
Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
3. Discussion
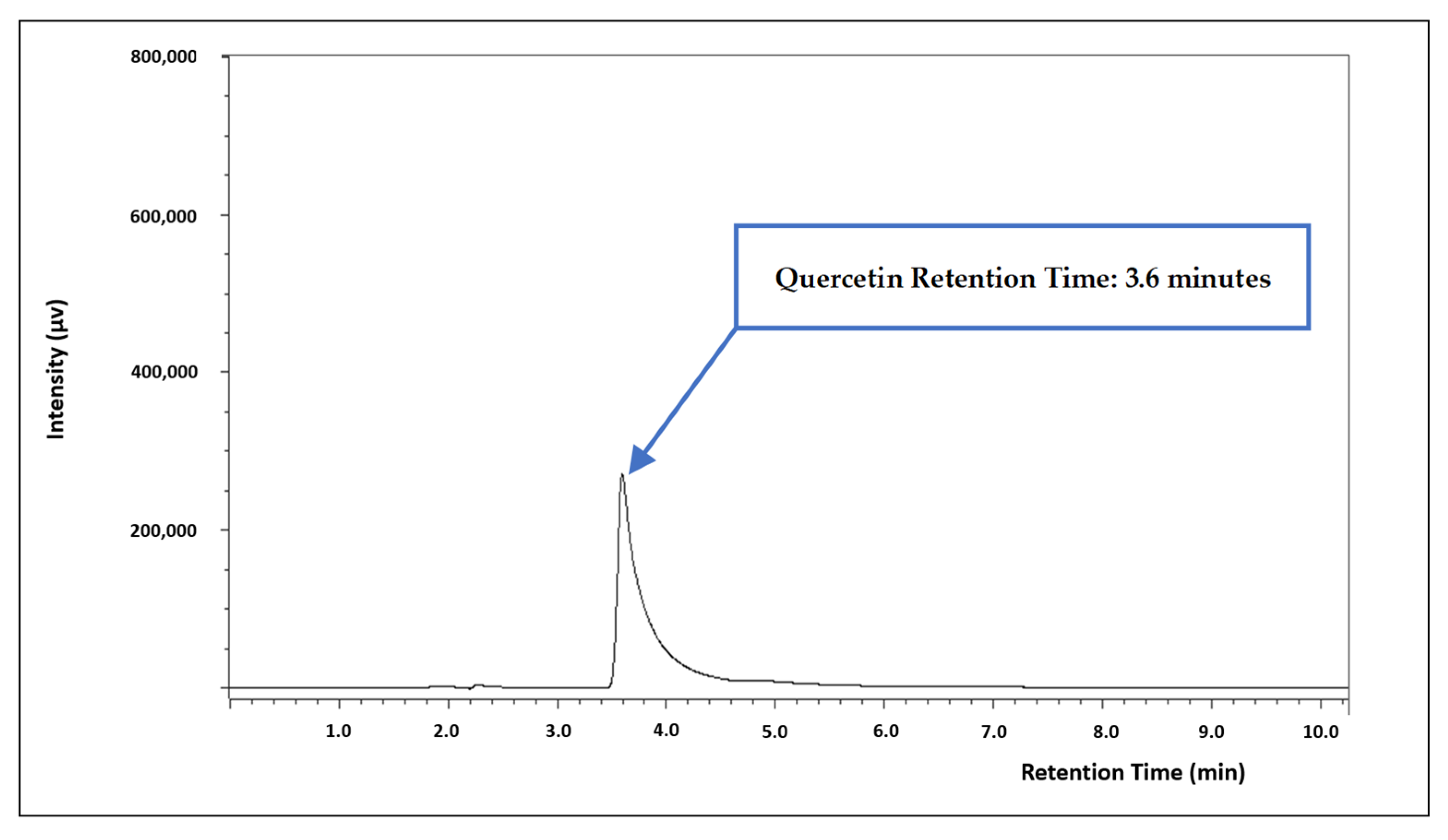
3.1. Chromatographic Conditions
3.2. Method Validation
3.2.1. Linearity
3.2.2. Detection and Quantification Limits
3.2.3. Precision
3.2.4. Accuracy
3.2.5. Specificity/Selectivity
3.2.6. Robustness
3.2.7. Stability
3.3. Overall Assessment of the Method Performance and Quercetin Quantification in Nanoparticles
3.4. Limitations
4. Materials and Methods
4.1. Reagents
4.2. Chromatographic Conditions
4.3. Standard Solution Preparation
4.4. Method Validation
4.4.1. Linearity and Range
4.4.2. Limit of Detection and Limit of Quantification
4.4.3. Precision
4.4.4. Accuracy
4.4.5. Specificity/Selectivity
4.4.6. Robustness
4.4.7. Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chew, Y.L.; Khor, M.A.; Lim, Y.Y. Choices of Chromatographic Methods as Stability Indicating Assays for Pharmaceutical Products: A Review. Heliyon 2021, 7, e06553. [Google Scholar] [CrossRef] [PubMed]
- Skoog, D.A.; Holler, F.J.; Crouch, S.R. Principles of Instrumental Analysis, 7th ed.; Cengage Learning: Boston, MA, USA, 2018; ISBN 978-1-305-57721-3. [Google Scholar]
- Harris, D.C. Quantitative Chemical Analysis, 8th ed.; W. H. Freeman and Company: New York, NY, USA, 2010; ISBN 978-1-4292-1815-3. [Google Scholar]
- Araujo, P. Key Aspects of Analytical Method Validation and Linearity Evaluation. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.N. Validation of Analytical Methods. In Calibration and Validation of Analytical Methods—A Sampling of Current Approaches; Stauffer, M.T., Ed.; InTech: Rijeka, Croatia, 2018; ISBN 978-1-78923-084-0. [Google Scholar]
- International Conference on Harmonization. Validation of Analytical Procedures: Q2(R2); International Conference on Harmonization: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Diao, P.; Shu, X.; Li, L.; Xiong, L. Quercetin and Quercitrin Attenuates the Inflammatory Response and Oxidative Stress in LPS-Induced RAW264.7 Cells: In vitro Assessment and a Theoretical Model. BioMed Res. Int. 2019, 2019, 7039802. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Y.; Mirzaei, S.; Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Khan, H.; Daglia, M. Quercetin and Its Nano-Scale Delivery Systems in Prostate Cancer Therapy: Paving the Way for Cancer Elimination and Reversing Chemoresistance. Cancers 2021, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Shams, S.G.E.; Eissa, R.G. Amelioration of Ethanol-Induced Gastric Ulcer in Rats by Quercetin: Implication of Nrf2/HO1 and HMGB1/TLR4/NF-ΚB Pathways. Heliyon 2022, 8, e11159. [Google Scholar] [CrossRef]
- D’Andrea, G. Quercetin: A Flavonol with Multifaceted Therapeutic Applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef]
- Pinheiro, R.G.R.; Pinheiro, M.; Neves, A.R. Nanotechnology Innovations to Enhance the Therapeutic Efficacy of Quercetin. Nanomaterials 2021, 11, 2658. [Google Scholar] [CrossRef]
