
Harnessing Folate-Functionalized Nasal Delivery of Dox–Erlo-Loaded Biopolymeric Nanoparticles in Cancer Treatment: Development, Optimization, Characterization, and Biodistribution Analysis

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
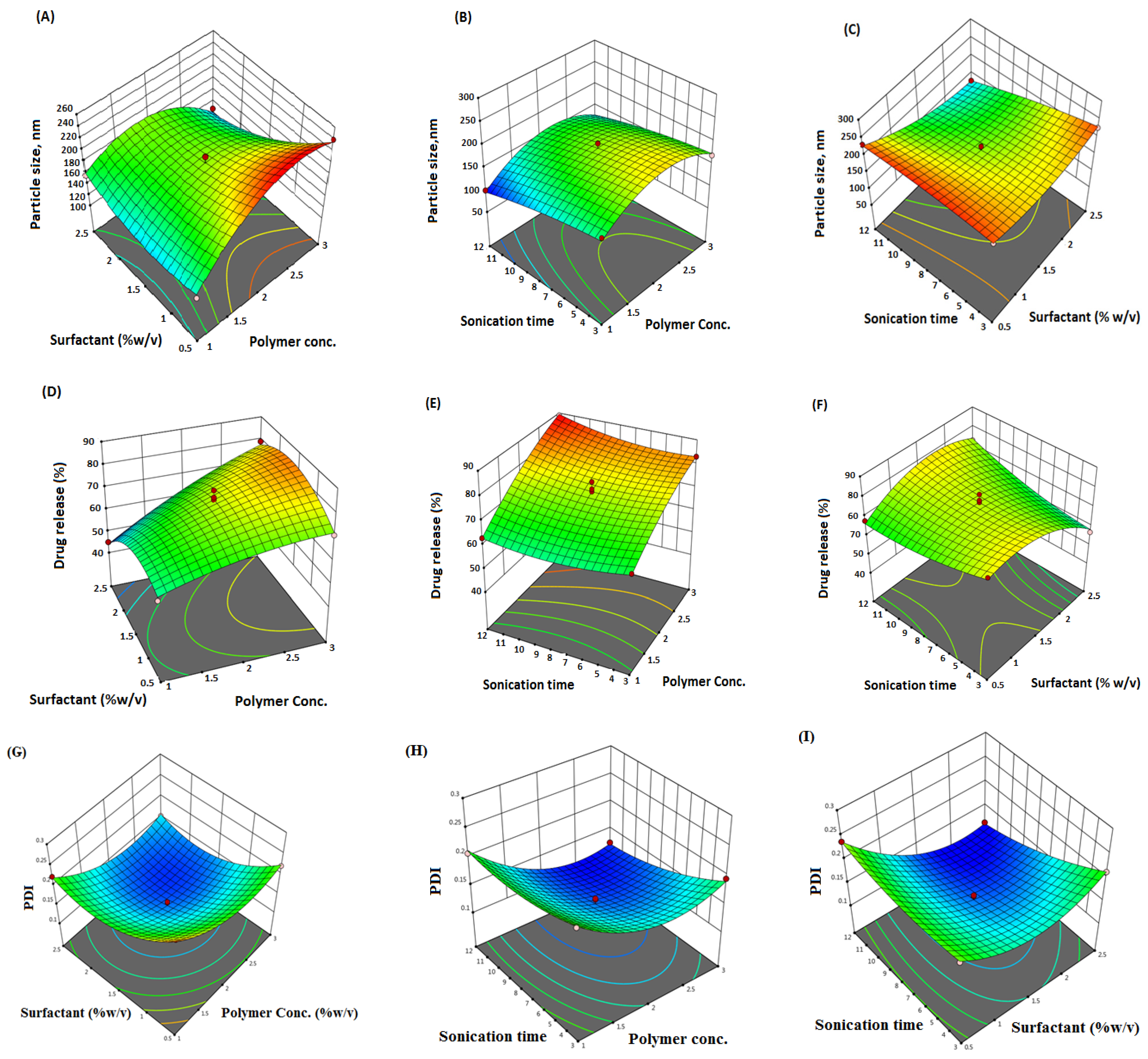
2.1. Formulation Optimization
2.2. Response 1: Effect on Particle Size
2.3. Response 2: Effect on % Drug Release
2.4. Response 3: Effect on the PDI
2.5. Characterization of Dox–ErloNPs
2.5.1. Particle Size and Zeta Potential
2.5.2. DSC of Dox–Erlo NPs
2.5.3. FT-IR Spectral Analysis
2.5.4. Proton Nuclear Magnetic Resonance (1H NMR)
2.5.5. X-ray Diffraction Analysis
2.5.6. In Vitro Drug Release
2.5.7. Kinetic Release Model
2.5.8. Hemolysis Study
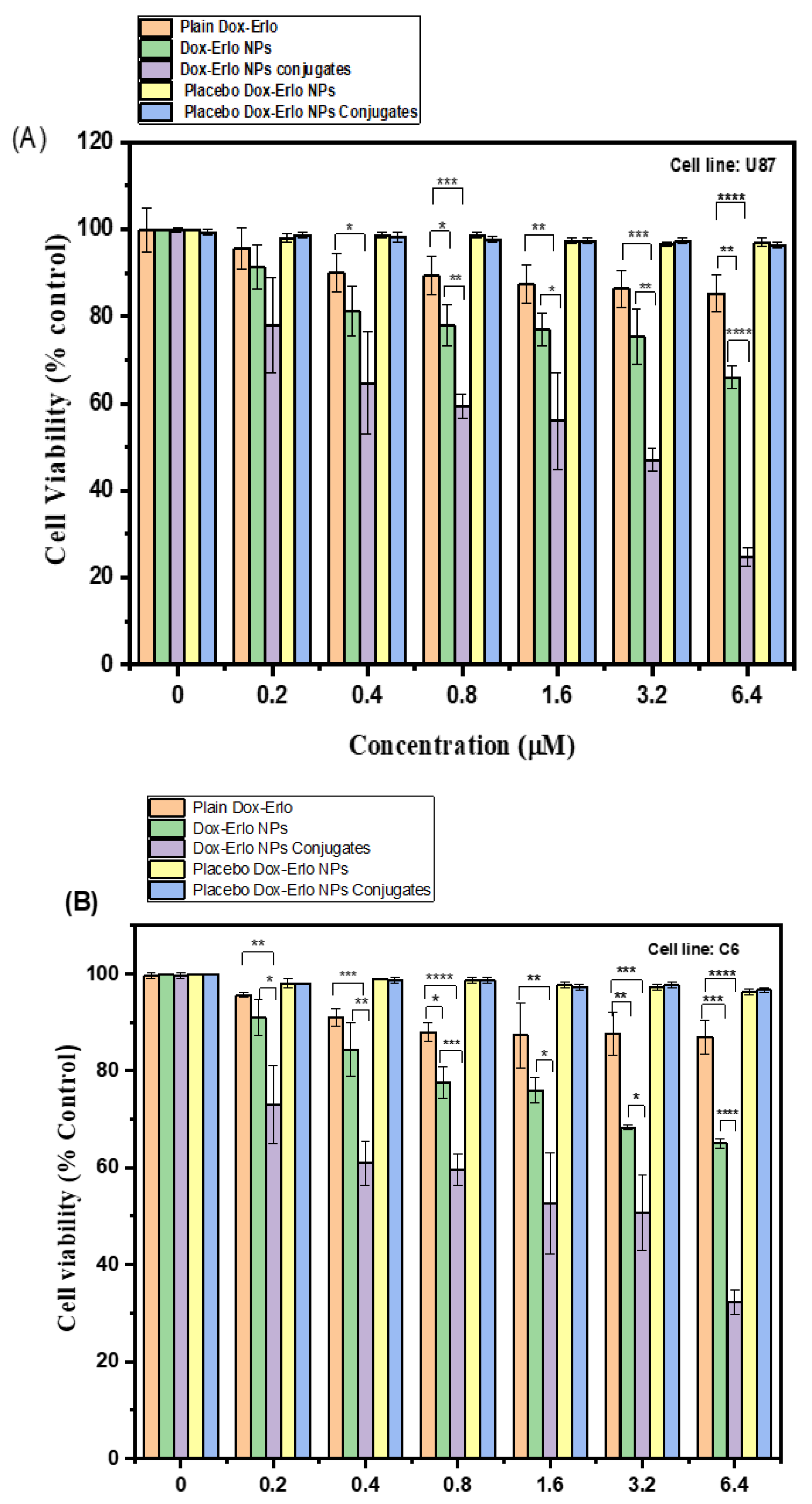
2.5.9. Cytotoxicity Assay
2.5.10. Biodistribution Study
2.5.11. Stability Study
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Cytotoxicity Study
Materials
4.3. Formulation Optimization Using Statistical Design
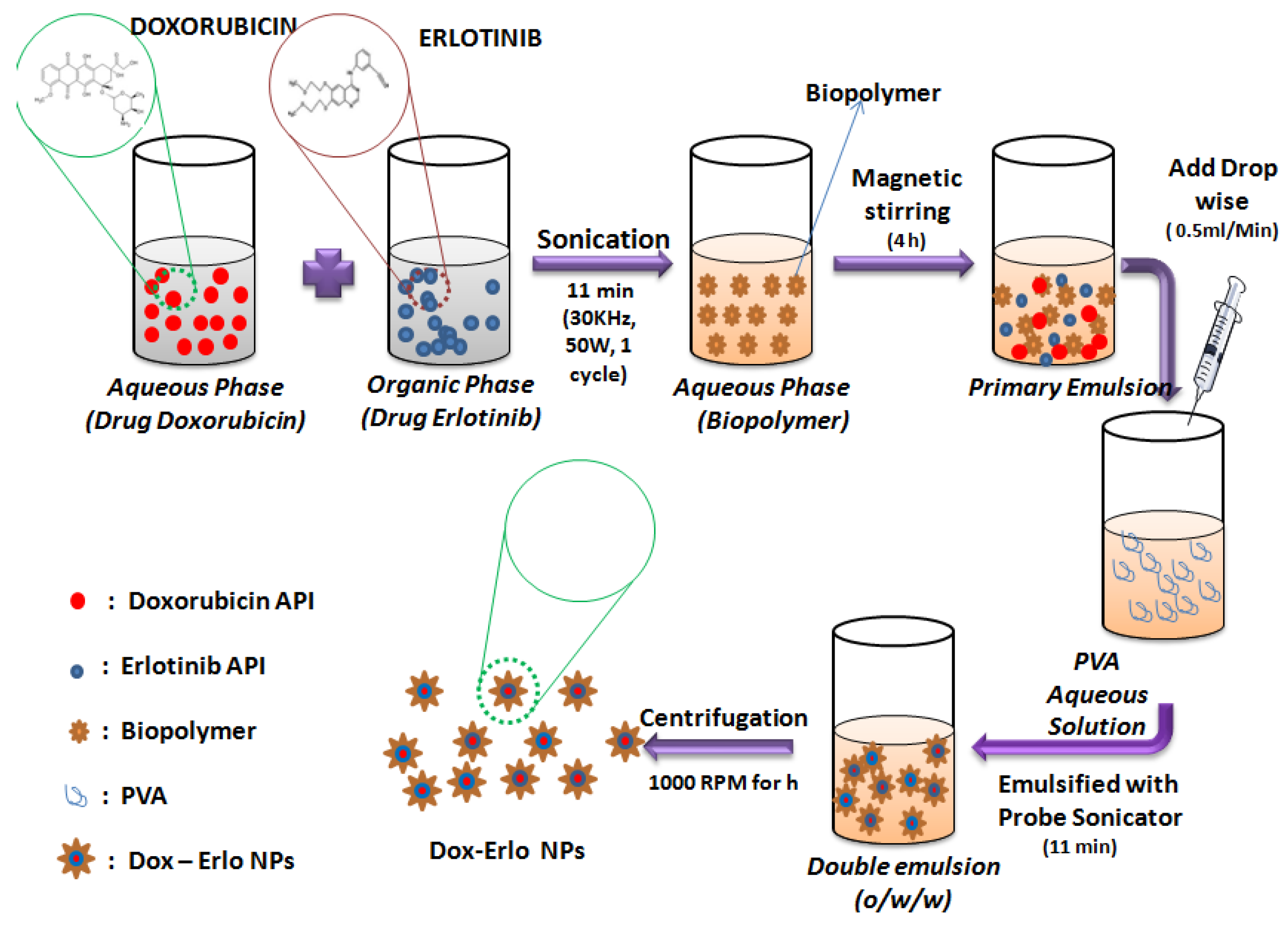
4.4. Preparation of Dox–Erlo-Loaded NPs
4.5. Surface Modification of Dox–Erlo Biopolymeric NPs
4.6. Characterization of Dox–ErloNanoparticles
4.6.1. Particle Analysis and Z-Average
4.6.2. Drug Entrapment and Loading in NPs
4.6.3. High-Resolution Transmission Electron Microscopy (HR-TEM)
4.6.4. Fourier Transform Infrared Spectroscopy (FT-IR)
4.6.5. Differential Scanning Calorimetry (DSC)
4.6.6. X-ray Diffraction (XRD)
4.6.7. Proton-Nucleic Magnetic Resonance (1H-NMR)
4.6.8. In Vitro Release Studies
4.6.9. Hemolysis Study
4.6.10. Cytotoxicity Study
4.6.11. Biodistribution Studies
4.6.12. Stability Study
4.6.13. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Amin, S.; Ahmad, M.Z.; Minhaj, M.A.; Mujtaba, M.A.; Ali, J. Molecular Targets and Nanoparticulate Systems Designed for the Improved Therapeutic Intervention in Glioblastoma Multiforme. Drug Res. 2021, 71, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Aaron, C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar]
