
Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Metformin Pharmacology
2.1. Pharmacokinetic Properties of Metformin
2.2. Mechanisms Involved in Glucoregulation
2.3. AMPK Signaling Pathway
2.4. Mitochondrial Respiratory Chain Complexes
2.5. mGPDH and Cytosolic Redox State
2.6. Interaction of Metformin Treatment, Metabolites, and Gut Microbiota
3. Mechanisms Involved in Central Nervous System Functions and Neuroprotection
3.1. Insulin Signaling
3.2. Antioxidant Mechanism of Metformin
3.3. Anti-Inflammatory Mechanisms of Metformin
3.4. Neurogenesis
3.5. Autophagy-Inducing Mechanism of Metformin
3.6. The Putative Relationship between Metformin and Aging
4. The Winding Path from Neuroprotective Mechanisms to Proving Benefits in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Parkinson Disease
4.3. Huntington
4.4. Epilepsy
4.5. Fragile X Syndrome
5. Concluding Remarks and Future Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicolucci, A.; Charbonnel, B.; Gomes, M.B.; Khunti, K.; Kosiborod, M.; Shestakova, M.V.; Shimomura, I.; Watada, H.; Chen, H.; Cid-Ruzafa, J.; et al. Treatment Patterns and Associated Factors in 14 668 People with Type 2 Diabetes Initiating a Second-line Therapy: Results from the Global DISCOVER Study Programme. Diabetes Obes. Metab. 2019, 21, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- WHO Model List of Essential Medicines; WHO: Geneva, Switzerland, 2021; p. 22.
- King, P.; Peacock, I.; Donnelly, R. The UK Prospective Diabetes Study (UKPDS): Clinical and Therapeutic Implications for Type 2 Diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef]
- Stroup, T.S.; Gray, N. Management of Common Adverse Effects of Antipsychotic Medications. World Psychiatry 2018, 17, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-R.; Zhang, F.-Y.; Gao, K.-M.; Ou, J.-J.; Shao, P.; Jin, H.; Guo, W.-B.; Chan, P.K.; Zhao, J.-P. Metformin Treatment of Antipsychotic-Induced Dyslipidemia: An Analysis of Two Randomized, Placebo-Controlled Trials. Mol. Psychiatry 2016, 21, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Samara, M.T.; Dold, M.; Gianatsi, M.; Nikolakopoulou, A.; Helfer, B.; Salanti, G.; Leucht, S. Efficacy, Acceptability, and Tolerability of Antipsychotics in Treatment-Resistant Schizophrenia. JAMA Psychiatry 2016, 73, 199. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Nazareth, I.; Petersen, I. Trends in Incidence, Prevalence and Prescribing in Type 2 Diabetes Mellitus between 2000 and 2013 in Primary Care: A Retrospective Cohort Study. BMJ Open 2016, 6, e010210. [Google Scholar] [CrossRef] [PubMed]
- Montvida, O.; Shaw, J.; Atherton, J.J.; Stringer, F.; Paul, S.K. Long-Term Trends in Antidiabetes Drug Usage in the U.S.: Real-World Evidence in Patients Newly Diagnosed With Type 2 Diabetes. Diabetes Care 2018, 41, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.; Dunne, F.P. Metformin for Pregnancy and beyond: The Pros and Cons. Diabet. Med. 2022, 39, e14700. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Mehendale, A.M. A Review on the Use of Metformin in Pregnancy and Its Associated Fetal Outcomes. Cureus 2022, 14, e30039. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Kristófi, R.; Eriksson, J.W. Metformin as an Anti-Inflammatory Agent: A Short Review. J. Endocrinol. 2021, 251, R11–R22. [Google Scholar] [CrossRef] [PubMed]
- Solymár, M.; Ivic, I.; Pótó, L.; Hegyi, P.; Garami, A.; Hartmann, P.; Pétervári, E.; Czopf, L.; Hussain, A.; Gyöngyi, Z.; et al. Metformin Induces Significant Reduction of Body Weight, Total Cholesterol and LDL Levels in the Elderly—A Meta-Analysis. PLoS ONE 2018, 13, e0207947. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The Effects of Metformin on Gut Microbiota and the Immune System as Research Frontiers. Diabetologia 2017, 60, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Silamiķele, L.; Silamiķelis, I.; Ustinova, M.; Kalniņa, Z.; Elbere, I.; Petrovska, R.; Kalniņa, I.; Kloviņš, J. Metformin Strongly Affects Gut Microbiome Composition in High-Fat Diet-Induced Type 2 Diabetes Mouse Model of Both Sexes. Front. Endocrinol. 2021, 12, 626359. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; et al. Global, Regional, and National Burden of Parkinson’s Disease, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Logroscino, G.; Piccininni, M.; Marin, B.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Alahdab, F.; Asgedom, S.W.; Awasthi, A.; Chaiah, Y.; et al. Global, Regional, and National Burden of Motor Neuron Diseases 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and Ethnic Estimates of Alzheimer’s Disease and Related Dementias in the United States (2015–2060) in Adults Aged ≥65 Years. Alzheimers Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Feigin, V.L.; Vos, T.; Alahdab, F.; Amit, A.M.L.; Bärnighausen, T.W.; Beghi, E.; Beheshti, M.; Chavan, P.P.; Criqui, M.H.; Desai, R.; et al. Burden of Neurological Disorders Across the US From 1990–2017. JAMA Neurol. 2021, 78, 165. [Google Scholar] [CrossRef]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why Has Therapy Development for Dementia Failed in the Last Two Decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef]
- Calcoen, D.; Elias, L.; Yu, X. What Does It Take to Produce a Breakthrough Drug? Nat. Rev. Drug Discov. 2015, 14, 161–162. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia Prevention, Intervention, and Care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- Golde, T.E. Disease-Modifying Therapies for Alzheimer’s Disease: More Questions than Answers. Neurotherapeutics 2022, 19, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Murasecco, I.; Mecocci, P. Diabetes Drugs in the Fight against Alzheimer’s Disease. Ageing Res. Rev. 2019, 54, 100936. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Popic, J.; Khoutorsky, A.; Sonenberg, N. Metformin for Treatment of Fragile X Syndrome and Other Neurological Disorders. Annu. Rev. Med. 2019, 70, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Sikich, L.; Reeves, G.; Johnson, J.; Keeton, C.; Spanos, M.; Kapoor, S.; Bussell, K.; Miller, L.; Chandrasekhar, T.; et al. Metformin Add-on vs. Antipsychotic Switch vs. Continued Antipsychotic Treatment plus Healthy Lifestyle Education in Overweight or Obese Youth with Severe Mental Illness: Results from the IMPACT Trial. World Psychiatry 2020, 19, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Narayan Dey, P.; Chen, C.; Monteiro, O.; et al. Metformin Reverses Early Cortical Network Dysfunction and Behavior Changes in Huntington’s Disease. Elife 2018, 7, e38744. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging Neuroprotective Effect of Metformin in Parkinson’s Disease: A Molecular Crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef]
- Sanati, M.; Aminyavari, S.; Afshari, A.R.; Sahebkar, A. Mechanistic Insight into the Role of Metformin in Alzheimer’s Disease. Life Sci. 2022, 291, 120299. [Google Scholar] [CrossRef] [PubMed]
- Chau, A.C.M.; Cheung, E.Y.W.; Chan, K.H.; Chow, W.S.; Shea, Y.F.; Chiu, P.K.C.; Mak, H.K.F. Impaired Cerebral Blood Flow in Type 2 Diabetes Mellitus—A Comparative Study with Subjective Cognitive Decline, Vascular Dementia and Alzheimer’s Disease Subjects. Neuroimage Clin. 2020, 27, 102302. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes Mellitus and Risk of Dementia: A Meta-Analysis of Prospective Observational Studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei Malazy, O.; Bandarian, F.; Qorbani, M.; Mohseni, S.; Mirsadeghi, S.; Peimani, M.; Larijani, B. The Effect of Metformin on Cognitive Function: A Systematic Review and Meta-Analysis. J. Psychopharmacol. 2022, 36, 666–679. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by Which Metformin Reduces Glucose Production in Type 2 Diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the Glucoregulatory Mechanisms of Metformin in Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Goodman, A.M. Efficacy of Metformin in Patients with Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 541–549. [Google Scholar] [CrossRef]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic Effects of Metformin in Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef]
