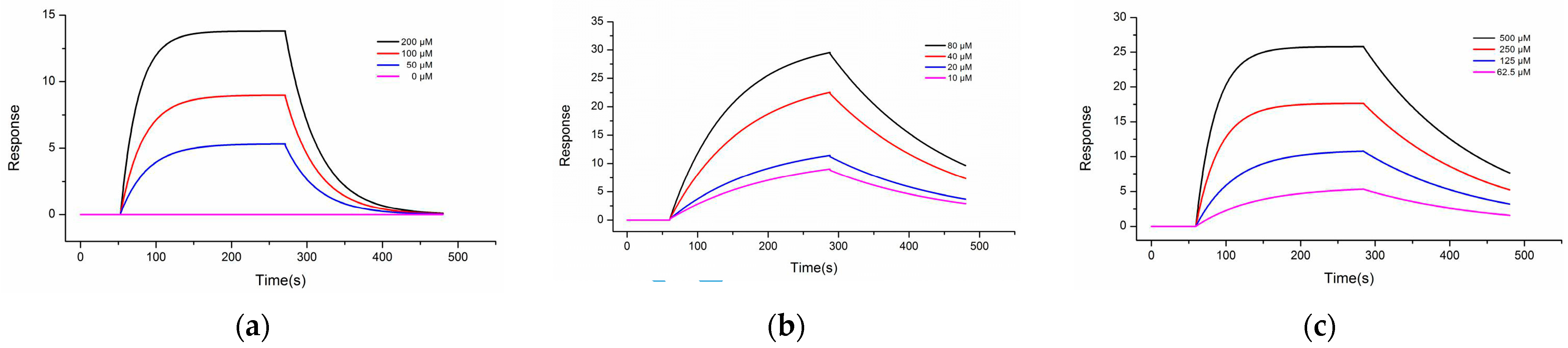
2.3. Surface Plasmon Resonance (SPR) Assay
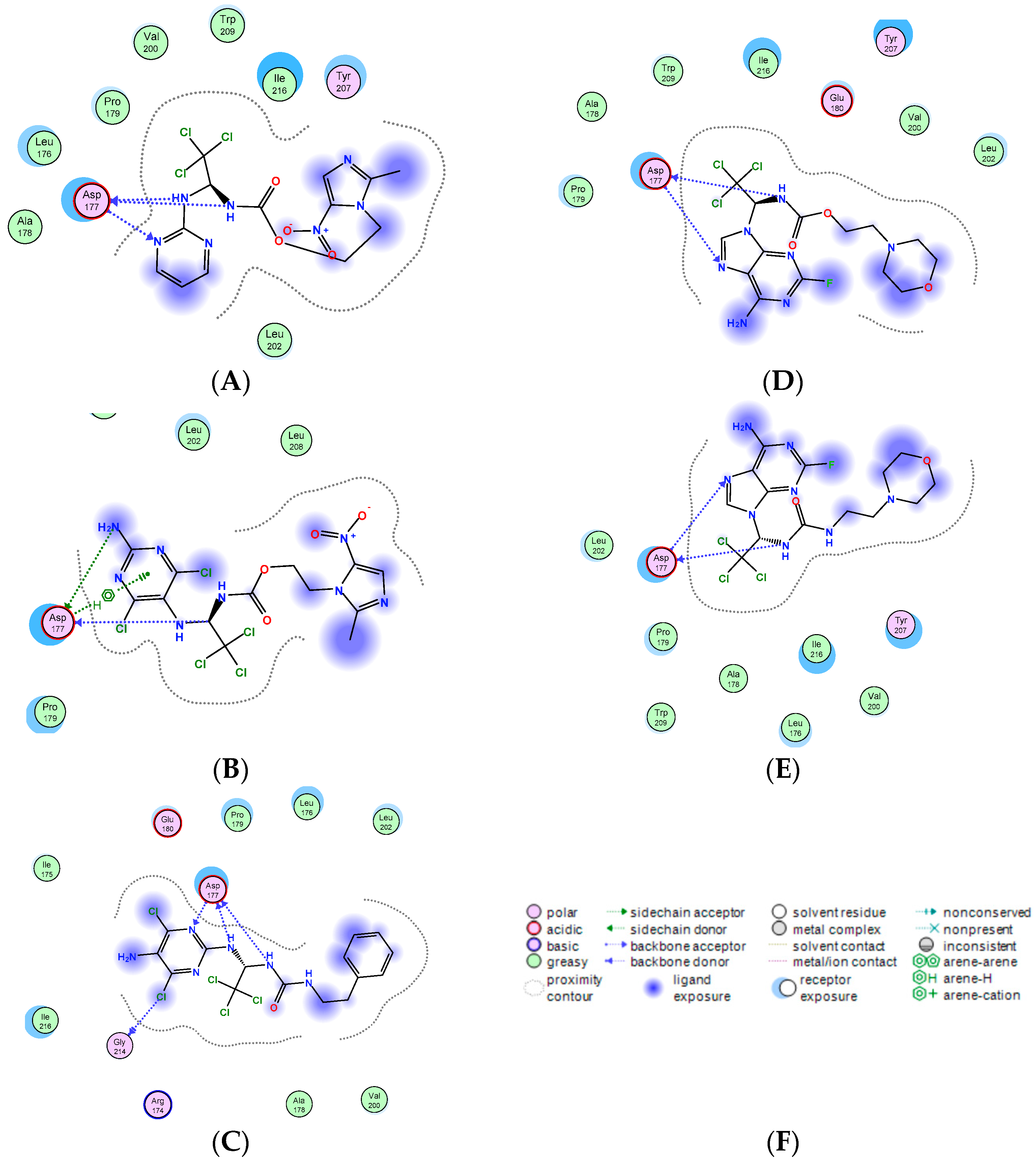
2.4. Molecular-Docking Simulation
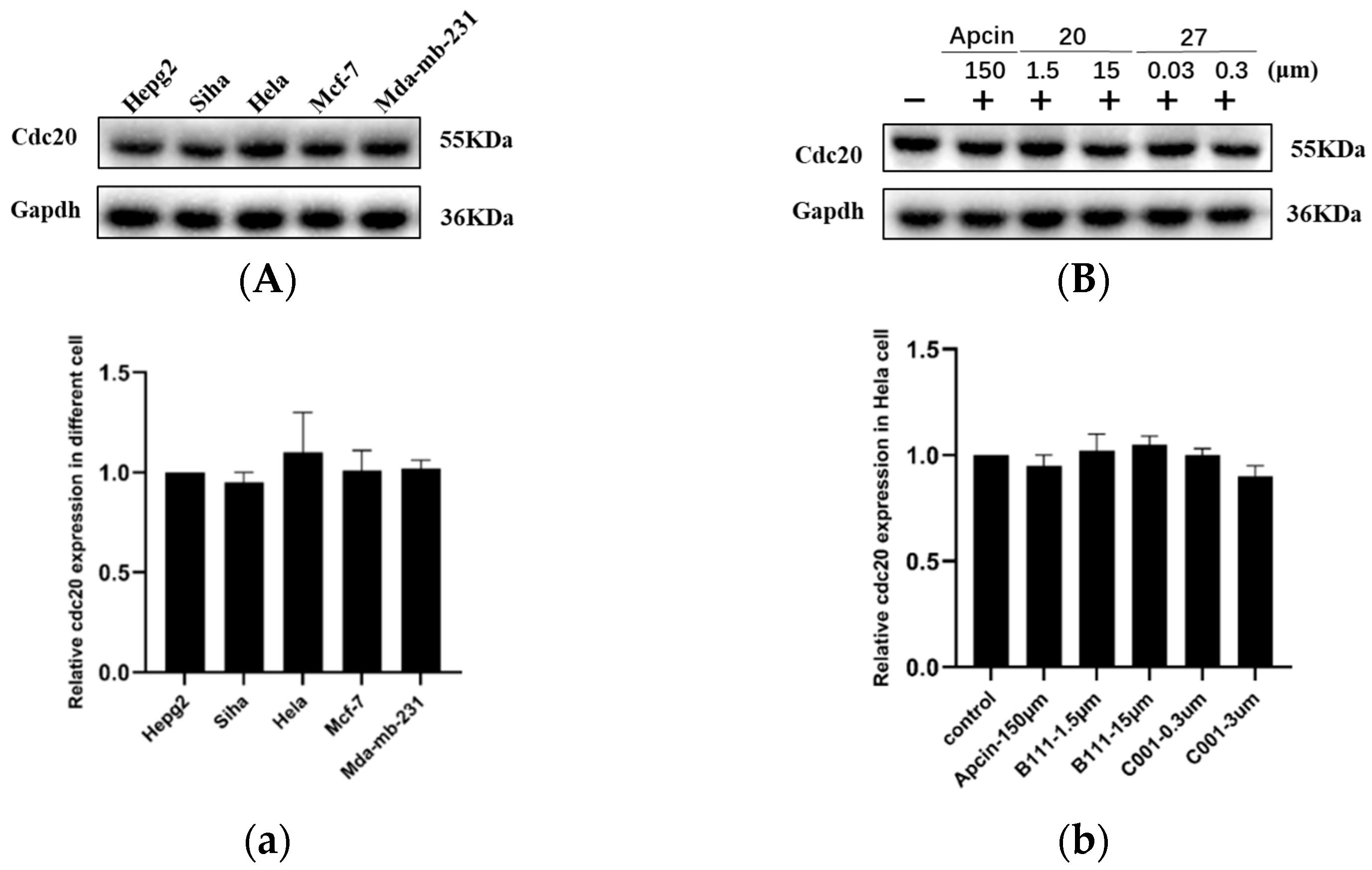
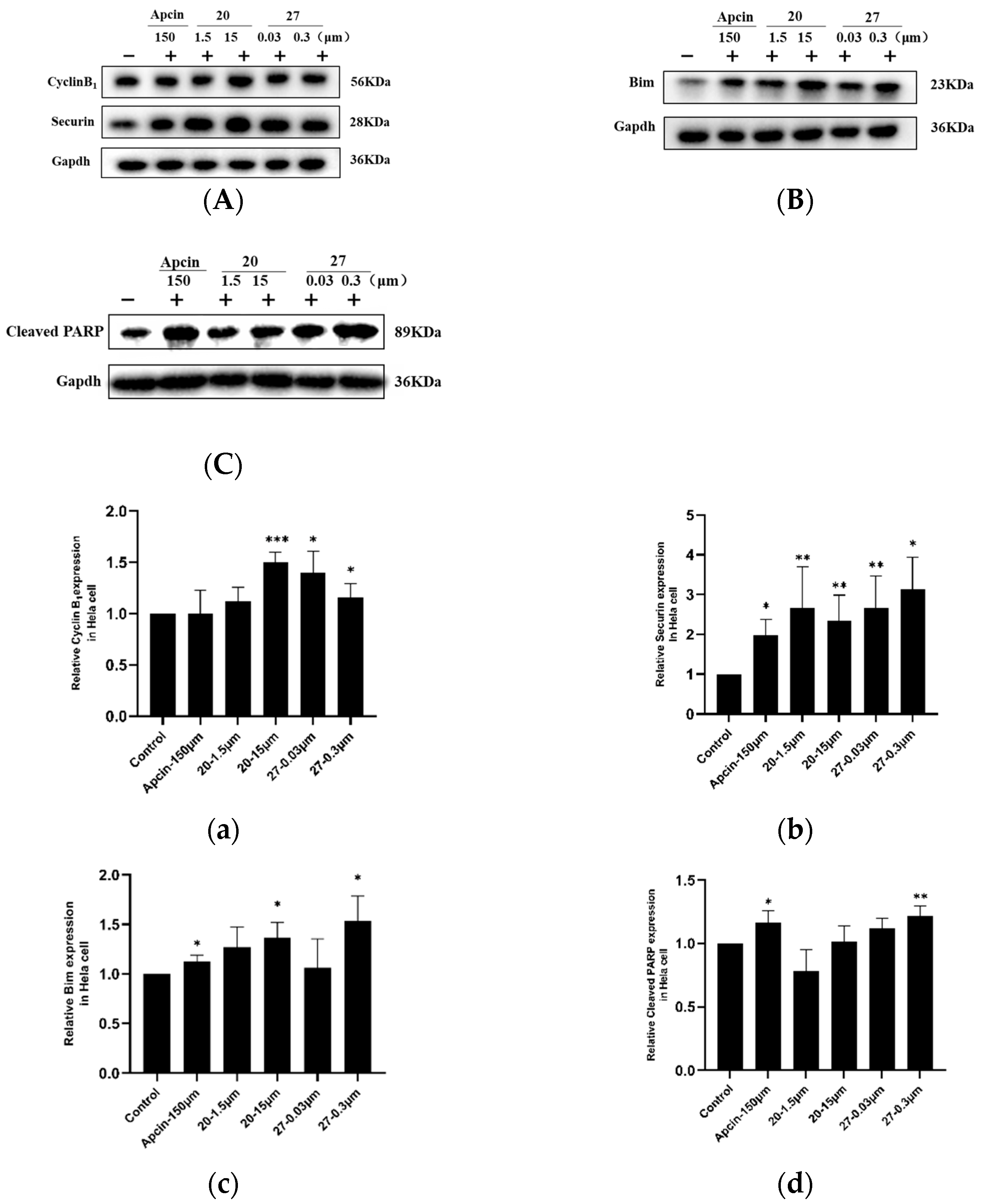
2.5. Western Blot
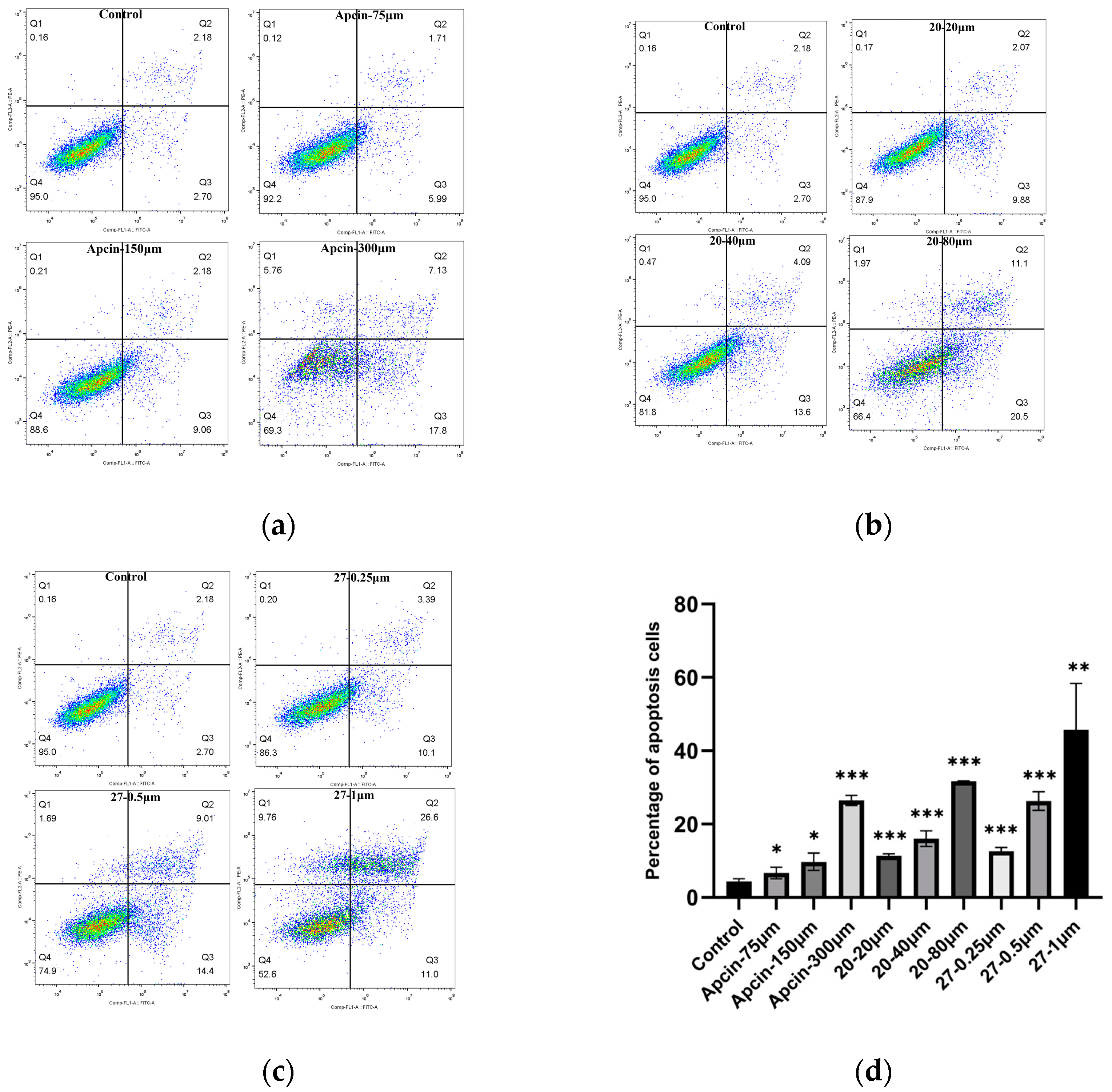
2.6. The Annexin V-FITC/PI Double-Staining Fluorescence Experiment
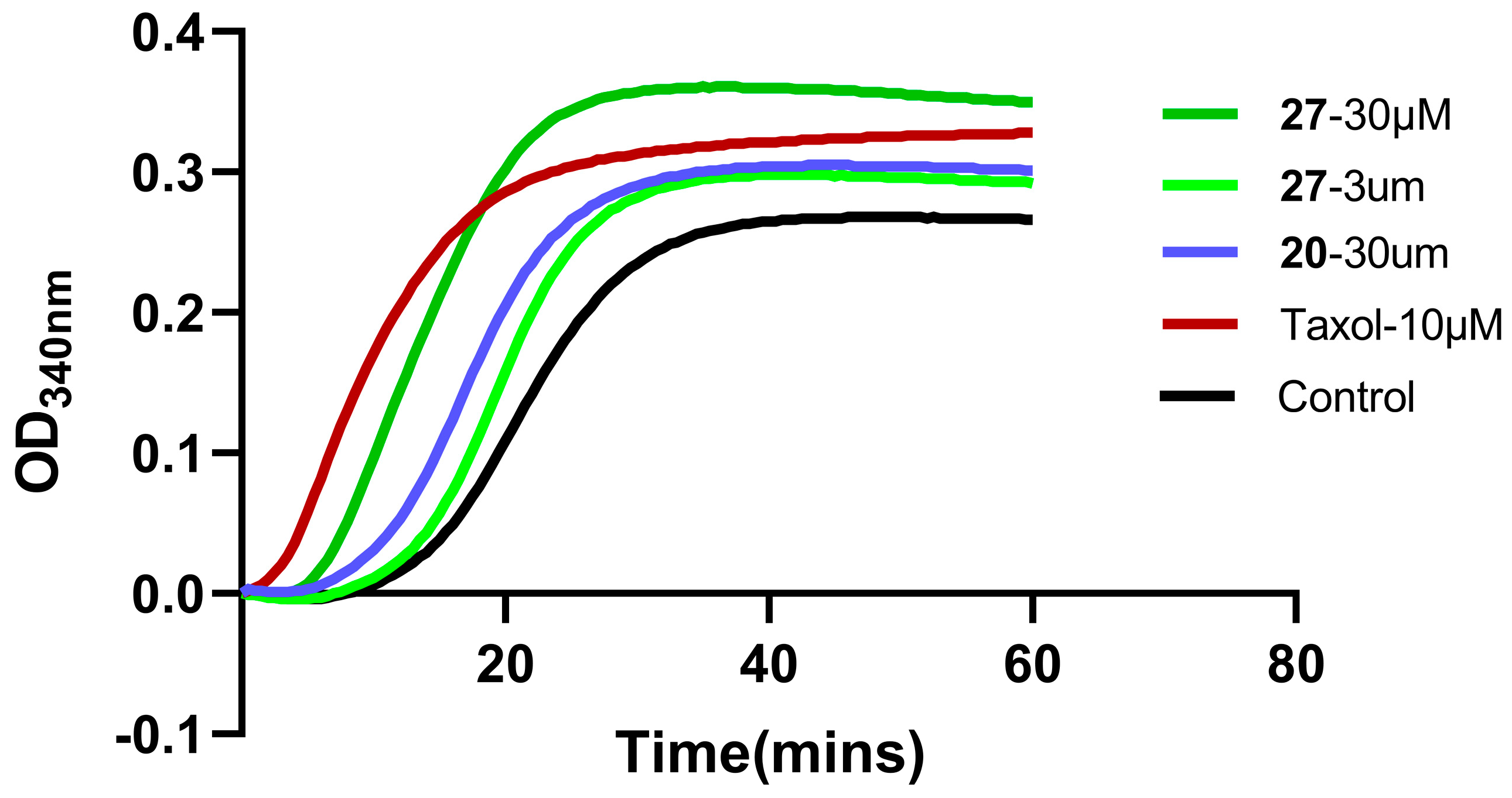
2.7. Microtubule Polymerization-Inhibition Experiment
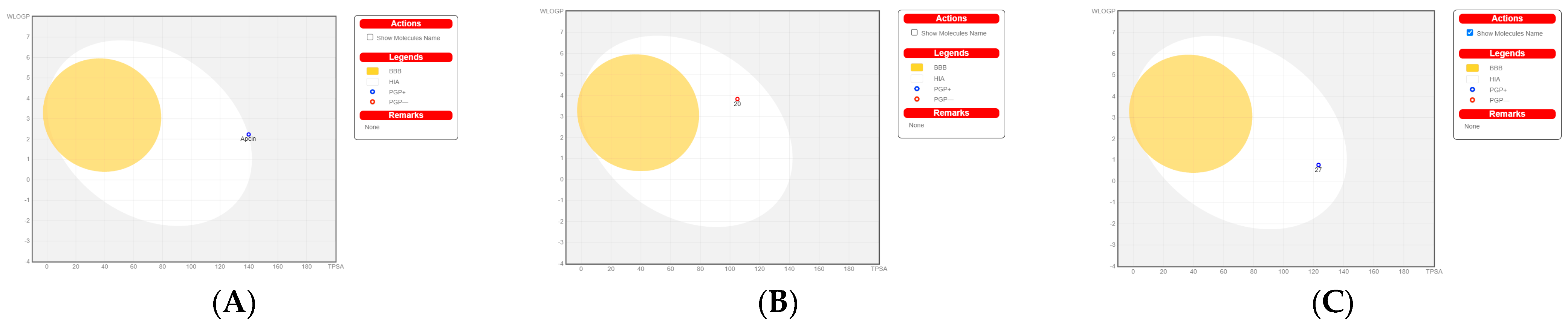
2.8. Bioled-Egg Model Analysis
3. Materials and Methods
3.1. Chemicals
3.1.1. General Procedure for the Synthesis of the Final Compounds 1–17
3.1.2. General Procedure for the Synthesis of the Final Compounds 18–30
3.2. Cell Culture and Cytotoxicity Assay
3.3. Surface Plasmon Resonance Analysis
3.4. Western Blot Assay
3.5. Cell-Apoptosis Assay
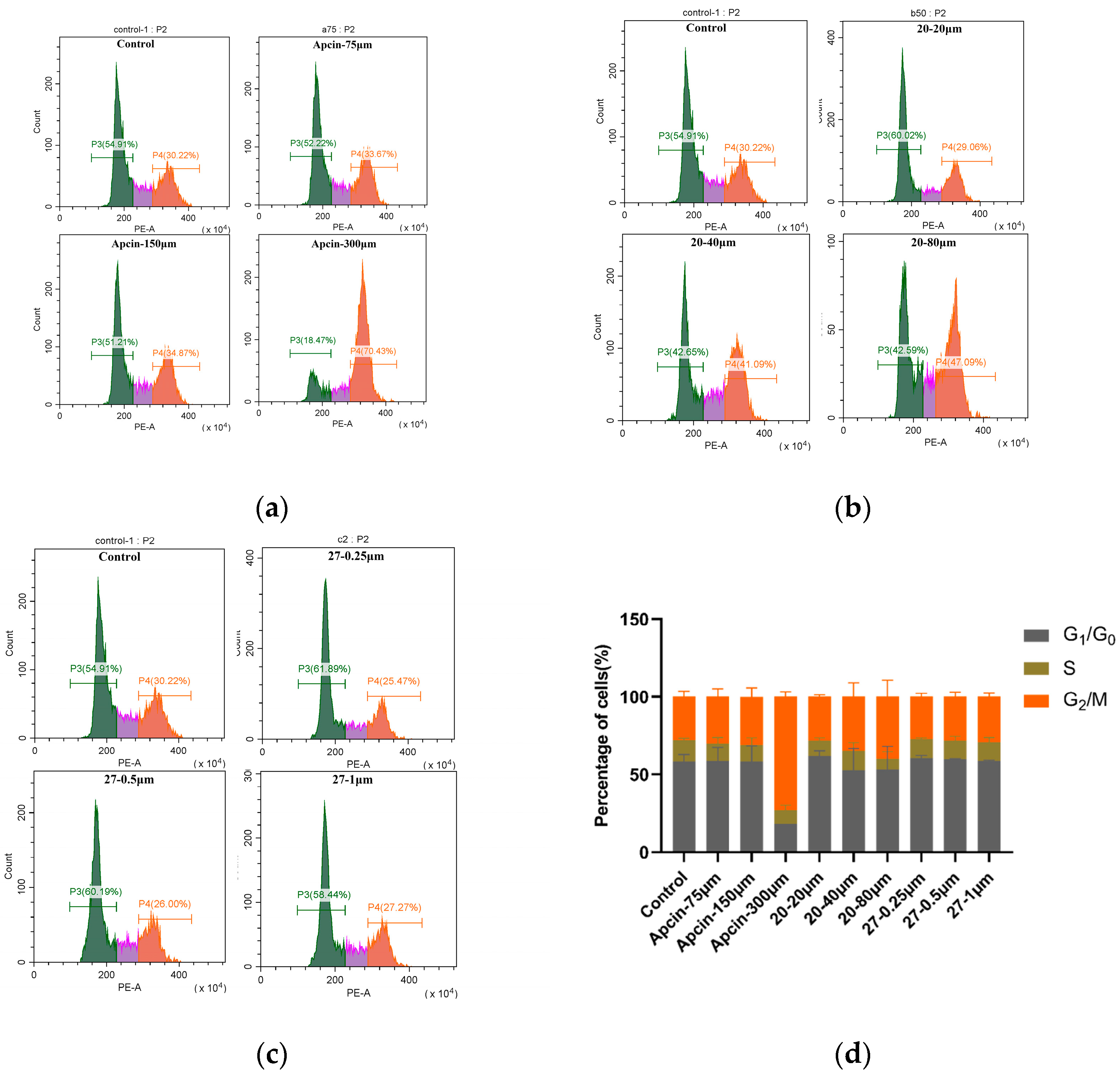
3.6. Cell-Cycle Assay
3.7. Tubulin Polymerization Assay In Vitro
3.8. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schrock, M.S.; Stromberg, B.R.; Scarberry, L.; Summer, M.K. APC/C ubiquitin ligase: Functions and mechanisms in tumorigenesis. Cancer Biol. 2020, 67, 80–91. [Google Scholar] [CrossRef]
- Brown, N.G.; VanderLinden, R.; Watson, E.; Weissmann, F.; Ordureau, A.; Wu, K.