- Clarke, J.; Butowski, N.; Chang, S. Recent Advances in Therapy for Glioblastoma. Arch. Neurol. 2010, 67, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Y.A.; Alfaifi, M.Y.; Shati, A.A.; Elbehairi, S.E.I.; Elshaarawy, R.F.M.; Kamal, I. Co-delivery of anticancer drugs via poly(ionic crosslinked chitosan-palladium) nanocapsules: Targeting more effective and sustainable cancer therapy. J. Drug Del. Sci. Technol. 2022, 69, 103151. [Google Scholar] [CrossRef]
- Jani, P.; Suman, S.; Subramanian, S.; Korde, A.; Gohel, D.; Singh, R.; Sawant, K. Development of mitochondrial targeted theranostic nanocarriers for treatment of gliomas. J. Drug Del. Sci. Technol. 2021, 64, 102648. [Google Scholar] [CrossRef]
- Akhter, M.H.; Madhav, N.S.; Ahmad, J. Epidermal growth factor receptor based active targeting: A paradigm shift towards advance tumor therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1188–1198. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Finch, A.; Solomou, G.; Wykes, V.; Pohl, U.; Bardella, C.; Watts, C. Advances in Research of Adult Gliomas. Int. J. Mol. Sci. 2021, 22, 924. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 11292. [Google Scholar] [CrossRef]
- Wong, I.Y.; Bhatia, S.N.; Toner, M. Nanotechnology: Emerging tools for biology and medicine. Genes. Dev. 2013, 27, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.H.; Khalilullah, H.; Gupta, M.; Alfaleh, M.A.; Alhakamy, N.A.; Riadi, Y.; Shadab, M. Impact of Protein Corona on the Biological Identity of Nanomedicine: Understanding the Fate of Nanomaterials in the Biological Milieu. Biomedicines 2021, 9, 1496. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.H.D.; Borm, P.J.A. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef]
- Ahmad, J.; Ameeduzzafar, M.Z.; Ahmad, J.; Akhter, M.H. Surface-Engineered Cancer Nanomedicine: Rational Design and Recent Progress. Curr. Pharm. Des. 2020, 26, 1181–1190. [Google Scholar] [CrossRef]
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Ahsan, M.J.; Mujtaba, M.A.; Amin, S. Nanocarriers in advanced drug targeting: Setting novel paradigm in cancer therapeutics. Artif. Cells Nanomed. Biotechnol. 2018, 46, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.H.; Beg, S.; Tarique, M.; Malik, A. Receptor-based targeting of engineered nanocarrier against solid tumors: Recent progress and challenges ahead. Biochim. Et Biophys. Acta-Gen. Subj. 2021, 1865, 129777. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.; Campos, E.V.R. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, J.; Tan, T.; Liu, M.; Zeng, Z.; Zeng, Y.; Zhang, L.; Fu, C.; Chen, D.; Xie, T. Nanoparticle Drug Delivery System for Glioma and Its Efficacy Improvement Strategies: A Comprehensive Review. Int. J. Nanomed. 2020, 5, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Luque-Michel, E.; Lemaire, L.; Blanco-Prieto, M.J. SPION and doxorubicin-loaded polymeric nanocarriers for glioblastoma theranostics. Drug Deliv. Transl. Res. 2021, 11, 515–523. [Google Scholar] [CrossRef]
- Alibolandi, M.; Farzad, S.A.; Mohammadi, M.; Abnous, K.; Taghdisi, S.M.; Kalalinia, F.; Ramezani, M. Tetrac-decorated chitosan-coated PLGA nanoparticles as a new platform for targeted delivery of SN38. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1003–1014. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Akhter, M.H.; Burzangi, A.S.; Alkreathy, H.; Alharthy, B.; Kotta, S.; Shadab, M.; Rashid, M.A.; Afzal, O.; Altamimi, A.S.A.; et al. Phytosterol-Loaded Surface-Tailored Bioactive-Polymer Nanoparticles for Cancer Treatment: Optimization, In Vitro Cell Viability, Antioxidant Activity, and Stability Studies. Gels 2022, 8, 219. [Google Scholar] [CrossRef] [PubMed]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Haponiuk, J.T.; Thomas, S.; Gopi, S. Biopolymer based nanomaterials in drug delivery systems: A review. Mater. Today Chem. 2018, 9, 43–55. [Google Scholar] [CrossRef]
- Sadasivuni, K.K.; Saha, P.; Adhikari, J.; Deshmukh, K.; Ahamed, M.B.; Cabibihan, J.J. Recent advances in mechanical properties of biopolymer composites: A review. Polym. Compos. 2020, 41, 32–59. [Google Scholar] [CrossRef]
- Venugopala, V.; Kumara, K.J.; Muralidharanc, S.; Parasuramanb, S.; Raja, P.V.; Kumara, K.V. Optimization and in-vivo evaluation of isradipine nanoparticles using Box-Behnken design surface response methodology. OpenNano 2016, 1, 1–15. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modelling on drug release from cnotrolled drug delivery system. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Akhter, M.H.; Ahmad, A.; Ali, J.; Mohan, G. Formulation and Development of CoQ10-Loaded s-SNEDDS for Enhancement of Oral Bioavailability. J. Pharm. Innov. 2014, 9, 121–131. [Google Scholar] [CrossRef]