- Cernea, S.; Dima, L.; Correll, C.U.; Manu, P. Pharmacological Management of Glucose Dysregulation in Patients Treated with Second-Generation Antipsychotics. Drugs 2020, 80, 1763–1781. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Metformin and Gut Microbiota: Their Interactions and Their Impact on Diabetes. Hormones 2019, 18, 141–144. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Lutz, O.M.D. Metformin and Aging: A Review. Gerontology 2019, 65, 581–590. [Google Scholar] [CrossRef]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the Gastrointestinal Tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of Organic Cation Transporter 1 in Hepatic and Intestinal Distribution of Metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, N.; Shima, H.; Asano, S.; Ishimoto, T.; Sugiura, T.; Matsubara, K.; Kusuhara, H.; Sugiyama, Y.; Sai, Y.; Miyamoto, K.; et al. Involvement of Carnitine/Organic Cation Transporter OCTN1/SLC22A4 in Gastrointestinal Absorption of Metformin. J. Pharm. Sci. 2013, 102, 3407–3417. [Google Scholar] [CrossRef] [PubMed]
- Haberkorn, B.; Fromm, M.F.; König, J. Transport of Drugs and Endogenous Compounds Mediated by Human OCT1: Studies in Single- and Double-Transfected Cell Models. Front. Pharmacol. 2021, 12, 662535. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Seki, M.; Hatakeyama, M.; Kurokawa, Y.; Uchiyama, H.; Takemura, M.; Yasugi, Y.; Kishimoto, H.; Tamai, I.; Wang, J.; et al. Multiple Transport Mechanisms Involved in the Intestinal Absorption of Metformin: Impact on the Nonlinear Absorption Kinetics. J. Pharm. Sci. 2022, 111, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.B.; Botton, M.R.; Struchiner, C.J.; Suarez-Kurtz, G. Influence of Pharmacogenetic Polymorphisms and Demographic Variables on Metformin Pharmacokinetics in an Admixed Brazilian Cohort. Br. J. Clin. Pharmacol. 2018, 84, 987–996. [Google Scholar] [CrossRef]
- Sundelin, E.; Jensen, J.B.; Jakobsen, S.; Gormsen, L.C.; Jessen, N. Metformin Biodistribution: A Key to Mechanisms of Action? J. Clin. Endocrinol. Metab. 2020, 105, 3374–3383. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Hougaard Christensen, M.M.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11 C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Gong, T.; Du, Y.; Wang, Y.; Ge, T.; Liu, J. Mechanism of Metformin Regulation in Central Nervous System: Progression and Future Perspectives. Biomed. Pharmacother. 2022, 156, 113686. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Serratosa, J.M.; Sánchez, M.P. Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. Int. J. Mol. Sci. 2021, 22, 5351. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Scherer, N.; Selzer, D.; Dings, C.; Hanke, N.; Dallmann, R.; Schwab, M.; Timmins, P.; Nock, V.; Lehr, T. Significant Impact of Time-of-Day Variation on Metformin Pharmacokinetics. Diabetologia 2023, 66, 1024–1034. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Dujic, T.; Zhou, K.; Yee, S.; van Leeuwen, N.; de Keyser, C.; Javorský, M.; Goswami, S.; Zaharenko, L.; Hougaard Christensen, M.; Out, M.; et al. Variants in Pharmacokinetic Transporters and Glycemic Response to Metformin: A Metgen Meta-Analysis. Clin. Pharmacol. Ther. 2017, 101, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Singh, R.; Singh, V.; Singh, H.; Kumari, P.; Chopra, H.; Sharma, R.; Nepovimova, E.; Valis, M.; Kuca, K.; et al. Metformin: Activation of 5′ AMP-Activated Protein Kinase and Its Emerging Potential beyond Anti-Hyperglycemic Action. Front. Genet. 2022, 13, 1022739. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic Loss of AMPK-Glycogen Binding Destabilises AMPK and Disrupts Metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef]
- Myers, R.W.; Guan, H.-P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic Pan-AMPK Activator MK-8722 Improves Glucose Homeostasis but Induces Cardiac Hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and Function of AMPK in Physiology and Diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.-H. AMPK, Mitochondrial Function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Shaw, R.J. AMPK: Restoring Metabolic Homeostasis over Space and Time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New Insights into Activation and Function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; DePinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Olivier, S.; Pochard, C.; Diounou, H.; Castillo, V.; Divoux, J.; Alcantara, J.; Leclerc, J.; Guilmeau, S.; Huet, C.; Charifi, W.; et al. Deletion of Intestinal Epithelial AMP-Activated Protein Kinase Alters Distal Colon Permeability but Not Glucose Homeostasis. Mol. Metab. 2021, 47, 101183. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef]
- Zhang, E.; Jin, L.; Wang, Y.; Tu, J.; Zheng, R.; Ding, L.; Fang, Z.; Fan, M.; Al-Abdullah, I.; Natarajan, R.; et al. Intestinal AMPK Modulation of Microbiota Mediates Crosstalk with Brown Fat to Control Thermogenesis. Nat. Commun. 2022, 13, 1135. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-Dose Metformin Targets the Lysosomal AMPK Pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.-W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Guo, H.; Zhang, C.-S.; Lin, S.-Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a Low-Energy Charge Signal Autonomously Initiates Assembly of AXIN-AMPK-LKB1 Complex for AMPK Activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-ATPase-Ragulator Complex Is a Common Activator for AMPK and MTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ou, Y.; Li, Y.; Hu, S.; Shao, L.-W.; Liu, Y. Metformin Extends C. elegans Lifespan through Lysosomal Pathway. Elife 2017, 6, e31268. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Blazina, I.; Selph, S. Diabetes Drugs for Nonalcoholic Fatty Liver Disease: A Systematic Review. Syst. Rev. 2019, 8, 295. [Google Scholar] [CrossRef]
- Pecinová, A.; Brázdová, A.; Drahota, Z.; Houštěk, J.; Mráček, T. Mitochondrial Targets of Metformin—Are They Physiologically Relevant? BioFactors 2019, 45, 703–711. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Giuliani, A.; Mensà, E.; Sabbatinelli, J.; De Nigris, V.; Rippo, M.R.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Pleiotropic Effects of Metformin: Shaping the Microbiome to Manage Type 2 Diabetes and Postpone Ageing. Ageing Res. Rev. 2018, 48, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zajda, A.; Huttunen, K.M.; Sikora, J.; Podsiedlik, M.; Markowicz-Piasecka, M. Is Metformin a Geroprotector? A Peek into the Current Clinical and Experimental Data. Mech. Ageing Dev. 2020, 191, 111350. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Kurelac, I.; Capristo, M.; Calvaruso, M.A.; Giorgio, V.; Bergamini, C.; Ghelli, A.; Nanni, P.; De Giovanni, C.; Carelli, V.; et al. Different MtDNA Mutations Modify Tumor Progression in Dependence of the Degree of Respiratory Complex I Impairment. Hum. Mol. Genet. 2014, 23, 1453–1466. [Google Scholar] [CrossRef]
- Feng, J.; Wang, X.; Ye, X.; Ares, I.; Lopez-Torres, B.; Martínez, M.; Martínez-Larrañaga, M.-R.; Wang, X.; Anadón, A.; Martínez, M.-A. Mitochondria as an Important Target of Metformin: The Mechanism of Action, Toxic and Side Effects, and New Therapeutic Applications. Pharmacol. Res. 2022, 177, 106114. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Bridges, H.R.; Blaza, J.N.; Yin, Z.; Chung, I.; Pollak, M.N.; Hirst, J. Structural Basis of Mammalian Respiratory Complex I Inhibition by Medicinal Biguanides. Science 2023, 379, 351–357. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.-M.; Cline, G.W.; Nakahara, K.; et al. Metformin, Phenformin, and Galegine Inhibit Complex IV Activity and Reduce Glycerol-Derived Gluconeogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2122287119. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef]