-P.; Zhang, W.; Yu, S.; Mercredi, P.; Harrison, J.; et al. Dual RING E3 architectures regulate multiubiquitination and ubiquitin chain elongation by APC/C. Cell 2016, 165, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.; McCormack, E.; Au, S.; Paul, A.; Willison, K.; Harper, W.; Barford, D. Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. EMBO J. 2003, 22, 786–796. [Google Scholar] [CrossRef]
- da Fonseca, P.; Kong, E.; Zhang, Z.; Schreiber, A.; Williams, M.; Morris, E.; Barford, D. Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature 2011, 470, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Vodermaier, H.; Maurer-Stroh, S.; Eisenhaber, F.; Peters, J.-M. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol. Cell 2005, 18, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Li, B.; Warrington, R.; Tomchick, D.; Yu, H.; Luo, X. Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. USA 2012, 109, 18419–18424. [Google Scholar] [CrossRef]
- Musacchio, A.; Salmon, E. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Sudakin, V.; Chan, G.K.; Yen, T.J. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 2001, 154, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, B.; Yu, H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef]
- Chang, D.; Ma, Y.; Ji, B.; Liu, Y.; Hwu, P.; Abbruzzese, J.; Logsdon, C.; Wang, H. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J. Hematol. Oncol. 2012, 5, 15. [Google Scholar] [CrossRef]