- Zhang, C.; Song, J.; Lou, L.; Qi, X.; Zhao, L.; Fan, B.; Sun, G.; Lv, Z.; Fan, Z.; Jiao, B.; et al. Doxorubicin-loaded nanoparticle coated with endothelial cells-derived exosomes for immunogenic chemotherapy of glioblastoma. Bioeng. Transl. Med. 2020, 3, e10203. [Google Scholar] [CrossRef]
- Amasya, G.; Ozturk, C.; Aksu, B.; Tarimci, N. QbD based formulation optimization of semi-solid lipid nanoparticles as nano-cosmeceuticals. J. Drug Deliv. Sci. Technol. 2021, 66, 102737. [Google Scholar] [CrossRef]
- Bellotti, E.; Cascone, M.G.; Barbani, N.; Rossin, D.; Rastaldo, R.; Giachino, C.; Cristallini, C. Targeting Cancer Cells Overexpressing Folate Receptors with New Terpolymer-Based Nanocapsules: Toward a Novel Targeted DNA Delivery System for Cancer Therapy. Biomedicines 2021, 9, 1275. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Uddin, N.; Girgis, S. Formulation and optimization of Biodegradable Insulin Loaded Nanoparticles. Int. J. Pharm. Sci. Health Care 2019, 3, 12–35. [Google Scholar] [CrossRef]
- Cheng, L.; Ma, H.; Shao, M.; Fan, Q.; Lv, H.; Peng, J.; Hao, T.; Li, D.; Zhao, C.; Zong, X. Synthesis of folate-chitosan nanoparticles loaded with ligustrazine to target folate receptor positive cancer cells. Mol. Med. Rep. 2017, 16, 1101–1108. [Google Scholar] [CrossRef]
- Parijat, P.; Kamal, D.; Harish, D. Erlotinib loaded chitosan nanoparticles: Formulation, physicochemical characterization and cytotoxic potential. Int. J. Biolog. Macromol. 2019, 139, 1304–1316. [Google Scholar]
- Zhou, X.; Tao, H.; Shi, K.H. Development of a nanoliposomal formulation of erlotinib for lung cancer and in vitro/in vivo antitumoral evaluation. Drug Des. Devel. Ther. 2017, 18, 1–8. [Google Scholar] [CrossRef]
- Sharma, G.; Modgil, A.; Layek, B.; Arora, K.; Sun, C.; Law, B.; Singh, J. Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: Biodistribution and transfection. J. Control. Release 2013, 167, 1–10. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Abbasalipourkabir, R.; Jalilian, F.A.; Asl, S.S.; Farmany, A.; Roshanaei, G.; Arabestani, M.R. Doxycycline-encapsulated solid lipid nanoparticles as promising tool against Brucella melitensis enclosed in macrophage: A pharmacodynamics study on J774A.1 cell line. Antimicrob. Resist Infect. Control 2019, 3, 8–62. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Li, S.; Liu, Z. Development and Evaluation of Multifunctional Poly (Lactic-co-glycolic acid) Nanoparticles Embedded in Carboxymethyl-Glucan Porous Microcapsules as a Novel Drug Delivery System for Gefitinib. Pharmaceutics 2019, 11, 469. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Langova, V.; Bailey, D.; Pattison, S.T.; Pattison, S.L.; Christensen, N.; Armstrong, L.R.; Brahmbhatt, V.N.; Smolarczyk, K.; Harrison, M.T.; et al. Targeted Doxorubicin Delivery to Brain Tumors via Minicells: Proof of Principle Using Dogs with Spontaneously Occurring Tumors as a Model. PLoS ONE 2016, 11, e0151832. [Google Scholar] [CrossRef]
- He, Y.; Su, Z.; Zhang, C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J. Control. Release 2016, 229, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef]
- Zhou, L.; Cheng, R.; Tao, H.; Ma, S.; Guo, W.; Meng, F.; Liu, H.; Liu, Z.; Zhong, Z. Endosomal pH-activatable poly(ethylene oxide)-graft-doxorubicin prodrugs: Synthesis, drug release, and biodistribution in tumor-bearing mice. Biomacromol 2011, 12, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chianga, C.F.; Chenc, L.F.; Liangad, P.C.; Hsiehb, W.Y.; Linae, W.L. Polymersomes conjugated with des-octanoyl ghrelin and folate as a BBB-penetrating cancer cell-targeting delivery system. Biomaterials 2014, 35, 4066–4081. [Google Scholar] [CrossRef]
- Tzeyung, A.S.; Shadab, M.; Bhattamisra, S.K.; Madheswaran, T.; Alhakamy, N.A.; Aldawsari, H.M.; Radhakrishnan, A.K. Fabrication, optimization, and Evaluation of Rotigotine-Loaded Chitosan Nanoparticles for Nose-To-Brain Delivery. Pharmaceutics 2019, 11, 26. [Google Scholar] [CrossRef]
- Pan, D.; Vargas-Morales, O.; Zern, B.; Anselmo, A.C.; Gupta, V.; Zakrewsky, M.; Mitragotri, S.; Muzykantov, V. The Effect of Polymeric Nanoparticles on Biocompatibility of Carrier Red Blood Cells. PLoS ONE 2016, 11, e0152074. [Google Scholar] [CrossRef]
- ICH Q1A (R2). Stability Testing Guidelines: Stability Testing of New Drug Substances and Products. ICH Step 5. CPMP/ICH/2736/99. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf (accessed on 13 July 2022).