- Baur, J.A.; Birnbaum, M.J. Control of Gluconeogenesis by Metformin: Does Redox Trump Energy Charge? Cell Metab. 2014, 20, 197–199. [Google Scholar] [CrossRef]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593.e7. [Google Scholar] [CrossRef]
- Xie, J.; Ye, J.; Cai, Z.; Luo, Y.; Zhu, X.; Deng, Y.; Feng, Y.; Liang, Y.; Liu, R.; Han, Z.; et al. GPD1 Enhances the Anticancer Effects of Metformin by Synergistically Increasing Total Cellular Glycerol-3-Phosphate. Cancer Res. 2020, 80, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism in Vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Alshawi, A.; Agius, L. Low Metformin Causes a More Oxidized Mitochondrial NADH/NAD Redox State in Hepatocytes and Inhibits Gluconeogenesis by a Redox-Independent Mechanism. J. Biol. Chem. 2019, 294, 2839–5691. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; Ansari, I.H.; Longacre, M.J.; Stoker, S.W. Metformin’s Therapeutic Efficacy in the Treatment of Diabetes Does Not Involve Inhibition of Mitochondrial Glycerol Phosphate Dehydrogenase. Diabetes 2021, 70, 1575–1580. [Google Scholar] [CrossRef]
- Calza, G.; Nyberg, E.; Mäkinen, M.; Soliymani, R.; Cascone, A.; Lindholm, D.; Barborini, E.; Baumann, M.; Lalowski, M.; Eriksson, O. Lactate-Induced Glucose Output Is Unchanged by Metformin at a Therapeutic Concentration—A Mass Spectrometry Imaging Study of the Perfused Rat Liver. Front. Pharmacol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Serino, M.; Chabo, C.; Blasco-Baque, V.; Amar, J. Gut Microbiota and Diabetes: From Pathogenesis to Therapeutic Perspective. Acta Diabetol. 2011, 48, 257–273. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of Mammals and Their Gut Microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Bäckhed, F. Host Responses to the Human Microbiome. Nutr. Rev. 2012, 70, S14–S17. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant Intestinal Microbiota in Individuals with Prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia Muciniphila and Several Short-Chain Fatty Acid–Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.V.; Duca, F.A.; Waise, T.M.Z.; Rasmussen, B.A.; Abraham, M.A.; Dranse, H.J.; Puri, A.; O’Brien, C.A.; Lam, T.K.T. Metformin Alters Upper Small Intestinal Microbiota That Impact a Glucose-SGLT1-Sensing Glucoregulatory Pathway. Cell Metab. 2018, 27, 101–117.e5. [Google Scholar] [CrossRef] [PubMed]
- Sansome, D.J.; Xie, C.; Veedfald, S.; Horowitz, M.; Rayner, C.K.; Wu, T. Mechanism of Glucose-lowering by Metformin in Type 2 Diabetes: Role of Bile Acids. Diabetes Obes. Metab. 2020, 22, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut Microbiota and Intestinal FXR Mediate the Clinical Benefits of Metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Osmanovic Barilar, J.; Knezovic, A.; Grünblatt, E.; Riederer, P.; Salkovic-Petrisic, M. Nine-Month Follow-up of the Insulin Receptor Signalling Cascade in the Brain of Streptozotocin Rat Model of Sporadic Alzheimer’s Disease. J. Neural Transm. 2015, 122, 565–576. [Google Scholar] [CrossRef]
- Shingo, A.S.; Kanabayashi, T.; Murase, T.; Kito, S. Cognitive Decline in STZ-3V Rats Is Largely Due to Dysfunctional Insulin Signalling through the Dentate Gyrus. Behav. Brain Res. 2012, 229, 378–383. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.S.; Mattson, M.P. PTEN Regulates Akt Kinase Activity in Hippocampal Neurons and Increases Their Sensitivity to Glutamate and Apoptosis. Neuromol. Med. 2002, 2, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, M.; Jiang, X.; Xia, Z.; Wang, Y.; An, D.; Wang, H.; Heng, B.; Liu, Y. Metformin Inhibits Aβ25-35 -induced Apoptotic Cell Death in SH-SY5Y Cells. Basic Clin. Pharmacol. Toxicol. 2019, 125, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Mantik, K.E.K.; Kim, S.; Gu, B.; Moon, S.; Kwak, H.-B.; Park, D.-H.; Kang, J.-H. Repositioning of Anti-Diabetic Drugs against Dementia: Insight from Molecular Perspectives to Clinical Trials. Int. J. Mol. Sci. 2023, 24, 11450. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, K.; Ma, C.; Wang, X.; Gong, Z.; Zhang, R.; Zang, D.; Cheng, Y. Evaluation of Metformin on Cognitive Improvement in Patients With Non-Dementia Vascular Cognitive Impairment and Abnormal Glucose Metabolism. Front. Aging Neurosci. 2018, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Kazkayasi, I.; Telli, G.; Nemutlu, E.; Uma, S. Intranasal Metformin Treatment Ameliorates Cognitive Functions via Insulin Signaling Pathway in ICV-STZ-Induced Mice Model of Alzheimer’s Disease. Life Sci. 2022, 299, 120538. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The Mechanisms of Action of Metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Herman, R.; Kravos, N.A.; Jensterle, M.; Janež, A.; Dolžan, V. Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. Int. J. Mol. Sci. 2022, 23, 1264. [Google Scholar] [CrossRef]
- Zeng, J.; Zhu, L.; Liu, J.; Zhu, T.; Xie, Z.; Sun, X.; Zhang, H. Metformin Protects against Oxidative Stress Injury Induced by Ischemia/Reperfusion via Regulation of the LncRNA-H19/MiR-148a-3p/Rock2 Axis. Oxid. Med. Cell. Longev. 2019, 2019, 8768327. [Google Scholar] [CrossRef]
- An, H.; Wei, R.; Ke, J.; Yang, J.; Liu, Y.; Wang, X.; Wang, G.; Hong, T. Metformin Attenuates Fluctuating Glucose-Induced Endothelial Dysfunction through Enhancing GTPCH1-Mediated ENOS Recoupling and Inhibiting NADPH Oxidase. J. Diabetes Complicat. 2016, 30, 1017–1024. [Google Scholar] [CrossRef]
- Xin, Z.; Ma, Z.; Jiang, S.; Wang, D.; Fan, C.; Di, S.; Hu, W.; Li, T.; She, J.; Yang, Y. FOXOs in the Impaired Heart: New Therapeutic Targets for Cardiac Diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Flachsbart, F.; Dose, J.; Gentschew, L.; Geismann, C.; Caliebe, A.; Knecht, C.; Nygaard, M.; Badarinarayan, N.; ElSharawy, A.; May, S.; et al. Identification and Characterization of Two Functional Variants in the Human Longevity Gene FOXO3. Nat. Commun. 2017, 8, 2063. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.; Loebel, M.; Steiner, S.; Bauer, S.; Karadeniz, Z.; Roeger, C.; Skurk, C.; Scheibenbogen, C.; Sotzny, F. Metformin Attenuates ROS via FOXO3 Activation in Immune Cells. Front. Immunol. 2021, 12, 581799. [Google Scholar] [CrossRef] [PubMed]
- Karami, F.; Jamaati, H.; Coleman-Fuller, N.; Zeini, M.S.; Hayes, A.W.; Gholami, M.; Salehirad, M.; Darabi, M.; Motaghinejad, M. Is Metformin Neuroprotective against Diabetes Mellitus-Induced Neurodegeneration? An Updated Graphical Review of Molecular Basis. Pharmacol. Rep. 2023, 75, 511–543. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Carvalho, C.; Santos, M.; Proenca, T.; Nunes, E.; Duarte, A.; Monteiro, P.; Seica, R.; Oliveira, C.; Moreira, P. Metformin Protects the Brain Against the Oxidative Imbalance Promoted by Type 2 Diabetes. Med. Chem. 2008, 4, 358–364. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Vento, M.; Baquero, M.; Cháfer-Pericás, C. Lipid Peroxidation in Neurodegeneration. Clin. Chim. Acta 2019, 497, 178–188. [Google Scholar] [CrossRef]
- Du, M.-R.; Gao, Q.-Y.; Liu, C.-L.; Bai, L.-Y.; Li, T.; Wei, F.-L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef]
- Rabieipoor, S.; Zare, M.; Ettcheto, M.; Camins, A.; Javan, M. Metformin Restores Cognitive Dysfunction and Histopathological Deficits in an Animal Model of Sporadic Alzheimer’s Disease. Heliyon 2023, 9, e17873. [Google Scholar] [CrossRef]
- Zhou, Y.; Ma, X.-Y.; Han, J.-Y.; Yang, M.; Lv, C.; Shao, Y.; Wang, Y.-L.; Kang, J.-Y.; Wang, Q.-Y. Metformin Regulates Inflammation and Fibrosis in Diabetic Kidney Disease through TNC/TLR4/NF-ΚB/MiR-155-5p Inflammatory Loop. World J. Diabetes 2021, 12, 19–46. [Google Scholar] [CrossRef]