- Wei, C.; Li, S.; Zhu, Y.; Chen, W.; Li, C.; Xu, R. Network Pharmacology Identify Intersection Genes of Quercetin and Alzheimer’s Disease as Potential Therapeutic Targets. Front. Aging Neurosci. 2022, 14, 902092. [Google Scholar] [CrossRef]
- Wang, W.; Sun, C.; Mao, L.; Ma, P.; Liu, F.; Yang, J.; Gao, Y. The Biological Activities, Chemical Stability, Metabolism and Delivery Systems of Quercetin: A Review. Trends Food Sci. Technol. 2016, 56, 21–38. [Google Scholar] [CrossRef]
- Alizadeh, S.R.; Ebrahimzadeh, M.A. Quercetin Derivatives: Drug Design, Development, and Biological Activities, a Review. Eur. J. Med. Chem. 2022, 229, 114068. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Deng, Y.; Xue, X.; Liao, L.; Zhou, M.; Peng, C.; Li, Y. Research Progress of Quercetin Delivery Systems. Curr. Pharm. Des. 2022, 28, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.; Abdallah, I.A.; Bedair, A.; Mansour, F.R. Sample Preparation Methods for Determination of Quercetin and Quercetin Glycosides in Diverse Matrices. Microchem. J. 2023, 194, 109233. [Google Scholar] [CrossRef]
- Mansour, F.R.; Abdallah, I.A.; Bedair, A.; Hamed, M. Analytical Methods for the Determination of Quercetin and Quercetin Glycosides in Pharmaceuticals and Biological Samples. Crit. Rev. Anal. Chem. 2023, 1–26. [Google Scholar] [CrossRef]
- Carvalho, D.; Pinho, C.; Oliveira, R.; Moreira, F.; Oliveira, A.I. Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022. Molecules 2023, 28, 7714. [Google Scholar] [CrossRef]
- Johnson, R. Assessment of Bias with Emphasis on Method Comparison. Clin. Biochem. Rev. 2008, 29, S37–S42. [Google Scholar]
- Abdelkawy, K.S.; Balyshev, M.E.; Elbarbry, F. A New Validated HPLC Method for the Determination of Quercetin: Application to Study Pharmacokinetics in Rats. Biomed. Chromatogr. 2017, 31, e3819. [Google Scholar] [CrossRef]
- Vasileiadou, A.; Karapanagiotis, I.; Zotou, A. Development and Validation of a Liquid Chromatographic Method with Diode Array Detection for the Determination of Anthraquinones, Flavonoids and Other Natural Dyes in Aged Silk. J. Chromatogr. A 2021, 1651, 462312. [Google Scholar] [CrossRef]
- Zu, Y.; Li, C.; Fu, Y.; Zhao, C. Simultaneous Determination of Catechin, Rutin, Quercetin, Kaempferol and Isorhamnetin in the Extract of Sea Buckthorn (Hippophae rhamnoides L.) Leaves by RP-HPLC with DAD. J. Pharm. Biomed. Anal. 2006, 41, 714–719. [Google Scholar] [CrossRef]
- Andrade-Filho, T.; Ribeiro, T.C.S.; Del Nero, J. The UV-Vis Absorption Spectrum of the Flavonol Quercetin in Methanolic Solution: A Theoretical Investigation. Eur. Phys. J. E 2009, 29, 253–259. [Google Scholar] [CrossRef] [PubMed]