- Zhao, Z.; Liu, W.; Jiang, Y.; Wan, Y.; Du, R.; Li, H. Solidification of heavy metals in lead smelting slag and development of cementitious materials. J. Clean. Prod. 2022, 359, 132134. [Google Scholar] [CrossRef]
- Xu, H.; Rahimpour, S.; Nesvick, C.L.; Zhang, X.; Ma, J.; Zhang, M.; Zhang, G.; Wang, L.; Yang, C.; Hong, C.S.; et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: Implications for bevacizumab antiangiogenic therapy. Oncotarget 2015, 6, 3592. [Google Scholar] [CrossRef]
- Kumar, S.; Besra, S.E. The Growth Suppressing Activity Of Spathodea Campanulata Bark On C6 & U87mg Involve Induction Of Apoptosis And Cell Cycle Arrest. World J. Pharm. Res. 2020, 9, 2517. [Google Scholar]
- Huang, C.; Luo, Y.; Zhao, J.; Yang, F.; Zhao, H.; Fan, W.; Ge, P. Shikonin Kills Glioma Cells through Necroptosis Mediated by RIP-1. PLoS ONE 2013, 8, e66326. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Rodrigues, B.S.; Sun, C.; Singh, J. Biodistribution of TAT or QLPVM coupled to receptor targeted liposomes for delivery of anticancer therapeutics to brain in vitro and in vivo. Nanomed. 2020, 23, 102112. [Google Scholar] [CrossRef] [PubMed]
- Shadab, M.; Alhakamy, N.A.; Aldawsari, H.M.; Husain, M.; Khan, N.; Alfaleh, M.A.; Asfour, H.Z.; Riadi, Y.; Bilgrami, A.L.; Akhter, M.H. Plumbagin-Loaded Glycerosome Gel as Topical Delivery System for Skin Cancer Therapy. Polymers 2021, 13, 923. [Google Scholar]
- Akhter, M.H.; Kumar, S.; Nomani, S. Sonication tailored enhance cytotoxicity of naringenin nanoparticle in pancreatic cancer: Design, optimization, and in vitro studies. Drug Dev. Ind. Pharm. 2020, 46, 659–672. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Lakkadwala, S.; Rodrigues, B.D.S.; Sun, C.; Singh, J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J. Cont. Rel. 2019, 307, 247–260. [Google Scholar] [CrossRef]
- Zhou, Z.; Kennell, C.; Jafari, M.; Lee, J.Y.; Ruiz-Torres, S.J.; Waltz, S.E.; Lee, J.H. Sequential delivery of erlotinib and doxorubicin for enhanced triple negative Breast cancer treatment using polymeric nanoparticle. Int. J. Pharm. 2017, 530, 300–307. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Level Used | ||
|---|---|---|---|
| Low | Medium | High | |
| (−1) | (0) | (+1) | |
| Polymer concentration (A), % w/v | 1.00 | 2.00 | 3.00 |
| PVA (B), % w/v | 0.50 | 1.50 | 2.50 |
| Sonication Time (C), min | 3.00 | 7.50 | 12.00 |
| Dependent variables | |||
| Particle size (R1) | Minimize | ||
| PDI (R3) | Minimize | ||
| Drug release (R2) | Maximize | ||
| Run Order | (A) | (B) | (C) | Actual Value of R1 | Predicted Value of R1 | Actual Value of R2 | Predicted Value of R2 | Actual Value of R3 | Predicted Value of R3 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.00 | 1.50 | 12.00 | 121.00 | 128.00 | 89.00 | 89.50 | 0.1230 | 0.1208 |
| 2 | 2.00 | 0.50 | 3.00 | 230.00 | 230.13 | 78.00 | 77.00 | 0.2230 | 0.2253 |
| 3 | 2.00 | 1.50 | 7.50 | 180.00 | 190.00 | 71.00 | 75.00 | 0.1430 | 0.1332 |
| 4 | 3.00 | 0.50 | 7.50 | 240.00 | 236.12 | 71.00 | 72.00 | 0.2210 | 0.2235 |