- Docrat, T.F.; Nagiah, S.; Chuturgoon, A.A. Metformin Protects against Neuroinflammation through Integrated Mechanisms of MiR-141 and the NF-ĸB-Mediated Inflammasome Pathway in a Diabetic Mouse Model. Eur. J. Pharmacol. 2021, 903, 174146. [Google Scholar] [CrossRef]
- Xu, T.; Wu, X.; Lu, X.; Liang, Y.; Mao, Y.; Loor, J.J.; Yang, Z. Metformin Activated AMPK Signaling Contributes to the Alleviation of LPS-Induced Inflammatory Responses in Bovine Mammary Epithelial Cells. BMC Vet. Res. 2021, 17, 97. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Huang, S.; Chen, Q.-B.; Zhang, D.; Li, W.; Ao, R.; Leung, F.C.-Y.; Zhang, Z.; Huang, J.; Tang, Y.; et al. Metformin Ameliorates Aβ Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 2315106. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin Treatment Prevents Amyloid Plaque Deposition and Memory Impairment in APP/PS1 Mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Tizazu, A.M.; Nyunt, M.S.Z.; Cexus, O.; Suku, K.; Mok, E.; Xian, C.H.; Chong, J.; Tan, C.; How, W.; Hubert, S.; et al. Metformin Monotherapy Downregulates Diabetes-Associated Inflammatory Status and Impacts on Mortality. Front. Physiol. 2019, 10, 572. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Kelly, S.; Ajodha, S.; Sahdev, A.; Bestwick, J.P.; Gabrovska, P.; Akanle, O.; Ajjan, R.; Kola, B.; Stadler, M.; et al. Metformin to Reduce Metabolic Complications and Inflammation in Patients on Systemic Glucocorticoid Therapy: A Randomised, Double-Blind, Placebo-Controlled, Proof-of-Concept, Phase 2 Trial. Lancet Diabetes Endocrinol. 2020, 8, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin Alleviates High Glucose-Induced ER Stress and Inflammation by Inhibiting the Interaction between Caveolin1 and AMPKα in Rat Astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef]
- Jin, Q.; Cheng, J.; Liu, Y.; Wu, J.; Wang, X.; Wei, S.; Zhou, X.; Qin, Z.; Jia, J.; Zhen, X. Improvement of Functional Recovery by Chronic Metformin Treatment Is Associated with Enhanced Alternative Activation of Microglia/Macrophages and Increased Angiogenesis and Neurogenesis Following Experimental Stroke. Brain Behav. Immun. 2014, 40, 131–142. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, W.; Li, H.; Pang, P.; Xue, F.; Wan, L.; Pei, L.; Yan, H. Metformin Restores Hippocampal Neurogenesis and Learning and Memory via Regulating Gut Microbiota in the Obese Mouse Model. Brain Behav. Immun. 2021, 95, 68–83. [Google Scholar] [CrossRef]
- Fang, M.; Jiang, H.; Ye, L.; Cai, C.; Hu, Y.; Pan, S.; Li, P.; Xiao, J.; Lin, Z. Metformin Treatment after the Hypoxia-Ischemia Attenuates Brain Injury in Newborn Rats. Oncotarget 2017, 8, 75308–75325. [Google Scholar] [CrossRef]
- Yuan, R.; Wang, Y.; Li, Q.; Zhen, F.; Li, X.; Lai, Q.; Hu, P.; Wang, X.; Zhu, Y.; Fan, H.; et al. Metformin Reduces Neuronal Damage and Promotes Neuroblast Proliferation and Differentiation in a Cerebral Ischemia/Reperfusion Rat Model. Neuroreport 2019, 30, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular Mechanisms of Cell Death in Neurological Diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Nozohouri, S.; Vaidya, B.; Abbruscato, T. Repurposing Metformin to Treat Age-Related Neurodegenerative Disorders and Ischemic Stroke. Life Sci. 2021, 274, 119343. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-L.; Luo, C.; Pu, D.; Zhang, G.-Q.; Zhao, Y.-X.; Sun, Y.; Zhao, K.-X.; Liao, Z.-Y.; Lv, A.-K.; Zhu, S.-Y.; et al. Metformin Attenuates Diabetes-Induced Tau Hyperphosphorylation in Vitro and in Vivo by Enhancing Autophagic Clearance. Exp. Neurol. 2019, 311, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Cheng, F.-F.; Liu, Y.-L.; Du, J.; Lin, J.-T. Metformin’s Mechanisms in Attenuating Hallmarks of Aging and Age-Related Disease. Aging Dis. 2022, 13, 970. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N. Effect of Buformin and Diphenylhydantoin on the Life Span, Estrous Function and Spontaneous Tumor Incidence in Rats. Vopr. Onkol. 1980, 26, 42–48. [Google Scholar]
- Chen, S.; Gan, D.; Lin, S.; Zhong, Y.; Chen, M.; Zou, X.; Shao, Z.; Xiao, G. Metformin in Aging and Aging-Related Diseases: Clinical Applications and Relevant Mechanisms. Theranostics 2022, 12, 2722–2740. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Deng, D.; Zhu, X. The Function, Mechanisms, and Clinical Applications of Metformin: Potential Drug, Unlimited Potentials. Arch. Pharm. Res. 2023, 46, 389–407. [Google Scholar] [CrossRef]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin Reduces All-Cause Mortality and Diseases of Ageing Independent of Its Effect on Diabetes Control: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 2021, 12, 933. [Google Scholar] [CrossRef]
- Halicka, H.D.; Zhao, H.; Li, J.; Lee, Y.-S.; Hsieh, T.-C.; Wu, J.M.; Darzynkiewicz, Z. Potential Anti-Aging Agents Suppress the Level of Constitutive MTOR- and DNA Damage-Signaling. Aging 2012, 4, 952–965. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Zhao, H.; Halicka, H.D.; Li, J.; Lee, Y.; Hsieh, T.; Wu, J.M. In Search of Antiaging Modalities: Evaluation of MTOR- and ROS/DNA Damage-signaling by Cytometry. Cytom. Part A 2014, 85, 386–399. [Google Scholar] [CrossRef]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. ROLE of IGF-1 System in the Modulation of Longevity: Controversies and New Insights From a Centenarians’ Perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Dazert, E.; Hall, M.N. MTOR Signaling in Disease. Curr. Opin. Cell Biol. 2011, 23, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Onken, B.; Driscoll, M. Metformin Induces a Dietary Restriction–Like State and the Oxidative Stress Response to Extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, P.S.; Blacker, D.; Blazer, D.G.; Ganguli, M.; Jeste, D.V.; Paulsen, J.S.; Petersen, R.C. Classifying Neurocognitive Disorders: The DSM-5 Approach. Nat. Rev. Neurol. 2014, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Q.; Li, W.-S.; Liu, Z.; Zhang, H.-L.; Ba, Y.-G.; Zhang, R.-X. Metformin Therapy and Cognitive Dysfunction in Patients with Type 2 Diabetes: A Meta-Analysis and Systematic Review. Medicine 2020, 99, e19378. [Google Scholar] [CrossRef] [PubMed]
- Valencia, W.M.; Palacio, A.; Tamariz, L.; Florez, H. Metformin and Ageing: Improving Ageing Outcomes beyond Glycaemic Control. Diabetologia 2017, 60, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin Regulates Metabolic and Nonmetabolic Pathways in Skeletal Muscle and Subcutaneous Adipose Tissues of Older Adults. Aging Cell 2018, 17, e12723. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Whitmer, R.A. Cognitive Dysfunction in Diabetes: How to Implement Emerging Guidelines. Diabetologia 2020, 63, 3–9. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive Decline and Dementia in Diabetes Mellitus: Mechanisms and Clinical Implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Gispen, W.H.; Biessels, G.-J. Cognition and Synaptic Plasticity in Diabetes Mellitus. Trends Neurosci. 2000, 23, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; das Carvalho, M.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s Disease and Type 2 Diabetes Mellitus: A Systematic Review of Proteomic Studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Yi, R.; Dong, Q.; Yao, L.; Liu, B. The Relationship between Diabetes-Related Cognitive Dysfunction and Leukoaraiosis. Acta Neurol. Belg. 2021, 121, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Jash, K.; Gondaliya, P.; Kirave, P.; Kulkarni, B.; Sunkaria, A.; Kalia, K. Cognitive Dysfunction: A Growing Link between Diabetes and Alzheimer’s Disease. Drug Dev. Res. 2020, 81, 144–164. [Google Scholar] [CrossRef]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Moheet, A.; Mangia, S.; Seaquist, E.R. Impact of Diabetes on Cognitive Function and Brain Structure. Ann. N. Y. Acad. Sci. 2015, 1353, 60–71. [Google Scholar] [CrossRef]
- Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20, 6176. [Google Scholar] [CrossRef]
- Tsukuda, K.; Mogi, M.; Li, J.-M.; Iwanami, J.; Min, L.-J.; Sakata, A.; Fujita, T.; Iwai, M.; Horiuchi, M. Diabetes-Associated Cognitive Impairment Is Improved by a Calcium Channel Blocker, Nifedipine. Hypertension 2008, 51, 528–533. [Google Scholar] [CrossRef]
- Khandelwal, M.; Manglani, K.; Upadhyay, P.; Azad, M.; Gupta, S. AdipoRon Induces AMPK Activation and Ameliorates Alzheimer’s like Pathologies and Associated Cognitive Impairment in APP/PS1 Mice. Neurobiol. Dis. 2022, 174, 105876. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.-J.; Nanduri, J. Hypoxia-Inducible Factors and Obstructive Sleep Apnea. J. Clin. Investig. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Li, Z.; Wang, Y.; Chen, Y.; Li, X.; Chen, K.; Shu, N.; Zhang, Z. Disrupted White Matter Network and Cognitive Decline in Type 2 Diabetes Patients. J. Alzheimers Dis. 2016, 53, 185–195. [Google Scholar] [CrossRef]
- Reijmer, Y.D.; Leemans, A.; Brundel, M.; Kappelle, L.J.; Biessels, G.J. Disruption of the Cerebral White Matter Network Is Related to Slowing of Information Processing Speed in Patients With Type 2 Diabetes. Diabetes 2013, 62, 2112–2115. [Google Scholar] [CrossRef]
- Areosa Sastre, A.; Vernooij, R.W.; González-Colaço Harmand, M.; Martínez, G. Effect of the Treatment of Type 2 Diabetes Mellitus on the Development of Cognitive Impairment and Dementia. Cochrane Database Syst. Rev. 2017, 2017, CD003804. [Google Scholar] [CrossRef]
- de Galan, B.E.; Zoungas, S.; Chalmers, J.; Anderson, C.; Dufouil, C.; Pillai, A.; Cooper, M.; Grobbee, D.E.; Hackett, M.; Hamet, P.; et al. Cognitive Function and Risks of Cardiovascular Disease and Hypoglycemia in Patients with Type 2 Diabetes: The Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) Trial. Diabetologia 2009, 52, 2328–2336. [Google Scholar] [CrossRef]
- Orkaby, A.R.; Cho, K.; Cormack, J.; Gagnon, D.R.; Driver, J.A. Metformin vs Sulfonylurea Use and Risk of Dementia in US Veterans Aged ≥65 Years with Diabetes. Neurology 2017, 89, 1877–1885. [Google Scholar] [CrossRef]
- Wang, C.-P.; Lorenzo, C.; Habib, S.L.; Jo, B.; Espinoza, S.E. Differential Effects of Metformin on Age Related Comorbidities in Older Men with Type 2 Diabetes. J. Diabetes Complicat. 2017, 31, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lin, L.; Zhang, Q.; Wu, J. Brain Gray Matter Changes in Type 2 Diabetes Mellitus: A Meta-Analysis of Whole-Brain Voxel-Based Morphometry Study. J. Diabetes Complicat. 2017, 31, 1698–1703. [Google Scholar] [CrossRef]
- Kitabchi, A.E.; Temprosa, M.; Knowler, W.C.; Kahn, S.E.; Haffner, S.M.; Andres, R.; Saudek, C.; Edelstein, S.L.; Arakaki, R.; Murphy, M.B.; et al. Role of Insulin Secretion and Sensitivity in the Evolution of Type 2 Diabetes in the Diabetes Prevention Program. Diabetes 2005, 54, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.; Temprosa, M.; Crandall, J.; Fowler, S.; Goldberg, R.; Horton, E.; Marcovina, S.; Mather, K.; Orchard, T.; Ratner, R.; et al. Intensive Lifestyle Intervention or Metformin on Inflammation and Coagulation in Participants With Impaired Glucose Tolerance. Diabetes 2005, 54, 1566–1572. [Google Scholar] [CrossRef]
- Orchard, T.J.; Temprosa, M.; Goldberg, R.; Haffner, S.; Ratner, R.; Marcovina, S.; Fowler, S. The Effect of Metformin and Intensive Lifestyle Intervention on the Metabolic Syndrome: The Diabetes Prevention Program Randomized Trial. Ann. Intern. Med. 2005, 142, 611. [Google Scholar] [CrossRef]
- Qiu, W.; Folstein, M. Insulin, Insulin-Degrading Enzyme and Amyloid-β Peptide in Alzheimer’s Disease: Review and Hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Gorgich, E.A.C.; Parsaie, H.; Yarmand, S.; Baharvand, F.; Sarbishegi, M. Long-Term Administration of Metformin Ameliorates Age-Dependent Oxidative Stress and Cognitive Function in Rats. Behav. Brain Res. 2021, 410, 113343. [Google Scholar] [CrossRef]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin Improves Learning and Memory in the SAMP8 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef]
- Son, S.M.; Shin, H.-J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-β Generation by β- and γ-Secretases via Autophagy Activation. J. Alzheimers Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Cheng, L.; Li, W.; Chen, Y.; Lin, Y.; Miao, Y. Autophagy and Diabetic Encephalopathy: Mechanistic Insights and Potential Therapeutic Implications. Aging Dis. 2022, 13, 447. [Google Scholar] [CrossRef]
- Aksoz, E.; Gocmez, S.S.; Sahin, T.D.; Aksit, D.; Aksit, H.; Utkan, T. The Protective Effect of Metformin in Scopolamine-Induced Learning and Memory Impairment in Rats. Pharmacol. Rep. 2019, 71, 818–825. [Google Scholar] [CrossRef]
- Samaras, K.; Makkar, S.; Crawford, J.D.; Kochan, N.A.; Wen, W.; Draper, B.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Metformin Use Is Associated With Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults With Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of Metformin on Neurodegenerative Disease among Elderly Adult US Veterans with Type 2 Diabetes Mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef]
- Zhou, J.-B.; Tang, X.; Han, M.; Yang, J.; Simó, R. Impact of Antidiabetic Agents on Dementia Risk: A Bayesian Network Meta-Analysis. Metabolism 2020, 109, 154265. [Google Scholar] [CrossRef]
- Battini, V.; Cirnigliaro, G.; Leuzzi, R.; Rissotto, E.; Mosini, G.; Benatti, B.; Pozzi, M.; Nobile, M.; Radice, S.; Carnovale, C.; et al. The Potential Effect of Metformin on Cognitive and Other Symptom Dimensions in Patients with Schizophrenia and Antipsychotic-Induced Weight Gain: A Systematic Review, Meta-Analysis, and Meta-Regression. Front. Psychiatry 2023, 14, 1215807. [Google Scholar] [CrossRef]
- Koo, B.K.; Kim, L.; Lee, J.; Moon, M.K. Taking Metformin and Cognitive Function Change in Older Patients with Diabetes. Geriatr. Gerontol. Int. 2019, 19, 755–761. [Google Scholar] [CrossRef]
- Geijselaers, S.L.C.; Sep, S.J.S.; Stehouwer, C.D.A.; Biessels, G.J. Glucose Regulation, Cognition, and Brain MRI in Type 2 Diabetes: A Systematic Review. Lancet Diabetes Endocrinol. 2015, 3, 75–89. [Google Scholar] [CrossRef]
- Exalto, L.G.; Biessels, G.J.; Karter, A.J.; Huang, E.S.; Katon, W.J.; Minkoff, J.R.; Whitmer, R.A. Risk Score for Prediction of 10 Year Dementia Risk in Individuals with Type 2 Diabetes: A Cohort Study. Lancet Diabetes Endocrinol. 2013, 1, 183–190. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and Metformin Counteract Altered Mitochondrial Function in the Insulin-Resistant Brain. JCI Insight 2019, 4, e130681. [Google Scholar] [CrossRef] [PubMed]
- Maniar, K.; Moideen, A.; Mittal, A.; Patil, A.; Chakrabarti, A.; Banerjee, D. A Story of Metformin-Butyrate Synergism to Control Various Pathological Conditions as a Consequence of Gut Microbiome Modification: Genesis of a Wonder Drug? Pharmacol. Res. 2017, 117, 103–128. [Google Scholar] [CrossRef]
- Ihara, M.; Saito, S. Drug Repositioning for Alzheimer’s Disease: Finding Hidden Clues in Old Drugs. J. Alzheimers Dis. 2020, 74, 1013–1028. [Google Scholar] [CrossRef]
- Jacobs, H.I.L.; Hopkins, D.A.; Mayrhofer, H.C.; Bruner, E.; van Leeuwen, F.W.; Raaijmakers, W.; Schmahmann, J.D. The Cerebellum in Alzheimer’s Disease: Evaluating Its Role in Cognitive Decline. Brain 2018, 141, 37–47. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Paunescu, H.; Dima, L.; Ghita, I.; Coman, L.; Ifteni, P.I.; Fulga, I.; Coman, O.A. A Systematic Review of Clinical Studies on the Effect of Psychoactive Cannabinoids in Psychiatric Conditions in Alzheimer Dementia. Am. J. Ther. 2020, 27, e249–e269. [Google Scholar] [CrossRef]
- Teodorescu, A.; Dima, L.; Ifteni, P.; Rogozea, L.M. Clozapine for Treatment-Refractory Behavioral Disturbance in Dementia. Am. J. Ther. 2018, 25, e320–e325. [Google Scholar] [CrossRef]