- De, R.; Jo, K.W.; Kim, K.-T. Influence of Molecular Structures on Fluorescence of Flavonoids and Their Detection in Mammalian Cells. Biomedicines 2022, 10, 1265. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Sánchez, R.; Ahuatl-García, F.; Dávila-Jiménez, M.M.; Elizalde-González, M.P.; Guevara-Villa, M.R.G. Chromatographic and Electrochemical Determination of Quercetin and Kaempferol in Phytopharmaceuticals. J. Pharm. Biomed. Anal. 2005, 38, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.F.; Yam, M.F.; Fung, Y.T.T.; Kiang, P.K.; Darwin, Y. HPLC Method for Simultaneous Quantitative Detection of Quercetin and Curcuminoids in Traditional Chinese Medicines. J. Pharmacopunct. 2014, 17, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Antal, D.S.; Schwaiger, S.; Ellmerer-Müller, E.P.; Stuppner, H. Cotinus coggygria Wood: Novel Flavanone Dimer and Development of an HPLC/UV/MS Method for the Simultaneous Determination of Fourteen Phenolic Constituents. Planta Med. 2010, 76, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Sree, K.S.N.; Dengale, S.J.; Mutalik, S.; Bhat, K. Dronedarone HCl-Quercetin Co-Amorphous System: Characterization and RP-HPLC Method Development for Simultaneous Estimation. J. AOAC Int. 2021, 104, 1232–1237. [Google Scholar] [CrossRef]
- de Sousa, J.P.B.; Brancalion, A.P.S.; Júnior, M.G.; Bastos, J.K. A Validated Chromatographic Method for the Determination of Flavonoids in Copaifera langsdorffii by HPLC. Nat. Prod. Commun. 2012, 7, 25–28. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Ghassabian, S. Linearity of Calibration Curves for Analytical Methods: A Review of Criteria for Assessment of Method Reliability. In Calibration and Validation of Analytical Methods—A Sampling of Current Approaches; InTechOpen: Rijeka, Croatia, 2018; pp. 109–128. [Google Scholar]
- Olszewska, M.A. New Validated High-Performance Liquid Chromatographic Method for Simultaneous Analysis of Ten Flavonoid Aglycones in Plant Extracts Using a C18 Fused-Core Column and Acetonitrile-Tetrahydrofuran Gradient. J. Sep. Sci. 2012, 35, 2174–2183. [Google Scholar] [CrossRef]
- Careri, M.; Corradini, C.; Elviri, L.; Nicoletti, I.; Zagnoni, I. Direct HPLC Analysis of Quercetin and Trans-Resveratrol in Red Wine, Grape, and Winemaking Byproducts. J. Agric. Food Chem. 2003, 51, 5226–5231. [Google Scholar] [CrossRef]
- Chen, H.-J.; Li, X.; Chen, J.-W.; Guo, S.; Cai, B.-C. Simultaneous Determination of Eleven Bioactive Compounds in Saururus chinensis from Different Harvesting Seasons by HPLC-DAD. J. Pharm. Biomed Anal. 2010, 51, 1142–1146. [Google Scholar] [CrossRef]
- Zhang, A.; Wan, L.; Wu, C.; Fang, Y.; Han, G.; Li, H.; Zhang, Z.; Wang, H. Simultaneous Determination of 14 Phenolic Compounds in Grape Canes by HPLC-DAD-UV Using Wavelength Switching Detection. Molecules 2013, 18, 14241–14257. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; He, Z.; He, F.; Wan, H. Determination of Quercetin, Plumbagin and Total Flavonoids in Drosera peltata Smith var. glabrata Y.Z.Ruan. Pharmacogn. Mag. 2012, 8, 263–267. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Herrador, M.Á. A Practical Guide to Analytical Method Validation, Including Measurement Uncertainty and Accuracy Profiles. TrAC—Trends Anal. Chem. 2007, 26, 227–238. [Google Scholar] [CrossRef]