| 5 | 1.00 | 1.50 | 3.00 | 159.00 | 152.00 | 65.00 | 64.50 | 0.2310 | 0.2332 |
| 6 | 1.00 | 1.50 | 12.00 | 100.00 | 96.25 | 63.00 | 63.00 | 0.2070 | 0.2117 |
| 7 | 2.00 | 2.50 | 3.00 | 227.00 | 230.13 | 57.00 | 58.50 | 0.2130 | 0.2132 |
| 8 | 2.00 | 1.50 | 7.50 | 201.00 | 190.00 | 74.00 | 75.00 | 0.1300 | 0.1332 |
| 9 | 2.00 | 1.50 | 7.50 | 189.00 | 190.00 | 79.00 | 75.00 | 0.1290 | 0.1332 |
| 10 | 3.00 | 1.50 | 3.00 | 170.00 | 173.75 | 83.00 | 83.00 | 0.1950 | 0.1903 |
| 11 | 2.00 | 1.50 | 7.50 | 200.00 | 190.00 | 76.00 | 75.00 | 0.1230 | 0.1332 |
| 12 | 2.00 | 2.50 | 12.00 | 130.00 | 129.88 | 73.00 | 74.00 | 0.1120 | 0.1097 |
| 13 | 1.00 | 0.50 | 7.50 | 136.00 | 142.87 | 59.00 | 60.50 | 0.2980 | 0.2935 |
| 14 | 1.00 | 2.50 | 7.50 | 156.00 | 159.88 | 45.00 | 44.00 | 0.2230 | 0.2205 |
| 15 | 3.00 | 2.50 | 7.50 | 127.00 | 120.12 | 79.00 | 77.50 | 0.1520 | 0.1565 |
| 16 | 2.00 | 0.50 | 12.00 | 232.00 | 228.88 | 68.00 | 66.50 | 0.2380 | 0.2377 |
| 17 | 2.00 | 1.50 | 7.50 | 180.00 | 190.00 | 75.00 | 75.00 | 0.1410 | 0.1332 |
| Quadratic Model | R–Squared | Adjusted R–Squared | Predicted R–Squared | SD | % CV |
|---|---|---|---|---|---|
| Response (R1) | 0.9768 | 0.9469 | 0.8327 | 9.94 | 5.68 |
| Response (R2) | 0.9740 | 0.9405 | 0.8524 | 2.60 | 3.68 |
| Response (R3) | 0.9914 | 0.9802 | 0.9502 | 0.0076 | 4.18 |
| Regression equation of the fitted quadratic model Particle size (R1) = +190.00 + 13.37 × A − 24.75 × B − 25.38 × C − 33.25 × A × B + 2.50 × A × C − 24.75 × B × C − 46.25 × A2 + 21.00 × B2 − 6.25 × C2 % Drug release (R2) = +75.00 + 11.25 × A − 2.75 × B +1.25×C + 5.50 × A × B + 2.00 × A × C + 6.50 × B × C − 2.75 × A2 − 8.75 × B2 + 2.75 × C2 PDI (R3) = +0.1332 − 0.0335 × A − 0.0350 × B − 0.0228 × C + 0.0015 × A × B − 0.0120 × A × C − 0.0290 × B × C + 0.0414 × A2 + 0.489 × B2 + 0.0144 × C2 | |||||
| Result of the Analysis of Variance | Particle Size (nm) | Drug Release (%) | PDI |
|---|---|---|---|
| 1. Regression analysis | |||
| Sum of squares | 29,090.22 | 1776.26 | 0.6131 |
| Degree of freedom (df) | 9 | 9 | 17 |
| Mean squares | 3232.25 | 197.36 | 0.0361 |
| F-value | 32.68 | 29.09 | 113.60 |
| p-Value | ˂0.0001 | ˂0.0001 | ˂0.0001 |
| 2. Lack-of-fit tests | |||
| Sum of squares | 270.25 | 13.50 | 0.0001 |
| df | 3 | 3 | 3 |
| Mean squares | 90.08 | 4.50 | 0.0000 |
| F-value | 0.8539 | 0.5294 | 0.5471 |
| p-Value | 0.5330 | 0.6858 | 0.6762 |
| Correlation of variation (% CV) | 5.68 | 3.68 | 4.18 |
| 3. Residual | |||
| Sum of squares | 692.25 | 47.50 | 0.0004 |
| df | 7 | 7 | 7 |
| Mean squares | 98.89 | 6.79 | 0.0001 |
| SD | 9.94 | 2.60 | 0.0076 |
| Variable Composition | Responses | Predicted Value | Experimental Value | % Error |
|---|---|---|---|---|
| A (2.94 % w/v) | R1 | 92.76 nm | 95.35 ± 10.25 nm | 2.79 |
| B (2.20 % w/v) | R2 | 89.91% | 79.203 ± 0.24% | 11.90 |
| C (11.39 min) | R3 | 0.102 | 0.109 | 6.8 |
| Erlo Release from Dox–Erlo-NP Conjugates at pH 5.4 | ||
| Zero order | 0.9002 | 1.65021 |
| First order | 0.9744 | −0.0355 |
| Higuchi matrix | 0.9096 | 9.7543 |