- Karra, H.; Repo, H.; Ahonen, I.; Löyttyniemi, E.; Pitkänen, R.; Lintunen, M.; Kuopio, T.; Söderström, M.; Kronqvist, P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br. J. Cancer 2014, 110, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Daigo, Y.; Aragaki, M.; Ishikawa, K.; Sato, M.; Kaji, M. Overexpression of KIAA0101 predicts poor prognosis in primary lung cancer patients. Lung Cancer 2012, 75, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-j.; Hu, K.-s.; Wang, D.-s.; Zeng, Z.-l.; Zhang, D.-s.; Chen, D.-l.; Bai, L.; Xu, R.-h. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J. Transl. Med. 2013, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Kim, H.-S.; Vassilopoulos, A.; Wang, R.-H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Ji, J.; et al. Knockout mouse models for sirtuins: Genome integrity, metabolism, and cancer. Cancer Res. 2011, 71, SY11-01. [Google Scholar] [CrossRef]
- Holland, A.J.; Taylor, S.S. CyclinB1-mediated inhbition of excess separase is required for timely dhromosone disjunction. J. Cell Sci. 2006, 119, 3325–3336. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.; Wandke, C.; Isenberg, N.; Geley, S. Dose-dependent effects of stab le cyclinB1 on progression through mitosis in human cells. EMBO 2006, 25, 2802–2813. [Google Scholar] [CrossRef]
- Listovsky, T.; Oren, Y.; Yudkovsky, Y.; Mahbubani, H.; Weiss, A.; Lebendiker, M.; Brandeis, M. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 2004, 23, 1619–1626. [Google Scholar] [CrossRef]
