
Computational and Experimental Drug Repurposing of FDA-Approved Compounds Targeting the Cannabinoid Receptor CB1

 ,
,  and
and
Abstract
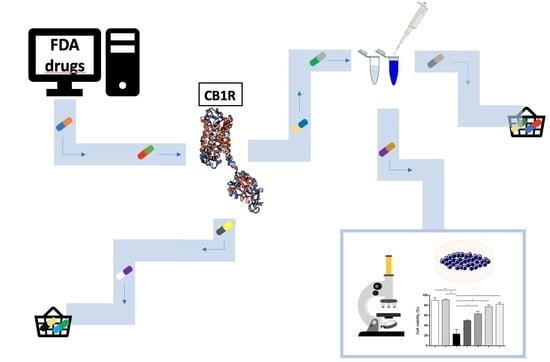
:
1. Introduction
2. Results
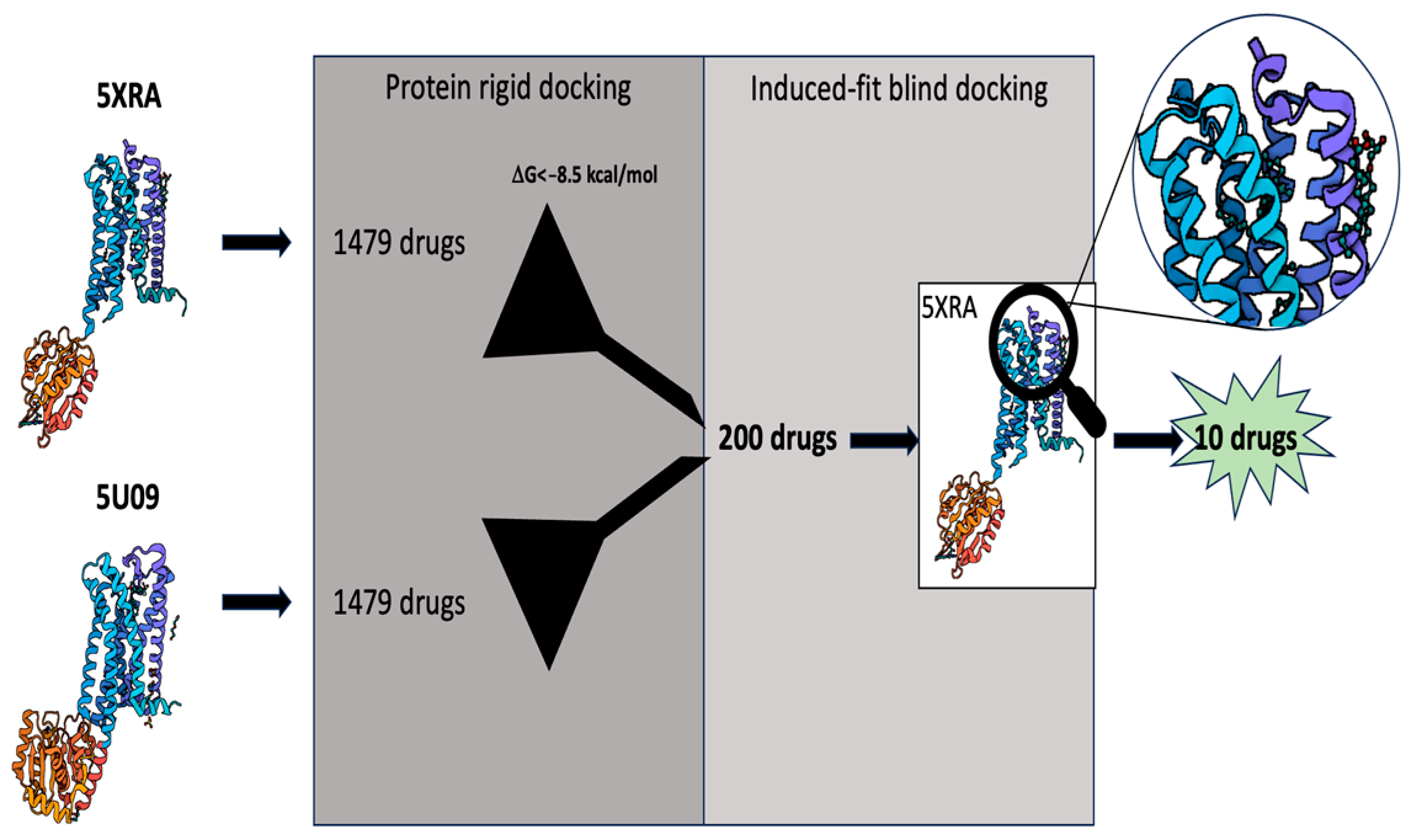
2.1. Virtual Screening
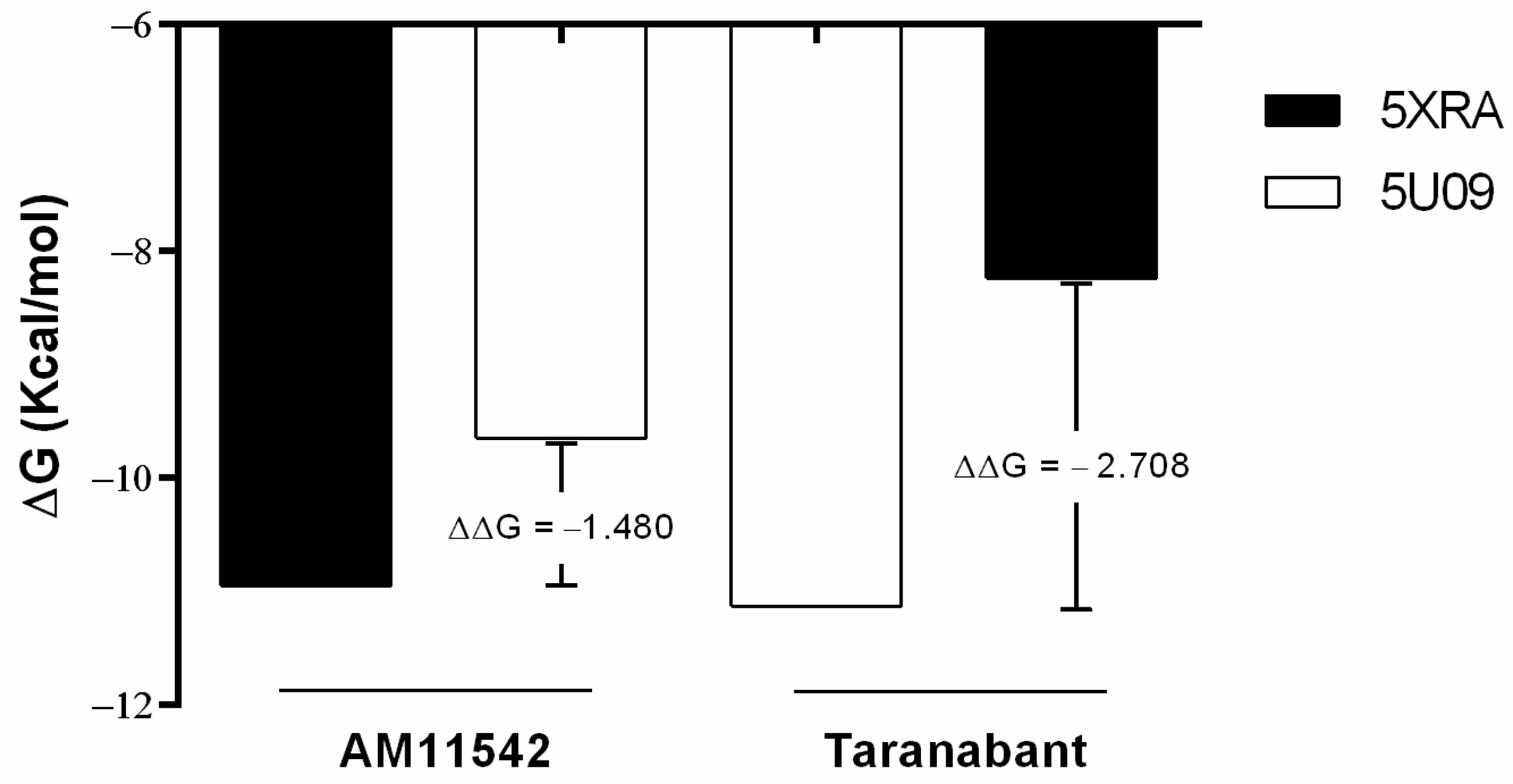
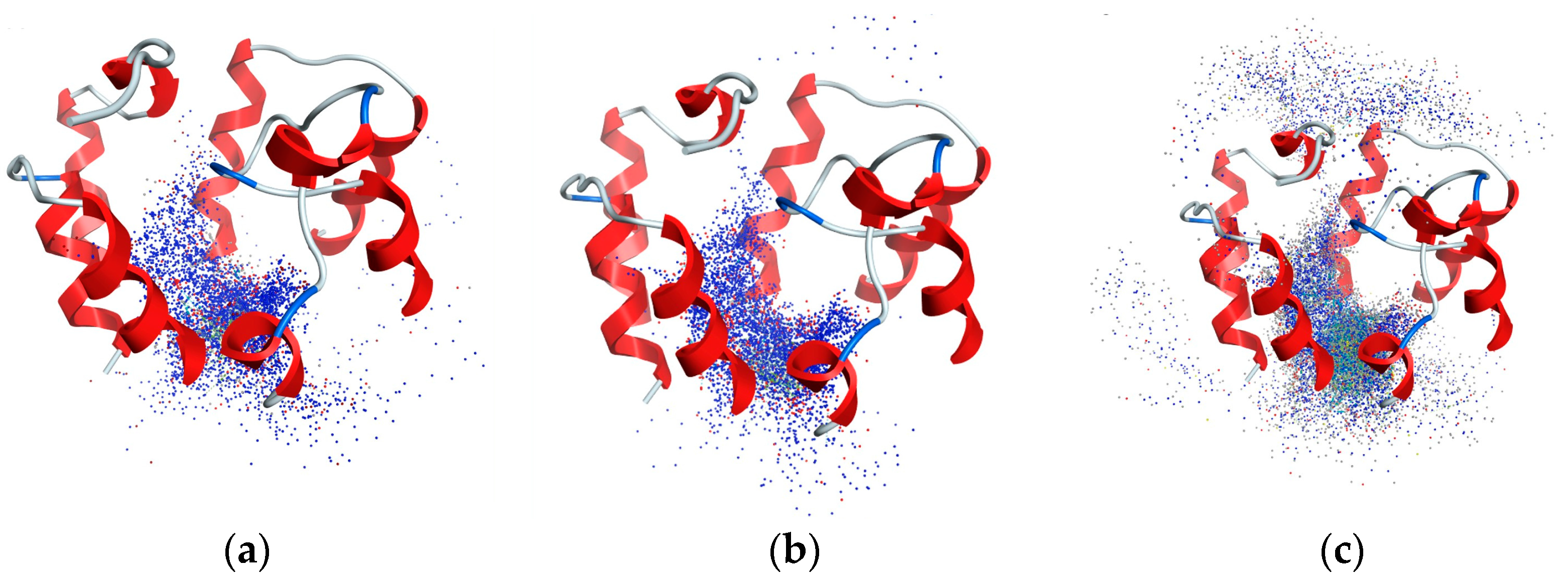
2.2. Analysis of CB1R Binding
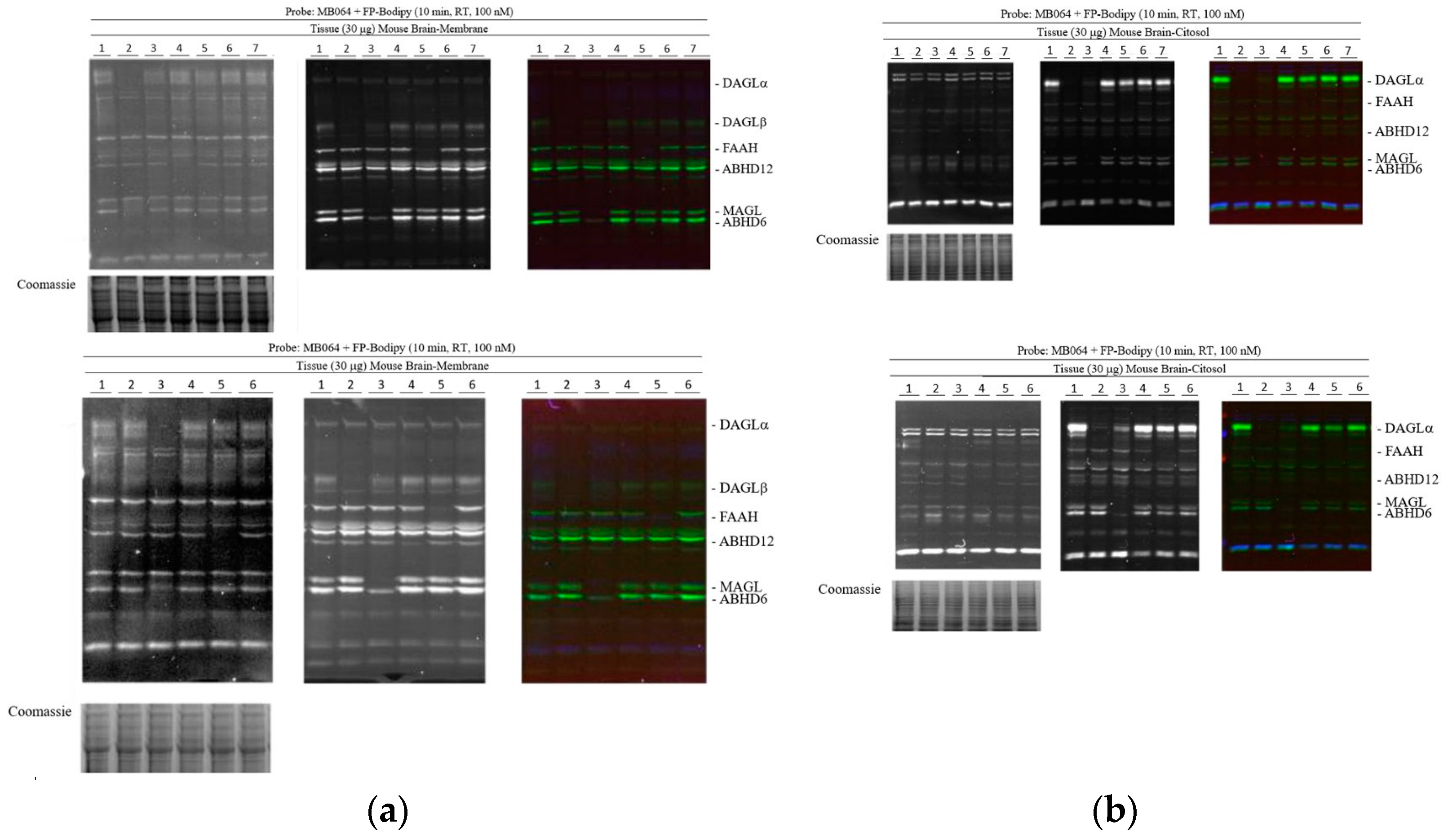
2.3. Activity-Based Protein Profiling
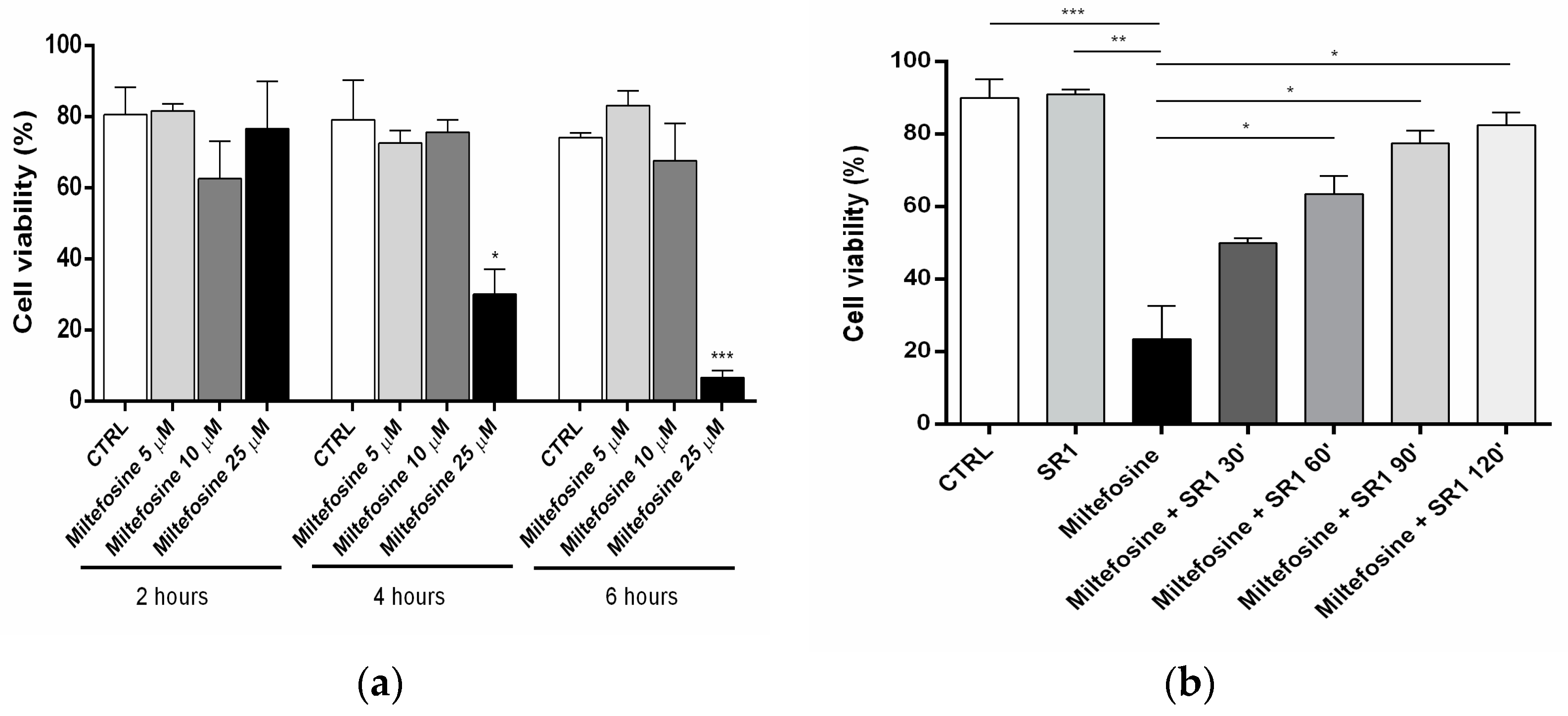
2.4. Cell Viability
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Virtual Screening
4.2.1. Proteins and Ligands Preparation
4.2.2. Re-Docking Validation
4.2.3. Protein Rigid Docking
4.2.4. Blind Induced-Fit Docking
4.3. Binding Assay
4.4. Activity-Based Protein Profiling (ABPP)
4.5. Cell Viability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Shah, S. Structure-Based Virtual Screening. In Protein Bioinformatics; Wu, C.H., Arighi, C.N., Ross, K.E., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1558, pp. 111–124. ISBN 978-1-4939-6781-0. [Google Scholar]
- Gambacorta, N.; Ciriaco, F.; Amoroso, N.; Altomare, C.D.; Bajorath, J.; Nicolotti, O. CIRCE: Web-Based Platform for the Prediction of Cannabinoid Receptor Ligands Using Explainable Machine Learning. J. Chem. Inf. Model. 