- Shakil, S. Molecular Interaction of Anti-Diabetic Drugs With Acetylcholinesterase and Sodium Glucose Co-Transporter 2. J. Cell. Biochem. 2017, 118, 3855–3865. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimers Dis. 2018, 62, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s Disease and Metabolic Syndrome: A Link from Oxidative Stress and Inflammation to Neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- de la Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes; Springer: Singapore, 2019; pp. 45–83. [Google Scholar]
- Li, L.; Cavuoto, M.; Biddiscombe, K.; Pike, K.E. Diabetes Mellitus Increases Risk of Incident Dementia in APOE Ɛ4 Carriers: A Meta-Analysis. J. Alzheimers Dis. 2020, 74, 1295–1308. [Google Scholar] [CrossRef]
- Poor, S.R.; Ettcheto, M.; Cano, A.; Sanchez-Lopez, E.; Manzine, P.R.; Olloquequi, J.; Camins, A.; Javan, M. Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals 2021, 14, 890. [Google Scholar] [CrossRef]
- Xue, F.; Du, H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells 2021, 10, 321. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 Diabetes Is Sporadic Alzheimer׳s Disease: Mini-Review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.-C.; Cheng, Y.-C.; Chen, S.-J.; Yen, C.-H.; Huang, R.-N. Metformin Activation of AMPK-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Mitochondrial Dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.-M.; Chen, Y.-L.; Pei, D.; Cheng, Y.-C.; Sun, B.; Nicol, C.J.; Yen, C.-H.; Chen, H.-M.; Liang, Y.-J.; Chiang, M.-C. The Neuroprotective Role of Metformin in Advanced Glycation End Product Treated Human Neural Stem Cells Is AMPK-Dependent. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lin, Y.; Dai, X.; Fang, W.; Wu, X.; Chen, X. Metformin Treatment Improves the Spatial Memory of Aged Mice in an APOE Genotype–Dependent Manner. FASEB J. 2019, 33, 7748–7757. [Google Scholar] [CrossRef]
- Liao, W.; Xu, J.; Li, B.; Ruan, Y.; Li, T.; Liu, J. Deciphering the Roles of Metformin in Alzheimer’s Disease: A Snapshot. Front. Pharmacol. 2022, 12, 728315. [Google Scholar] [CrossRef]
- Khezri, M.R.; Yousefi, K.; Mahboubi, N.; Hodaei, D.; Ghasemnejad-Berenji, M. Metformin in Alzheimer’s Disease: An Overview of Potential Mechanisms, Preclinical and Clinical Findings. Biochem. Pharmacol. 2022, 197, 114945. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Yu, Y.-S.; Yu, P.-T.; Fang, H.-W.; Chang, Y.-C.; Wu, K.C.W. Lipid-Assisted Synthesis of Magnesium-Loaded Hydroxyapatite as a Potential Bone Healing Material. J. Taiwan Inst. Chem. Eng. 2021, 129, 40–51. [Google Scholar] [CrossRef]
- Chen, W.-B.; Chen, J.; Liu, Z.-Y.; Luo, B.; Zhou, T.; Fei, E.-K. Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons. Brain Sci. 2020, 10, 706. [Google Scholar] [CrossRef]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased Localization of APP-C99 in Mitochondria-associated ER Membranes Causes Mitochondrial Dysfunction in Alzheimer Disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal Tau, Mitochondrial Dysfunction, Impaired Axonal Transport of Mitochondria, and Synaptic Deprivation in Alzheimer’s Disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef]
- Pilipenko, V.; Narbute, K.; Pupure, J.; Langrate, I.K.; Muceniece, R.; Kluša, V. Neuroprotective Potential of Antihyperglycemic Drug Metformin in Streptozocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Eur. J. Pharmacol. 2020, 881, 173290. [Google Scholar] [CrossRef] [PubMed]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin Treatment Alters Memory Function in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ping, F.; Jiang, N.; Li, Y. Association between Metformin and Neurodegenerative Diseases of Observational Studies: Systematic Review and Meta-Analysis. BMJ Open Diabetes Res. Care 2020, 8, e001370. [Google Scholar] [CrossRef]
- Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.-M.; Hartikainen, S. Metformin and Risk of Alzheimer’s Disease Among Community-Dwelling People With Diabetes: A National Case-Control Study. J. Clin. Endocrinol. Metab. 2020, 105, e963–e972. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Barera, A.; Vasto, S.; Di Carlo, M. Metformin Increases APP Expression and Processing via Oxidative Stress, Mitochondrial Dysfunction and NF-ΚB Activation: Use of Insulin to Attenuate Metformin’s Effect. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1046–1059. [Google Scholar] [CrossRef]
- Kuhla, A.; Brichmann, E.; Rühlmann, C.; Thiele, R.; Meuth, L.; Vollmar, B. Metformin Therapy Aggravates Neurodegenerative Processes in ApoE–/– Mice. J. Alzheimers Dis. 2019, 68, 1415–1427. [Google Scholar] [CrossRef]
- Peng, L.; Fang, X.; Xu, F.; Liu, S.; Qian, Y.; Gong, X.; Zhao, X.; Ma, Z.; Xia, T.; Gu, X. Amelioration of Hippocampal Insulin Resistance Reduces Tau Hyperphosphorylation and Cognitive Decline Induced by Isoflurane in Mice. Front. Aging Neurosci. 2021, 13, 686506. [Google Scholar] [CrossRef]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, Other Antidiabetic Drugs, and Risk of Alzheimer’s Disease: A Population-Based Case-Control Study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased Risk of Cognitive Impairment in Patients With Diabetes Is Associated With Metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef]
- Wu, C.; Ouk, M.; Wong, Y.Y.; Anita, N.Z.; Edwards, J.D.; Yang, P.; Shah, B.R.; Herrmann, N.; Lanctôt, K.L.; Kapral, M.K.; et al. Relationships between Memory Decline and the Use of Metformin or DPP4 Inhibitors in People with Type 2 Diabetes with Normal Cognition or Alzheimer’s Disease, and the Role APOE Carrier Status. Alzheimers Dement. 2020, 16, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.L.; Nelson, P.T.; Kryscio, R.J.; Schmitt, F.A.; Fardo, D.W.; Woltjer, R.L.; Cairns, N.J.; Yu, L.; Dodge, H.H.; Xiong, C.; et al. Diabetes Is Associated with Cerebrovascular but Not Alzheimer’s Disease Neuropathology. Alzheimers Dement. 2016, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Silverman, J.M.; Davis, K.L.; Marin, D.; Grossman, H.Z.; Schmeidler, J.; Purohit, D.P.; Perl, D.P.; Davidson, M.; Mohs, R.C.; et al. Type 2 Diabetes Is Negatively Associated With Alzheimer’s Disease Neuropathology. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, J.A. Different Patterns of Cerebral Injury in Dementia With or Without Diabetes. Arch. Neurol. 2009, 66, 315. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Piperi, C.; Papavassiliou, A.G. High-Mobility Group Box 1 in Parkinson’s Disease: From Pathogenesis to Therapeutic Approaches. J. Neurochem. 2018, 146, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Biomarkers of Parkinson’s Disease. In Biomarkers in Toxicology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 895–909. [Google Scholar]
- Vilaça-Faria, H.; Salgado, A.J.; Teixeira, F.G. Mesenchymal Stem Cells-Derived Exosomes: A New Possible Therapeutic Strategy for Parkinson’s Disease? Cells 2019, 8, 118. [Google Scholar] [CrossRef]
- Huang, J.; Yang, J.; Shen, Y.; Jiang, H.; Han, C.; Zhang, G.; Liu, L.; Xu, X.; Li, J.; Lin, Z.; et al. HMGB1 Mediates Autophagy Dysfunction via Perturbing Beclin1-Vps34 Complex in Dopaminergic Cell Model. Front. Mol. Neurosci. 2017, 10, 13. [Google Scholar] [CrossRef]