- Choudhary, A.; Kant, V.; Jangir, B.L.; Joshi, V.G. Quercetin Loaded Chitosan Tripolyphosphate Nanoparticles Accelerated Cutaneous Wound Healing in Wistar Rats. Eur. J. Pharmacol. 2020, 880, 173172. [Google Scholar] [CrossRef]
- Ribani, M.; Grespan Bottoli, C.B.; Collins, C.H.; Fontes Jardim, I.C.S.; Costa Melo, L.F. Validação Em Métodos Cromatográficos e Eletroforéticos. Quím. Nova 2004, 27, 771–780. [Google Scholar] [CrossRef]
- Satheeshkumar, N.; Shantikumar, S.; Komali, M. Identification and Quantification of Aldose Reductase Inhibitory Flavonoids in Herbal Formulation and Extract of Gymnema sylvestre Using HPLC-PDA and LC-MS/MS. Chromatogr. Res. Int. 2014, 2014, 518175. [Google Scholar] [CrossRef]
- Dabeek, W.M.; Marra, M.V. Dietary Quercetin and Kaempferol: Bioavailability and Potential Cardiovascular-Related Bioactivity in Humans. Nutrients 2019, 11, 2288. [Google Scholar] [CrossRef]
- Jan, S.; Ahmad, J.; Dar, M.M.; Wani, A.A.; Tahir, I.; Kamili, A.N. Development and Validation of a Reverse Phase HPLC-DAD Method for Separation, Detection & Quantification of Rutin and Quercetin in Buckwheat (Fagopyrum spp.). J. Food Sci. Technol. 2022, 59, 2875–2883. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Gowthamarajan, K.; Priyanka Dwarampudi, L.; Bhaskaran, M.; Kadiyala, M. Analytical Method Development, Validation and Forced Degradation Studies for Rutin, Quercetin, Curcumin, and Piperine by RP-UFLC Method. Drug Dev. Ind. Pharm. 2021, 47, 562–568. [Google Scholar] [CrossRef]
- Srivastava, M.; Singh, M.; Maurya, P.; Srivastava, N.; Gupta, N.; Shanker, K. Simultaneous Quantification of Five Bioactive Phenylethanoid, Iridoid, and Flavonol Glycosides in Duranta erecta L.: Ultra Performance Liquid Chromatography Method Validation and Uncertainty Measurement. J. Pharm. Biomed. Anal. 2019, 174, 711–717. [Google Scholar] [CrossRef]
- Gul, H.; Awais, M.; Saddick, S.; Ahmed, Y.; Sher Khan, F.; Ahmed, E.; Afzal, U.; Naqvi, S.; Asghar Khan, M.; Gulfraz, M.; et al. Quantification of Biochemical Compounds in Bauhinia variegata Linn Flower Extract and Its Hepatoprotective Effect. Saudi J. Biol. Sci. 2021, 28, 247–254. [Google Scholar] [CrossRef]
- Karabat, R.R.; Rasheed, A.S.; Hassan, M.J.M. A New Method in Some German Grape Wines Using Zic-Hilic Technology with UV Detection to Separate and Identify Quercetin. Plant Arch. 2020, 20, 2692–2696. [Google Scholar]
- Abdulrazak, S.; Nuhu, A.A.; Yashim, Z.I. Quantitative Determination of Quercetin in Ginkgo biloba Leaf Extract (EGb 761) Using GC-MS. Nigerian J. Sci. Res. 2017, 16, 441–445. [Google Scholar]