2023, 63, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ou-Yang, S.; Lu, J.; Kong, X.; Liang, Z.; Luo, C.; Jiang, H. Computational Drug Discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Stasiulewicz, A.; Lesniak, A.; Setny, P.; Bujalska-Zadrożny, M.; Sulkowska, J.I. Identification of CB1 Ligands among Drugs, Phytochemicals and Natural-Like Compounds: Virtual Screening and In Vitro Verification. ACS Chem. Neurosci. 2022, 13, 2991–3007. [Google Scholar] [CrossRef]
- Catella-Lawson, F.; Reilly, M.P.; Kapoor, S.C.; Cucchiara, A.J.; DeMarco, S.; Tournier, B.; Vyas, S.N.; FitzGerald, G.A. Cyclooxygenase Inhibitors and the Antiplatelet Effects of Aspirin. N. Engl. J. Med. 2001, 345, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L.; Macintyre, D.E. Proceedings: Inhibition of Human Platelet Aggregation by Aspirin In Vitro and Ex Vivo. Br. J. Pharmacol. 1974, 52, 451P. [Google Scholar] [PubMed]
- Menter, D.G.; Bresalier, R.S. An Aspirin a Day: New Pharmacological Developments and Cancer Chemoprevention. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Joharatnam-Hogan, N.; Hatem, D.; Cafferty, F.H.; Petrucci, G.; Cameron, D.A.; Ring, A.; Kynaston, H.G.; Gilbert, D.C.; Wilson, R.H.; Hubner, R.A.; et al. Thromboxane Biosynthesis in Cancer Patients and Its Inhibition by Aspirin: A Sub-Study of the Add-Aspirin Trial. Br. J. Cancer 2023, 129, 706–720. [Google Scholar] [CrossRef]
- Virk, H.U.H.; Escobar, J.; Rodriguez, M.; Bates, E.R.; Khalid, U.; Jneid, H.; Birnbaum, Y.; Levine, G.N.; Smith, S.C.; Krittanawong, C. Dual Antiplatelet Therapy: A Concise Review for Clinicians. Life 2023, 13, 1580. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.T. Thalidomide Embryopathy: A Model for the Study of Congenital Incomitant Horizontal Strabismus. Trans. Am. Ophthalmol. Soc. 1991, 89, 623–674. [Google Scholar] [PubMed]