- Huang, P.; Le, X.; Huang, F.; Yang, J.; Yang, H.; Ma, J.; Hu, G.; Li, Q.; Chen, Z. Discovery of a dual tubulin polymerization and cell division cycle 20 homologue inhibitor via structural modification on Apcin. J. Med. Chem. 2020, 63, 4685–4700. [Google Scholar] [CrossRef] [PubMed]
- Harley, M.; Allan, L.; Sanderson, H.; Clarke, P. Phosphorylation of Mcl-1 by CDK1–cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Tan, M.; Yang, J.; Inuzuka, H.; Dai, X.; Wu, T.; Liu, J.; Shaik, S.; Chen, G.; Deng, J.; et al. APC Cdc20 suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev. Cell 2014, 29, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Guillamot, M.; de Cárcer, G.; Eguren, M.; Trickey, M.; García-Higuera, I.; Moreno, S.; Yamano, H.; Cañamero, M.; Malumbres, M. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α, δ phosphatase. Cancer Cell 2010, 18, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Richeson, K.V.; Bodrug, T.; Sackton, K.L.; Yamaguchi, M.; Paulo, J.A.; Gygi, S.P.; Schulman, B.A.; Brown, N.G.; King, R.W. Paradoxical mitotic exit induced by a small molecule inhibitor of APC/CCdc20. Nat. Chem. Biol. 2020, 16, 546–555. [Google Scholar] [CrossRef]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y. Improving scoring-docking-screeningpowers of protein–ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Structure | Number | Structure |