- Peasari, J.R.; Motamarry, S.S.; Varma, K.S.; Anitha, P.; Potti, R.B. Chromatographic Analysis of Phytochemicals in Costus igneus and Computational Studies of Flavonoids. Inform. Med. Unlocked 2018, 13, 34–40. [Google Scholar] [CrossRef]
- Desmiaty, Y.; Alatas, F. Determination of Quercetin in Hibiscus sabdariffa L. Calyces by High-Performance Liquid Chromatography (HPLC). In Proceeding of the International Seminar on Chemistry, West Java, Indonesia, 30–31 October 2008; pp. 385–388. [Google Scholar]
- Ahmed, M.F.; Rao, A.S. Simultaneous Determination of Phenolic Compounds in Brassica oleracea L. Int. J. Pharm. Pharm. Sci. 2014, 6, 534–537. [Google Scholar]
- Wirth, M.J.; Smith, E.A.; Anthony, S.R. Measurement and Simulation of Tailing Zones of a Cationic Dye in Analytical-Scale Reversed Phase Chromatography. J. Chromatogr. A 2004, 1034, 69–75. [Google Scholar] [CrossRef]
- Khan, M.N.; Ul Haq, F.; Rahman, S.; Ali, A.; Musharraf, S.G. Metabolite Distribution and Correlation Studies of Ziziphus jujuba and Ziziphus nummularia Using LC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2020, 178, 112918. [Google Scholar] [CrossRef]
- Jia, Q.; Zhang, S.; Zhang, H.; Yang, X.; Cui, X.; Su, Z.; Hu, P. A Comparative Study on Polyphenolic Composition of Berries from the Tibetan Plateau by UPLC-Q-Orbitrap MS System. Chem. Biodivers. 2020, 17, e2000033. [Google Scholar] [CrossRef]
- Sharma, S.; Joshi, R.; Kumar, D. Quantitative Analysis of Flavonols, Flavonol Glycoside and Homoisoflavonoids in Polygonatum verticillatum Using UHPLC-DAD-QTOF-IMS and Evaluation of Their Antioxidant Potential. Phytochem. Anal. 2020, 31, 333–339. [Google Scholar] [CrossRef]
- Chavan, S.D.; Desai, D.M. Analytical Method Validation: A Brief Review. World J. Adv. Res. Rev. 2022, 16, 389–402. [Google Scholar] [CrossRef]
- Hejniak, J.; Baranowska, I.; Stencel, S.; Bajkacz, S. Separation and Determination of Selected Polyphenols from Medicinal Plants. J. Chromatogr. Sci. 2019, 57, 17–26. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Compound—Calibration Curve | Linearity Range (µg/mL) | Calibration Equation | r2 | r |
|---|---|---|---|---|
| QUE—nonadjusted | 0.14–245 | y = 15,485x + 11,659 | 0.9976 | 0.9988 |
| QUE—adjusted [1] | 0.14–5 | y = 15,262x − 271.48 | 0.9994 | 0.9997 |
| QUE—adjusted [2] | 5–245 | y = 15,317x + 42,292 | 0.9954 | 0.9977 |
| QUE Concentration (µg/mL) | QUE—Nonadjusted | QUE—Adjusted [1] | QUE—Adjusted [2] | |||
|---|---|---|---|---|---|---|
| [Obtained] | Accordance (%) | [Obtained] | Accordance (%) | [Obtained] | Accordance (%) | |
| 0.14 | −0.62 | −442 | 0.15 | 107 | - | - |
| 0.35 | −0.37 | −106 | 0.41 | 117 | - | - |
| 0.57 | −0.29 | −51 | 0.49 | 86 | - | - |
| 2.8 | 1.98 | 71 | 2.8 | 100 | - | - |
| 5 | 4.16 | 83 | 5 | 100 | 2.2 | 44 |
| 65 | 64.25 | 100 | - | - | 63 | 97 |
| 125 | 130.06 | 104 | - | - | 129 | 103 |
| 185 | 193.26 | 104 | - | - | 193 | 104 |
| 245 | 236.40 | 96 | - | - | 234 | 96 |