- van der Brug, M.P.; Singleton, A.; Gasser, T.; Lewis, P.A. Parkinson’s Disease: From Human Genetics to Clinical Trials. Sci. Transl. Med. 2015, 7, 305ps20. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin Resistance and Parkinson’s Disease: A New Target for Disease Modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s Disease Dementia: Convergence of α-Synuclein, Tau and Amyloid-β Pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Compta, Y.; Parkkinen, L.; Kempster, P.; Selikhova, M.; Lashley, T.; Holton, J.L.; Lees, A.J.; Revesz, T. The Significance of α-Synuclein, Amyloid-β and Tau Pathologies in Parkinson’s Disease Progression and Related Dementia. Neurodegener. Dis. 2014, 13, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward Clinical Application of the Keap1–Nrf2 Pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Park, P.-H.; Choi, D.-Y. Metformin Attenuates Rotenone-Induced Oxidative Stress and Mitochondrial Damage via the AKT/Nrf2 Pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.-K.; Go, J.; Park, H.-Y.; Choi, Y.-K.; Seo, Y.J.; Choi, J.H.; Rhee, M.; Lee, T.G.; Lee, C.-H.; Kim, K.-S. Metformin Regulates Astrocyte Reactivity in Parkinson’s Disease and Normal Aging. Neuropharmacology 2020, 175, 108173. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Wu, S.-L.; Chen, T.-C.; Chuang, C.-S. Antidiabetic Agents for Treatment of Parkinson’s Disease: A Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 4805. [Google Scholar] [CrossRef]
- Wang, D.-X.; Chen, A.-D.; Wang, Q.-J.; Xin, Y.-Y.; Yin, J.; Jing, Y.-H. Protective Effect of Metformin against Rotenone-Induced Parkinsonism in Mice. Toxicol. Mech. Methods 2020, 30, 350–357. [Google Scholar] [CrossRef]
- Pérez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin Lowers Ser-129 Phosphorylated α-Synuclein Levels via MTOR-Dependent Protein Phosphatase 2A Activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef]
- Ryu, Y.-K.; Park, H.-Y.; Go, J.; Choi, D.-H.; Kim, Y.-H.; Hwang, J.H.; Noh, J.-R.; Lee, T.G.; Lee, C.-H.; Kim, K.-S. Metformin Inhibits the Development of L-DOPA-Induced Dyskinesia in a Murine Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 5715–5726. [Google Scholar] [CrossRef] [PubMed]
- Tayara, K.; Espinosa-Oliva, A.M.; García-Domínguez, I.; Ismaiel, A.A.; Boza-Serrano, A.; Deierborg, T.; Machado, A.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Divergent Effects of Metformin on an Inflammatory Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 440. [Google Scholar] [CrossRef]
- Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.J.; Montes-de-Oca-Luna, R.; Rodriguez-Rocha, H.; Garcia-Garcia, A. Metformin and Trehalose-Modulated Autophagy Exerts a Neurotherapeutic Effect on Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 7253–7273. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide Once Weekly versus Placebo in Parkinson’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Ismaiel, A.A.K.; Espinosa-Oliva, A.M.; Santiago, M.; García-Quintanilla, A.; Oliva-Martín, M.J.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Metformin, besides Exhibiting Strong in Vivo Anti-Inflammatory Properties, Increases Mptp-Induced Damage to the Nigrostriatal Dopaminergic System. Toxicol. Appl. Pharmacol. 2016, 298, 19–30. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Chu, X.; Cook, J.A.; Custodio, J.M.; Galetin, A.; Giacomini, K.M.; Lee, C.A.; Paine, M.F.; Ray, A.S.; Ware, J.A.; et al. ITC Commentary on Metformin Clinical Drug-Drug Interaction Study Design That Enables an Efficacy- and Safety-Based Dose Adjustment Decision. Clin. Pharmacol. Ther. 2018, 104, 781–784. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Li, L.; Li, Q.; Ren, K.; Sun, X.; Li, J. Metformin Treatment and Homocysteine: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8, 798. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Han, K.-D.; Kwon, H.; Park, S.-E.; Park, Y.-G.; Kim, Y.-H.; Yoo, S.-J.; Rhee, E.-J.; Lee, W.-Y. Association Between Glycemic Status and the Risk of Parkinson Disease: A Nationwide Population-Based Study. Diabetes Care 2020, 43, 2169–2175. [Google Scholar] [CrossRef]
- Kuan, Y.-C.; Huang, K.-W.; Lin, C.-L.; Hu, C.-J.; Kao, C.-H. Effects of Metformin Exposure on Neurodegenerative Diseases in Elderly Patients with Type 2 Diabetes Mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 77–83. [Google Scholar] [CrossRef]
- Brakedal, B.; Flønes, I.; Reiter, S.F.; Torkildsen, Ø.; Dölle, C.; Assmus, J.; Haugarvoll, K.; Tzoulis, C. Glitazone Use Associated with Reduced Risk of Parkinson’s Disease. Mov. Disord. 2017, 32, 1594–1599. [Google Scholar] [CrossRef]
- Liu, W.; Tang, J. Association between Diabetes Mellitus and Risk of Parkinson’s Disease: A Prisma-compliant Meta-analysis. Brain Behav. 2021, 11, e02082. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, J.; Jiang, J.; Liu, F.; Zhang, Y. Do Oral Antidiabetic Medications Alter the Risk of Parkinson’s Disease? An Updated Systematic Review and Meta-Analysis. Neurol. Sci. 2023, 44, 4193–4203. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Jung, J.; Falk, J.; Herrmann, W.; Geisel, J.; Friesenhahn-Ochs, B.; Lammert, F.; Fassbender, K.; Kostopoulos, P. Serum Vitamin B12 Not Reflecting Vitamin B12 Status in Patients with Type 2 Diabetes. Biochimie 2013, 95, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Feng, H.; Peng, S.; Xiao, J.; Zhang, J. Association of Plasma Homocysteine, Vitamin B12 and Folate Levels with Cognitive Function in Parkinson’s Disease: A Meta-Analysis. Neurosci. Lett. 2017, 636, 190–195. [Google Scholar] [CrossRef]
- Vinueza Veloz, A.F.; Carpio Arias, T.V.; Vargas Mejía, J.S.; Tapia Veloz, E.C.; Piedra Andrade, J.S.; Nicolalde Cifuentes, T.M.; Heredia Aguirre, S.I.; Vinueza Veloz, M.F. Cognitive Function and Vitamin B12 and D among Community-Dwelling Elders: A Cross-Sectional Study. Clin. Nutr. ESPEN 2022, 50, 270–276. [Google Scholar] [CrossRef]
- Christine, C.W.; Auinger, P.; Saleh, N.; Tian, M.; Bottiglieri, T.; Arning, E.; Tran, N.K.; Ueland, P.M.; Green, R. Relationship of Cerebrospinal Fluid Vitamin B12 Status Markers With Parkinson’s Disease Progression. Mov. Disord. 2020, 35, 1466–1471. [Google Scholar] [CrossRef]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-Term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association Between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 678649. [Google Scholar] [CrossRef]
- Morena, E.; Romano, C.; Marconi, M.; Diamant, S.; Buscarinu, M.C.; Bellucci, G.; Romano, S.; Scarabino, D.; Salvetti, M.; Ristori, G. Peripheral Biomarkers in Manifest and Premanifest Huntington’s Disease. Int. J. Mol. Sci. 2023, 24, 6051. [Google Scholar] [CrossRef]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-Mediated Clearance of Huntingtin Aggregates Triggered by the Insulin-Signaling Pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef]
- Ortega, Z.; Lucas, J.J. Ubiquitin—Proteasome System Involvement in Huntington’s Disease. Front. Mol. Neurosci. 2014, 7, 77. [Google Scholar] [CrossRef]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 Activates AMPK-Dependent Cytoprotective Autophagy in TRAIL-Treated Epithelial Cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The Energy Sensing LKB1–AMPK Pathway Regulates P27kip1 Phosphorylation Mediating the Decision to Enter Autophagy or Apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK Activation Protects from Neuronal Dysfunction and Vulnerability across Nematode, Cellular and Mouse Models of Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Croce, K.R.; Yamamoto, A. A Role for Autophagy in Huntington’s Disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin Treatment Reduces Motor and Neuropsychiatric Phenotypes in the ZQ175 Mouse Model of Huntington Disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin Intake Associates with Better Cognitive Function in Patients with Huntington’s Disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed]
- Landwehrmeyer, G.B.; Fitzer-Attas, C.J.; Giuliano, J.D.; Gonçalves, N.; Anderson, K.E.; Cardoso, F.; Ferreira, J.J.; Mestre, T.A.; Stout, J.C.; Sampaio, C. Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington’s Disease. Mov. Disord. Clin. Pract. 2017, 4, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in Adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Mazarati, A.M.; Lewis, M.L.; Pittman, Q.J. Neurobehavioral Comorbidities of Epilepsy: Role of Inflammation. Epilepsia 2017, 58, 48–56. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Shah, S.; Kumari, Y.; Othman, I. Role of Inflammation in Epilepsy and Neurobehavioral Comorbidities: Implication for Therapy. Eur. J. Pharmacol. 2018, 837, 145–155. [Google Scholar] [CrossRef]