|---|---|---|---|
| 1 |  | 16 |  |
| 2 |  | 17 |  |
| 3 |  | 18 |  |
| 4 |  | 19 |  |
| 5 |  | 20 |  |
| 6 |  | 21 |  |
| 7 |  | 22 |  |
| 8 |  | 23 |  |
| 9 |  | 24 |  |
| 10 |  | 25 |  |
| 11 |  | 26 |  |
| 12 |  | 27 |  |
| 13 |  | 28 |  |
| 14 |  | 29 |  |
| 15 |  | 30 |  |
| Compound | IC50 ± SD (μM) | |||
|---|---|---|---|---|
| Mda-mb-231 | Hepg2 | Mcf-7 | Hela | |
| 1 | >100 | >100 | >100 | >100 |
| 2 | >100 | 66.26 ± 1.91 | 79.30 ± 20.34 | 86.71 ± 4.25 |
| 3 | >100 | >100 | >100 | >100 |
| 4 | >100 | >100 | >100 | >100 |
| 5 | >100 | >100 | >100 | >100 |
| 6 | >100 | >100 | >100 | >100 |
| 7 | 34.71 ± 1.23 | 20.69 ± 3.60 | 21.34 ± 4.52 | 34.21 ± 5.16 |
| 8 | >100 | >100 | >100 | >100 |
| 9 | >100 | >100 | >100 | >100 |
| 10 | >100 | >100 | >100 | >100 |
| 11 | >100 | >100 | >100 | >100 |
| 12 | >100 | 27.11 ± 5.27 | 27.72 ± 2.13 | 43.54 ± 20.35 |
| 13 | >100 | >100 | >100 | >100 |
| 14 | >100 | 51.81 ± 2.43 | 44.70 ± 7.62 | 79.61 ± 45.23 |
| 15 | >100 | >100 | >100 | >100 |
| 16 | >100 | >100 | >100 | >100 |
| 17 | 25.18 ± 10.77 | >100 | >100 | 60.00 ± 7.55 |
| 18 | 50.38 ± 3.07 | 30.59 ± 0.10 | 33.81 ± 1.91 | 36.68 ± 1.23 |
| 19 | 68.9 ± 2.67 | 27.76 ± 1.85 | 23.50 ± 4.17 | 34.04 ± 0.02 |
| 20 | 30.45 ± 13.01 | 31.19 ± 8.37 | 34.95 ± 3.06 | 24.71 ± 2.01 |
| 21 | >100 | >100 | >100 | >100 |
| 22 | 27.81 ± 0.17 | 26.13 ± 0.05 | 24.81 ± 9.49 | 31.91 ± 3.29 |