| Compound—Calibration Curve | m | sm | Slope RSD (%) | 95% Confidence Interval | |
|---|---|---|---|---|---|
| Minimum | Maximum | ||||
| QUE—nonadjusted | 15,485 | 289.47 | 1.87 | −65,423 | 88,741 |
| QUE—adjusted [1] | 15,262 | 211.13 | 1.38 | −2006 | 1463 |
| QUE—adjusted [2] | 15,317 | 601.69 | 3.93 | −247,002 | 331,585 |
| QUE Concentration (µg/mL) | Intraday Precision—RSD (%) | Interday Precision—RSD (%) |
|---|---|---|
| 0.35 | 5.66 | 9.42 |
| 0.57 | 5.47 | 8.19 |
| 5 | 6.74 | 6.87 |
| 125 | 2.41 | 7.38 |
| 185 | 2.64 | 7.18 |
| QUE Concentration (µg/mL) | Obtained Concentration | Accuracy (%) |
|---|---|---|
| 0.35 | 0.31 | 88.6 |
| 0.49 | 0.51 | 104.1 |
| 0.57 | 0.52 | 91.2 |
| 49 | 52 | 106.1 |
| 125 | 121 | 96.8 |
| 196 | 217 | 110.7 |
| Method | Parameter | Parameter Value | Peak Area | % of Peak Area in Relation to Optimized Method | Retention Time (min) | Difference of Retention Time in Relation to Optimized Method (min) |
|---|---|---|---|---|---|---|
| Optimized | pH | 3.32 | 7,304,837 | N/A | 3.8 | N/A |
| variation for robustness evaluation | 3.43 | 6,741,931 | 92.29 | 3.8 | 0.0 min | |
| Optimized | Flow rate | 1.3 mL/min | 6,111,868 | N/A | 3.7 | N/A |
| variation for robustness evaluation | 1.5 mL/min | 4,299,546 | 70.35 | 2.4 | −1.3 min |
| Condition | Concentration (µg/mL) | Day 0 | Day 5 | Day 7 | ||
|---|---|---|---|---|---|---|
| Mean (n = 3) | Mean (n = 3) | Stability (%) | Mean (n = 3) | Stability (%) | ||
| −20 °C | 0.57 | 7129 | 7929 | 111.22 | 8737 | 122.57 |
| 5 | 71,284 | 78,463 | 110.07 | 77,477 | 108.69 | |
| 125 | 2,170,741 | 2,347,331 | 108.14 | 2,155,391 | 99.29 | |
| 4 °C | 0.57 | 8048 | 8758 | 108.82 | 8697 | 111.41 |
| 5 | 74,799 | 83,905 | 112.17 | 78,945 | 105.54 | |
| 125 | 2,123,647 | 2,318,601 | 109.18 | 2,255,046 | 106.19 | |
| Room temperature | 0.57 | 7799 | 6674 | 85.58 | 5336 | 68.42 |
| 5 | 73,338 | 75,772 | 103.32 | 67,525 | 92.07 | |
| 125 | 2,246,811 | 2,474,717 | 110.14 | 2,378,043 | 105.84 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, D.; Jesus, Â.; Pinho, C.; Oliveira, R.F.; Moreira, F.; Oliveira, A.I. Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles. Pharmaceuticals 2023, 16, 1736. https://doi.org/10.3390/ph16121736
Carvalho D, Jesus Â, Pinho C, Oliveira RF, Moreira F, Oliveira AI. Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles. Pharmaceuticals. 2023; 16(12):1736. https://doi.org/10.3390/ph16121736
Chicago/Turabian StyleCarvalho, Daniel, Ângelo Jesus, Cláudia Pinho, Rita Ferraz Oliveira, Fernando Moreira, and Ana Isabel Oliveira. 2023. "Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles" Pharmaceuticals 16, no. 12: 1736. https://doi.org/10.3390/ph16121736
APA StyleCarvalho, D., Jesus, Â., Pinho, C., Oliveira, R. F., Moreira, F., & Oliveira, A. I. (2023). Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles. Pharmaceuticals, 16(12), 1736. https://doi.org/10.3390/ph16121736