- Vargesson, N. Thalidomide-Induced Teratogenesis: History and Mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Fabro, S.; Smith, R.L.; Williams, R.T. Toxicity and Teratogenicity of Optical Isomers of Thalidomide. Nature 1967, 215, 296. [Google Scholar] [CrossRef] [PubMed]
- Franks, M.E.; Macpherson, G.R.; Figg, W.D. Thalidomide. Lancet 2004, 363, 1802–1811. [Google Scholar] [CrossRef]
- Langtry, H.D.; Markham, A. Sildenafil: A Review of Its Use in Erectile Dysfunction. Drugs 1999, 57, 967–989. [Google Scholar] [CrossRef]
- Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Tocilizumab in the Treatment of Rheumatoid Arthritis: An Evidence-Based Review and Patient Selection. Drug Des. Devel. Ther. 2018, 13, 57–70. [Google Scholar] [CrossRef]
- Freitas, E.; Guttman-Yassky, E.; Torres, T. Baricitinib for the Treatment of Alopecia Areata. Drugs 2023, 83, 761–770. [Google Scholar] [CrossRef]
- von Hentig, N. Repositioning HIV Protease Inhibitors and Nucleos(t)Ide RNA Polymerase Inhibitors for the Treatment of SARS-CoV-2 Infection and COVID-19. Eur. J. Clin. Pharmacol. 2021, 77, 1297–1307. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Agoramoorthy, G.; Lee, S.-S. The Drug Repurposing for COVID-19 Clinical Trials Provide Very Effective Therapeutic Combinations: Lessons Learned from Major Clinical Studies. Front. Pharmacol. 