- Powell, G.; Ziso, B.; Larner, A. The Overlap between Epilepsy and Alzheimer’s Disease and the Consequences for Treatment. Expert Rev. Neurother. 2019, 19, 653–661. [Google Scholar] [CrossRef]
- Garg, N.; Joshi, R.; Medhi, B. Cracking Novel Shared Targets between Epilepsy and Alzheimer’s Disease: Need of the Hour. Rev. Neurosci. 2018, 29, 425–442. [Google Scholar] [CrossRef]
- Asadollahi, M.; Atazadeh, M.; Noroozian, M. Seizure in Alzheimer’s Disease: An Underestimated Phenomenon. Am. J. Alzheimers Dis. Other Dement. 2019, 34, 81–88. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron–Glia Interactions in the Pathophysiology of Epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Mehrabi, S.; Sanadgol, N.; Barati, M.; Shahbazi, A.; Vahabzadeh, G.; Barzroudi, M.; Seifi, M.; Gholipourmalekabadi, M.; Golab, F. Evaluation of Metformin Effects in the Chronic Phase of Spontaneous Seizures in Pilocarpine Model of Temporal Lobe Epilepsy. Metab. Brain Dis. 2018, 33, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Xu, X.; Xu, F.; Zhang, W.; Zhang, W.; Liu, L.; Wang, W. Metformin Protects against Seizures, Learning and Memory Impairments and Oxidative Damage Induced by Pentylenetetrazole-Induced Kindling in Mice. Biochem. Biophys. Res. Commun. 2014, 448, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, B.; Zheng, F.; Li, Y.; Zhang, Y.; Hu, Y.; Wang, X. Chronic Metformin Treatment Facilitates Seizure Termination. Biochem. Biophys. Res. Commun. 2017, 484, 450–455. [Google Scholar] [CrossRef] [PubMed]
- del Rubio Osornio, M.C.; Custodio Ramírez, V.; Calderón Gámez, D.; Paz Tres, C.; Carvajal Aguilera, K.G.; Phillips Farfán, B.V. Metformin Plus Caloric Restriction Show Anti-Epileptic Effects Mediated by MTOR Pathway Inhibition. Cell. Mol. Neurobiol. 2018, 38, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Eldosoky, M.; El-Shafey, M.; El-Mesery, M.; Ali, A.N.; Abbas, K.M.; Abulseoud, O.A. Effects of Metformin on Apoptosis and α-Synuclein in a Rat Model of Pentylenetetrazole-Induced Epilepsy. Can. J. Physiol. Pharmacol. 2019, 97, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Yimer, E.M.; Surur, A.; Wondafrash, D.Z.; Gebre, A.K. The Effect of Metformin in Experimentally Induced Animal Models of Epileptic Seizure. Behav. Neurol. 2019, 2019, 6234758. [Google Scholar] [CrossRef]
- Singh, R.; Sarangi, S.C.; Singh, S.; Tripathi, M. A Review on Role of Metformin as a Potential Drug for Epilepsy Treatment and Modulation of Epileptogenesis. Seizure 2022, 101, 253–261. [Google Scholar] [CrossRef]
- Nandini, H.S.; Paudel, Y.N.; Krishna, K.L. Envisioning the Neuroprotective Effect of Metformin in Experimental Epilepsy: A Portrait of Molecular Crosstalk. Life Sci. 2019, 233, 116686. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Constanti, A.; Russo, E.; De Sarro, G. MTOR Pathway Inhibition as a New Therapeutic Strategy in Epilepsy and Epileptogenesis. Pharmacol. Res. 2016, 107, 333–343. [Google Scholar] [CrossRef]
- Meng, X.-F.; Yu, J.-T.; Song, J.-H.; Chi, S.; Tan, L. Role of the MTOR Signaling Pathway in Epilepsy. J. Neurol. Sci. 2013, 332, 4–15. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Hardie, D.G. Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Demaré, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a Potential Therapeutic for Neurological Disease: Mobilizing AMPK to Repair the Nervous System. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, Independent of AMPK, Inhibits MTORC1 in a Rag GTPase-Dependent Manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Rüber, T.; David, B.; Lüchters, G.; Nass, R.D.; Friedman, A.; Surges, R.; Stöcker, T.; Weber, B.; Deichmann, R.; Schlaug, G.; et al. Evidence for Peri-Ictal Blood–Brain Barrier Dysfunction in Patients with Epilepsy. Brain 2018, 141, 2952–2965. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.S.; Soldner, E.L.B.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Vijayanti, S.; Saha, L. Targeting Crosstalk between Nuclear Factor (Erythroid-Derived 2)-like 2 and Nuclear Factor Kappa Beta Pathway by Nrf2 Activator Dimethyl Fumarate in Epileptogenesis. Int. J. Neurosci. 2018, 128, 987–994. [Google Scholar] [CrossRef]
- Anderson, V.C.; Lenar, D.P.; Quinn, J.F.; Rooney, W.D. The Blood-Brain Barrier and Microvascular Water Exchange in Alzheimer’s Disease. Cardiovasc. Psychiatry Neurol. 2011, 2011, 615829. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, O.; Feintuch, A.; Benifla, M.; Cohen, A.; Friedman, A.; Shelef, I. Blood-Brain Barrier Breakdown Following Traumatic Brain Injury: A Possible Role in Posttraumatic Epilepsy. Cardiovasc. Psychiatry Neurol. 2011, 2011, 765923. [Google Scholar] [CrossRef] [PubMed]
- Serlin, Y.; Levy, J.; Shalev, H. Vascular Pathology and Blood-Brain Barrier Disruption in Cognitive and Psychiatric Complications of Type 2 Diabetes Mellitus. Cardiovasc. Psychiatry Neurol. 2011, 2011, 609202. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Metformin Attenuates Blood-Brain Barrier Disruption in Mice Following Middle Cerebral Artery Occlusion. J. Neuroinflamm. 2014, 11, 177. [Google Scholar] [CrossRef]
- Ahmadian, S.R.; Ghasemi-Kasman, M.; Pouramir, M.; Sadeghi, F. Arbutin Attenuates Cognitive Impairment and Inflammatory Response in Pentylenetetrazol-Induced Kindling Model of Epilepsy. Neuropharmacology 2019, 146, 117–127. [Google Scholar] [CrossRef]
- Goel, R.; Saxena, P. Pycnogenol Protects against Pentylenetetrazole-Induced Oxidative Stress and Seizures in Mice. Curr. Clin. Pharmacol. 2019, 14, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Mallick, A.A.; Edwards, H.; Cortina-Borja, M.; Laugharne, M.; Likeman, M.; O’Callaghan, F.J.K. The Metformin in Tuberous Sclerosis (MiTS) Study: A Randomised Double-Blind Placebo-Controlled Trial. EClinicalMedicine 2021, 32, 100715. [Google Scholar] [CrossRef]
- Bisulli, F.; Muccioli, L.; d’Orsi, G.; Canafoglia, L.; Freri, E.; Licchetta, L.; Mostacci, B.; Riguzzi, P.; Pondrelli, F.; Avolio, C.; et al. Treatment with Metformin in Twelve Patients with Lafora Disease. Orphanet. J. Rare Dis. 2019, 14, 149. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.L.; Basit, H.; Shah, M.; Los, E. Fragile X Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V.; et al. Insulin Signaling Misregulation Underlies Circadian and Cognitive Deficits in a Drosophila Fragile X Model. Mol. Psychiatry 2017, 22, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin Ameliorates Core Deficits in a Mouse Model of Fragile X Syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as Targeted Treatment in Fragile X Syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef]
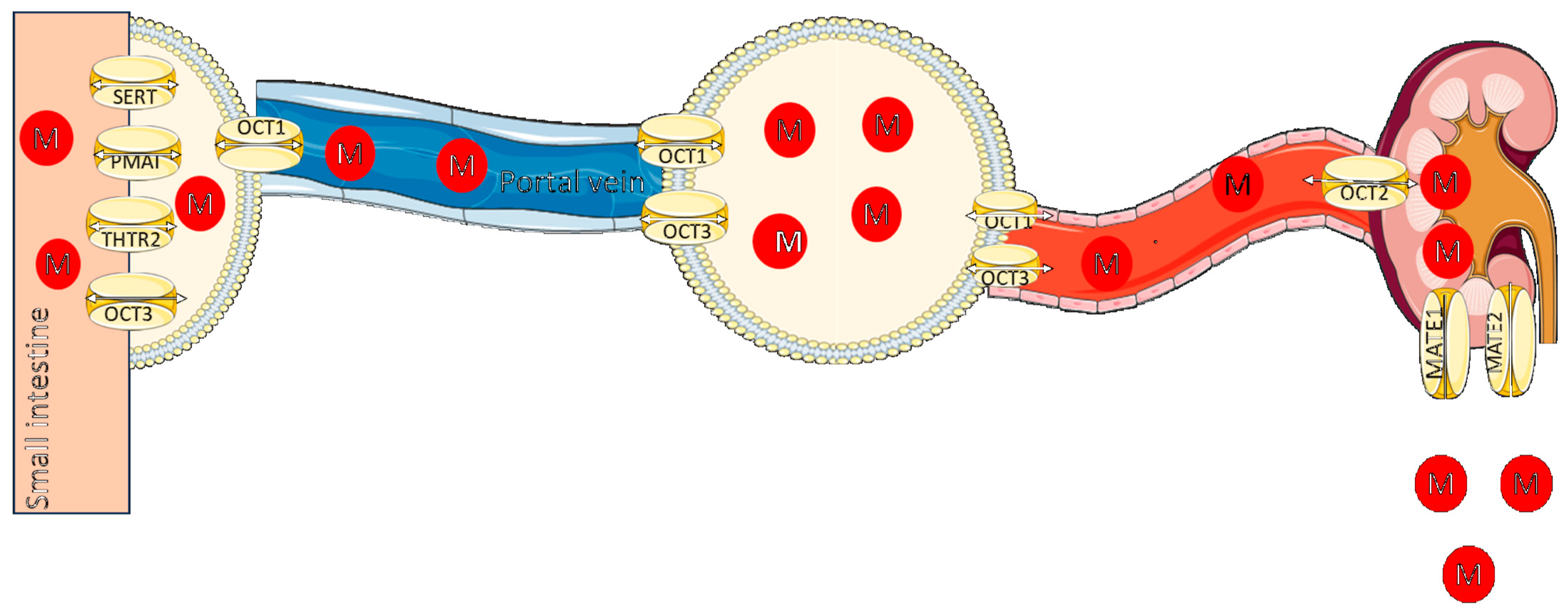

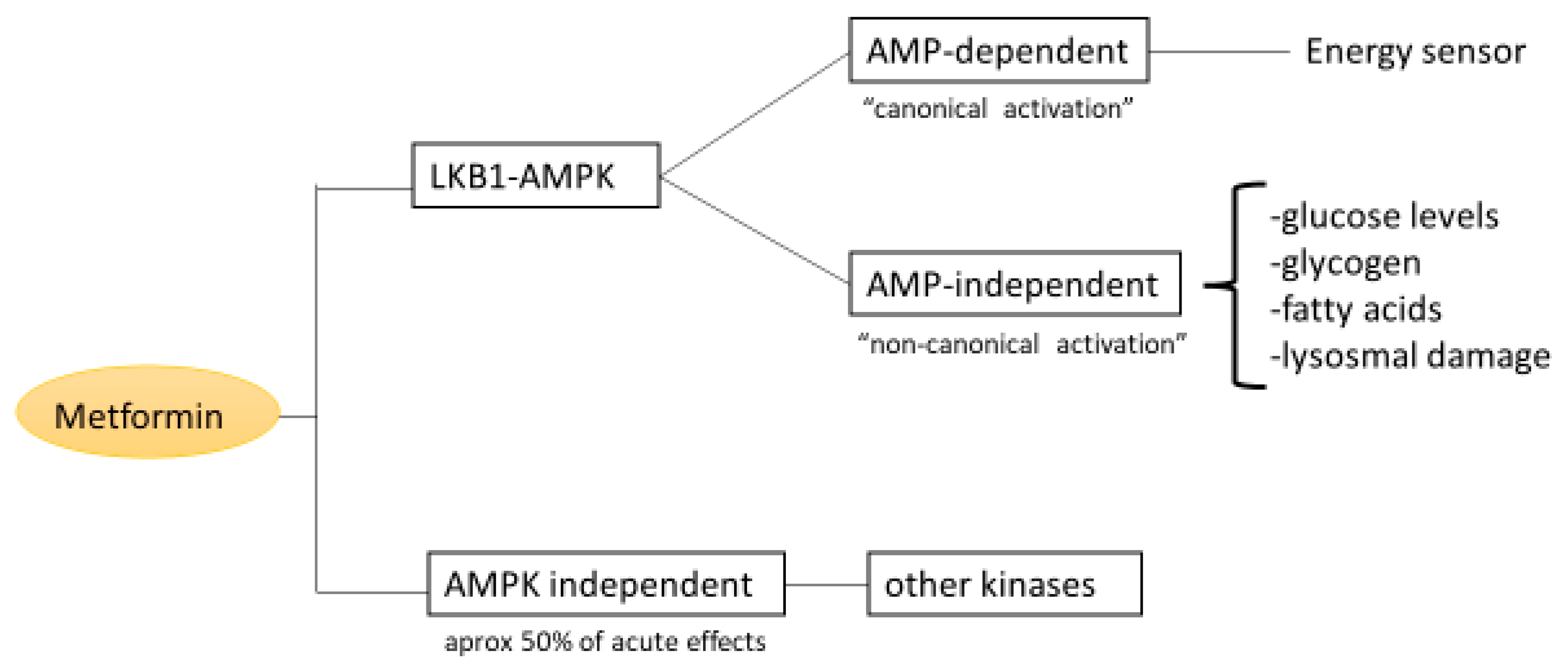

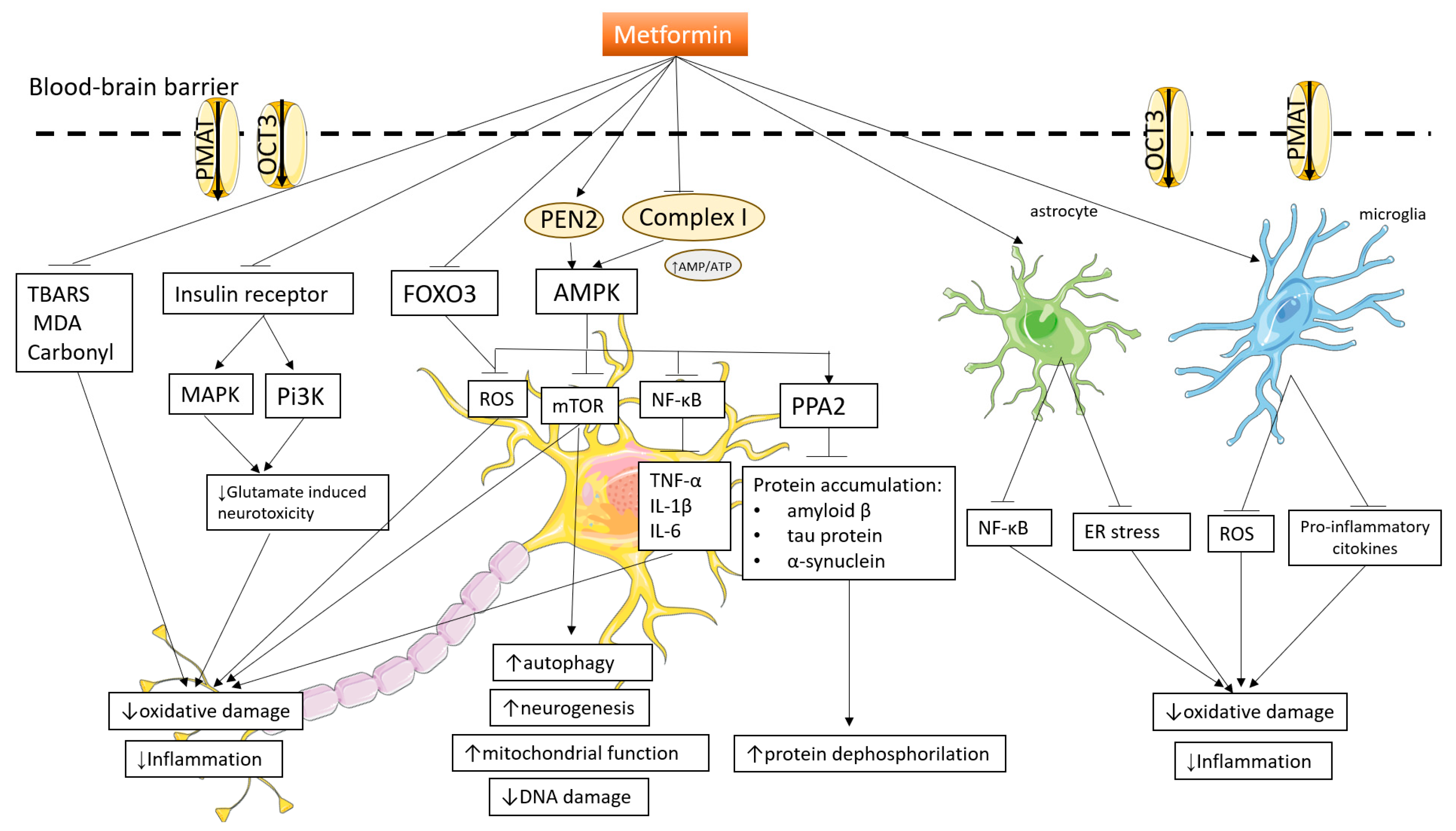
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isop, L.M.; Neculau, A.E.; Necula, R.D.; Kakucs, C.; Moga, M.A.; Dima, L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 1714. https://doi.org/10.3390/ph16121714
Isop LM, Neculau AE, Necula RD, Kakucs C, Moga MA, Dima L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals. 2023; 16(12):1714. https://doi.org/10.3390/ph16121714
Chicago/Turabian StyleIsop, Laura Mihaela, Andrea Elena Neculau, Radu Dan Necula, Cristian Kakucs, Marius Alexandru Moga, and Lorena Dima. 2023. "Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders" Pharmaceuticals 16, no. 12: 1714. https://doi.org/10.3390/ph16121714
APA StyleIsop, L. M., Neculau, A. E., Necula, R. D., Kakucs, C., Moga, M. A., & Dima, L. (2023). Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals, 16(12), 1714. https://doi.org/10.3390/ph16121714