| Korsmeyer–Peppas | 0.9793 | 3.0543 |
| Hixson–Crowell | 0.9593 | 0.0090 |
| Dox release from Dox–Erlo-NP conjugates at pH 5.4 | ||
| Model Fitting | R2 | k |
| Zero order | 0.8541 | 1.2361 |
| First order | 0.9231 | −0.0195 |
| Higuchi Matrix | 0.9233 | 8.3559 |
| Korsmeyer–Peppas | 0.9751 | 2.5251 |
| Hixson–Crowell | 0.9025 | 0.0056 |
| Erlo Release from Dox–Erlo-NP Conjugates at pH 7.4 | ||
| Model Fitting | R2 | k |
| Zero order | 0.8704 | 1.3919 |
| First order | 0.9537 | −0.0244 |
| Higuchi Matrix | 0.9190 | 8.8754 |
| Korsmeyer–Peppas | 0.9782 | 2.7330 |
| Hixson–Crowell | 0.9306 | 0.0067 |
| Dox release from Dox–Erlo NPs conjugates at pH 7.4 | ||
| Model Fitting | R2 | k |
| Zero order | 0.8718 | 1.1629 |
| First order | 0.9294 | −0.0183 |
| Higuchi Matrix | 0.9062 | 8.3554 |
| Korsmeyer–Peppas | 0.9709 | 2.9451 |
| Hixson–Crowell | 0.9125 | 0.0052 |
| Sampling Period (in Days) | Particle Size (nm) | Zeta Potential (mV) | % Entrapment Efficiency | |||
|---|---|---|---|---|---|---|
| (25 ± 2 °C, 65 ± 5% RH) | (40 ± 2 °C, 75 ± 5% RH) | (25 ± 2 °C, 65 ± 5% RH) | (40 ± 2 °C, 75 ± 5% RH) | (25 ± 2 °C, 65 ± 5% RH) | (40 ± 2 °C, 75 ± 5% RH) | |
| 0 | 95.35 ± 10.23 | 95.35 ± 10.33 | −18.1 ± 2.40 | −18.1 ± 2.40 | 80 ± 2.3% | 80 ± 4.6% |
| 30 | 99.39 ± 11.03 | 100.46 ± 9.2 | −18.3 ± 3.40 | −19.3 ± 2.31 | 79.3 ± 3.4% | 79 ± 3.2% |
| 60 | 104.22 ± 13.44 | 106.25 ± 14.25 | −19.2 ± 3.24 | −20.2 ± 2.05 | 78 ± 3.8% | 77 ± 4.4% |
| 90 | 109.45 ± 12.48 | 115.33 ± 12.38 | −21.1 ± 4.01 | −20.4± 3.20 | 76 ± 5.3% | 73 ± 3.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farheen, M.; Akhter, M.H.; Chitme, H.; Akhter, M.S.; Tabassum, F.; Jaremko, M.; Emwas, A.-H. Harnessing Folate-Functionalized Nasal Delivery of Dox–Erlo-Loaded Biopolymeric Nanoparticles in Cancer Treatment: Development, Optimization, Characterization, and Biodistribution Analysis. Pharmaceuticals 2023, 16, 207. https://doi.org/10.3390/ph16020207
Farheen M, Akhter MH, Chitme H, Akhter MS, Tabassum F, Jaremko M, Emwas A-H. Harnessing Folate-Functionalized Nasal Delivery of Dox–Erlo-Loaded Biopolymeric Nanoparticles in Cancer Treatment: Development, Optimization, Characterization, and Biodistribution Analysis. Pharmaceuticals. 2023; 16(2):207. https://doi.org/10.3390/ph16020207
Chicago/Turabian StyleFarheen, Ms, Md Habban Akhter, Havagiray Chitme, Md Sayeed Akhter, Fauzia Tabassum, Mariusz Jaremko, and Abdul-Hamid Emwas. 2023. "Harnessing Folate-Functionalized Nasal Delivery of Dox–Erlo-Loaded Biopolymeric Nanoparticles in Cancer Treatment: Development, Optimization, Characterization, and Biodistribution Analysis" Pharmaceuticals 16, no. 2: 207. https://doi.org/10.3390/ph16020207
APA StyleFarheen, M., Akhter, M. H., Chitme, H., Akhter, M. S., Tabassum, F., Jaremko, M., & Emwas, A.-H. (2023). Harnessing Folate-Functionalized Nasal Delivery of Dox–Erlo-Loaded Biopolymeric Nanoparticles in Cancer Treatment: Development, Optimization, Characterization, and Biodistribution Analysis. Pharmaceuticals, 16(2), 207. https://doi.org/10.3390/ph16020207