2021, 12, 704205. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Maccarrone, M.; Di Marzo, V.; Gertsch, J.; Grether, U.; Howlett, A.C.; Hua, T.; Makriyannis, A.; Piomelli, D.; Ueda, N.; Van Der Stelt, M. Goods and Bads of the Endocannabinoid System as a Therapeutic Target: Lessons Learned after 30 Years. Pharmacol. Rev. 2023, 75, 885–958. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G Protein-Coupled Receptor Ligand Identification Using β-Arrestin PathHunterTM Assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-Resolution Crystal Structure of the Human CB1 Cannabinoid Receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal Structures of Agonist-Bound Human Cannabinoid Receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.; Johnson, M.; Melvin, L.; De Costa, B.; Rice, K. Characterization and Localization of Cannabinoid Receptors in Rat Brain: A Quantitative In Vitro Autoradiographic Study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Cavuoto, P.; McAinch, A.J.; Hatzinikolas, G.; Janovská, A.; Game, P.; Wittert, G.A. The Expression of Receptors for Endocannabinoids in Human and Rodent Skeletal Muscle. Biochem. Biophys. Res. Commun. 2007, 364, 105–110. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Endocannabinoids in the Gut. Cannabis Cannabinoid Res. 2016, 1, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Malenczyk, K.; Jazurek, M.; Keimpema, E.; Silvestri, C.; Janikiewicz, J.; Mackie, K.; Di Marzo, V.; Redowicz, M.J.; Harkany, T.; Dobrzyn, A. CB1 Cannabinoid Receptors Couple to Focal Adhesion Kinase to Control Insulin Release. J. Biol. Chem. 2013, 288, 32685–32699. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 Receptor Is Required for Development of Diet-Induced Steatosis, Dyslipidemia, and Insulin and Leptin Resistance in Mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and Characterization of a Cannabinoid Receptor in Rat Brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef]
- Baggelaar, M.P.; Maccarrone, M.; Van Der Stelt, M. 2-Arachidonoylglycerol: A Signaling Lipid with Manifold Actions in the Brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef]
- Götz, M.R.; Collado, J.A.; Fernández-Ruiz, J.; Fiebich, B.L.; García-Toscano, L.; Gómez-Cañas, M.; Koch, O.; Leha, A.; Muñoz, E.; Navarrete, C.; et al. Structure–Effect Relationships of Novel Semi-Synthetic Cannabinoid Derivatives. Front. Pharmacol. 2019, 10, 1284. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid Receptor Ligands: Clinical and Neuropharmacological Considerations, Relevant to Future Drug Discovery and Development. Expert Opin. Investig. Drugs 2000, 9, 1553–1571. [Google Scholar] [CrossRef]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C.E. Pharmacological Evaluation of New Constituents of “Spice”: Synthetic Cannabinoids Based on Indole, Indazole, Benzimidazole and Carbazole Scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef]
- King, A. Neuropsychiatric Adverse Effects Signal the End of the Line for Rimonabant. Nat. Rev. Cardiol. 2010, 7, 602. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.B.; Morris, M.J. Depression and Anxiety with Rimonabant. Lancet 2007, 370, 1671–1672. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.J.; Banister, S.D.; Irizarry, L.; Trecki, J.; Schwartz, M.; Gerona, R. “Zombie” Outbreak Caused by the Synthetic Cannabinoid AMB-FUBINACA in New York. N. Engl. J. Med. 2017, 376, 235–242. [Google Scholar] [CrossRef]
- Kumar, K.K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, A.; Haron, M.H.; Klein, M.L.; Elokely, K. Understanding the Dynamics of the Structural States of Cannabinoid Receptors and the Role of Different Modulators. Life 2022, 12, 2137. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.J.; Kuc, R.E.; Davenport, A.P. Radioligand Binding Assays and Their Analysis. In Receptor Binding Techniques; Davenport, A.P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 897, pp. 31–77. ISBN 978-1-61779-908-2. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Jakowiecki, J.; Orzeł, U.; Chawananon, S.; Miszta, P.; Filipek, S. The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules 2020, 25, 1930. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Franks, L.N.; Ford, B.M.; Prather, P.L. Selective Estrogen Receptor Modulators: Cannabinoid Receptor Inverse Agonists with Differential CB1 and CB2 Selectivity. Front. Pharmacol. 2016, 7, 503. [Google Scholar] [CrossRef]