| 23 | >100 | 67.92 ± 6.74 | 60.40 ± 3.91 | >100 |
| 24 | 58.56 ± 18.38 | 32.36 ± 5.00 | 29.15 ± 3.46 | >100 |
| 25 | 52.21 ± 0.63 | 32.48 ± 6.31 | 37.31 ± 1.63 | 41.62 ± 8.81 |
| 26 | 61.65 ± 11.11 | 32.76 ± 3.94 | 44.82 ± 8.78 | 44.73 ± 4.60 |
| 27 | 0.32 ± 0.04 | 0.24 ± 0.11 | 0.27 ± 0.06 | 0.06 ± 0.02 |
| 28 | 0.61 ± 0.12 | 0.28 ± 0.13 | 0.41 ± 0.05 | 0.17 ± 0.08 |
| 29 | 0.37 ± 0.07 | 0.13 ± 0.05 | 0.23 ± 0.09 | 0.08 ± 0.01 |
| 30 | 73.02 ± 42.68 | 58.01 ± 10.56 | 72.40 ± 30.90 | >100 |
| 9f | 1.40 ± 0.22 | 0.41 ± 0.36 | 0.61 ± 0.12 | 0.10 ± 0.02 |
| Apcin | >100 | >100 | >100 | 181.88 ± 12.49 |
| 7b# | / | 13.6 ± 3.1 | 114.0 ± 5.6 | 27.1 ± 21.0 |
| 7d# | / | 25.6 ± 6.1 | 159.5 ± 5.0 | 63.2 ± 0.9 |
| Compounds Number | CLogP | Toxicity Probability (Value) | |||
|---|---|---|---|---|---|
| Hepatotoxicity | Reproductive Toxicity | Nephrotoxicity | Acute Oral Toxicity | ||
| 1 | 0.82 | 0.8446 (+) | 0.8778 (+) | 0.8365 (-) | 0.5778 (III) |
| 2 | 2.76 | 0.7250 (+) | 0.8444 (+) | 0.6910 (-) | 0.6629 (III) |
| 3 | 2.57 | 0.8282 (+) | 0.8222 (+) | 0.6370 (-) | 0.6566 (III) |
| 4 | 1.12 | 0.6500 (+) | 0.8556 (+) | 0.6890 (-) | 0.6584 (III) |
| 5 | 1.53 | 0.8282 (+) | 0.8125 (+) | 0.5677 (+) | 0.6821 (III) |
| 6 | 2.09 | 0.8375 (+) | 0.6000 (+) | 0.5278 (+) | 0.6001 (III) |
| 7 | 3.78 | 0.7909 (+) | 0.8556 (+) | 0.5000 (+) | 0.6326 (III) |
| 8 | 3.46 | 0.8250 (+) | 0.6333 (+) | 0.7849 (+) | 0.6416 (III) |