- Soli, M.; Bertaccini, A.; Carparelli, F.; Gotti, R.; Cavrini, V.; Andrisano, V.; Martorana, G. Vasoactive cocktails for erectile dysfunction: Chemical stability of PGE1, paraverine and phentolamine. J. Urol. 1998, 160, 551–555. [Google Scholar] [CrossRef]
- Barglow, K.T.; Cravatt, B.F. Activity-Based Protein Profiling for the Functional Annotation of Enzymes. Nat. Methods 2007, 4, 822–827. [Google Scholar] [CrossRef]
- Van Rooden, E.J.; Florea, B.I.; Deng, H.; Baggelaar, M.P.; Van Esbroeck, A.C.M.; Zhou, J.; Overkleeft, H.S.; Van Der Stelt, M. Mapping In Vivo Target Interaction Profiles of Covalent Inhibitors Using Chemical Proteomics with Label-Free Quantification. Nat. Protoc. 2018, 13, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.P.A.; Van Der Vliet, D.; Bakker, A.T.; Jiang, M.; Grimm, S.H.; Campiani, G.; Butini, S.; Van Der Stelt, M. Development of a Multiplexed Activity-Based Protein Profiling Assay to Evaluate Activity of Endocannabinoid Hydrolase Inhibitors. ACS Chem. Biol. 2018, 13, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Mouscadet, J.-F.; Tchertanov, L. Raltegravir: Molecular Basis of Its Mechanism of Action. Eur. J. Med. Res. 2009, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the Folate Receptor α in Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Wieder, T.; Orfanos, C.E.; Geilen, C.C. Induction of Ceramide-Mediated Apoptosis by the Anticancer Phospholipid Analog, Hexadecylphosphocholine. J. Biol. Chem. 1998, 273, 11025–11031. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Maccarrone, M. Cannabinoids and Cancer. Cancers 2021, 13, 4458. [Google Scholar] [CrossRef]