| 9 | 3.31 | 0.6750 (+) | 0.8444 (+) | 0.6964 (-) | 0.4618 (III) |
| 10 | 2.56 | 0.8125 (+) | 0.5889 (+) | 0.5186 (+) | 0.5953 (III) |
| 11 | 3.54 | 0.7949 (+) | 0.6667 (+) | 0.5442 (+) | 0.6387 (III) |
| 12 | 4.24 | 0.6574 (+) | 0.8667 (+) | 0.6711 (-) | 0.6240 (III) |
| 13 | 1.10 | 0.7875 (+) | 0.8000 (+) | 0.4630 (+) | 0.5615 (III) |
| 14 | 2.19 | 0.8000 (+) | 0.8111 (+) | 0.6556 (+) | 0.5431 (III) |
| 15 | 2.62 | 0.7324 (+) | 0.8444 (+) | 0.6566 (-) | 0.6356 (III) |
| 16 | 3.28 | 0.7324 (+) | 0.8556 (+) | 0.6775 (-) | 0.6327 (III) |
| 17 | 3.67 | 0.7449 (+) | 0.8556 (+) | 0.6353 (-) | 0.6327 (III) |
| 18 | 4.16 | 0.6824 (+) | 0.8444 (+) | 0.7312 (-) | 0.6618 (III) |
| 19 | 3.76 | 0.6750 (+) | 0.9000 (+) | 0.4574 (+) | 0.6006 (III) |
| 20 | 3.37 | 0.6875 (+) | 0.8667 (+) | 0.6345 (-) | 0.5737 (III) |
| 21 | 1.98 | 0.6500 (+) | 0.8444 (+) | 0.5688 (-) | 0.6185 (III) |
| 22 | 4.03 | 0.6324 (+) | 0.8556 (+) | 0.6663 (-) | 0.6558 (III) |
| 23 | 2.82 | 0.7199 (+) | 0.8444 (+) | 0.7814 (-) | 0.6667 (III) |
| 24 | 2.88 | 0.6532 (+) | 0.8778 (+) | 0.6251 (-) | 0.6189 (III) |
| 25 | 3.00 | 0.6074 (+) | 0.8333 (+) | 0.7259 (-) | 0.6482 (III) |
| 26 | 3.05 | 0.7199 (+) | 0.8333 (+) | 0.7440 (-) | 0.6482 (III) |
| 27 | 0.89 | 0.5375 (+) | 0.9222 (+) | 0.7872 (-) | 0.6717 (III) |
| 28 | 2.53 | 0.5500 (+) | 0.9556 (+) | 0.8026 (-) | 0.6142 (III) |
| 29 | 0.73 | 0.7000 (+) | 0.9000 (+) | 0.8905 (-) | 0.5652 (III) |
| 30 | 3.47 | 0.7034 (+) | 0.8556 (+) | 0.4722 (+) | 0.6156 (III) |
| Apcin | 1.22 | 0.8177 (+) | 0.8556 (+) | 0.7157 (-) | 0.5846 (III) |
| 7d | 1.94 | 0.8052 (+) | 0.8778 (+) | 0.7515 (+) | 0.5856 (III) |
| 9f | 1.25 | 0.5198 (+) | 0.9444 (+) | 0.6703 (-) | 0.6240 (III) |
| Compounds | Ka (1/M * S) | Kd (1/S) | Kd (μM) | IC50 (μM) Hela |
|---|---|---|---|---|
| 20 | 726 | 5.78 × 10−3 | 79.6 | 24.71 ± 2.01 |
| 27 | 64.0 | 6.21 × 10−3 | 97.0 | 0.06 ± 0.02 |
| Apcin | 985 | 2.33 × 10−2 | 236 | 181.88 ± 12.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Le, X.; Hu, G.; Li, Q.; Chen, Z. Discovery of Ureido-Based Apcin Analogues as Cdc20-specific Inhibitors against Cancer. Pharmaceuticals 2023, 16, 304. https://doi.org/10.3390/ph16020304
He Y, Le X, Hu G, Li Q, Chen Z. Discovery of Ureido-Based Apcin Analogues as Cdc20-specific Inhibitors against Cancer. Pharmaceuticals. 2023; 16(2):304. https://doi.org/10.3390/ph16020304
Chicago/Turabian StyleHe, Yiqin, Xiangyang Le, Gaoyun Hu, Qianbin Li, and Zhuo Chen. 2023. "Discovery of Ureido-Based Apcin Analogues as Cdc20-specific Inhibitors against Cancer" Pharmaceuticals 16, no. 2: 304. https://doi.org/10.3390/ph16020304
APA StyleHe, Y., Le, X., Hu, G., Li, Q., & Chen, Z. (2023). Discovery of Ureido-Based Apcin Analogues as Cdc20-specific Inhibitors against Cancer. Pharmaceuticals, 16(2), 304. https://doi.org/10.3390/ph16020304