- De Miguel, R.; Montejano, R.; Stella-Ascariz, N.; Arribas, J.R. A Safety Evaluation of Raltegravir for the Treatment of HIV. Expert Opin. Drug Saf. 2018, 17, 217–223. [Google Scholar] [CrossRef]
- Ismail, M.; Hasan, H.; El-Orfali, Y.; Ismail, H.; Khawaja, G. Anti-Inflammatory, Antioxidative, and Hepatoprotective Effects of Trans Δ 9-Tetrahydrocannabinol/Sesame Oil on Adjuvant-Induced Arthritis in Rats. Evid. Based Complement. Alternat. Med. 2018, 2018, 9365464. [Google Scholar] [CrossRef]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral Miltefosine for Indian Visceral Leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef]
- Maccarrone, M.; Di Rienzo, M.; Battista, N.; Gasperi, V.; Guerrieri, P.; Rossi, A.; Finazzi-Agrò, A. The Endocannabinoid System in Human Keratinocytes. J. Biol. Chem. 2003, 278, 33896–33903. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Baker, D. RosettaLigand Docking with Full Ligand and Receptor Flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef] [PubMed]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The Performance of Current Methods in Ligand–Protein Docking. Curr. Sci. 2002, 83, 845–856. [Google Scholar]
- Kalinowsky, L.; Weber, J.; Balasupramaniam, S.; Baumann, K.; Proschak, E. A Diverse Benchmark Based on 3D Matched Molecular Pairs for Validating Scoring Functions. ACS Omega 2018, 3, 5704–5714. [Google Scholar] [CrossRef]
- Fezza, F.; Oddi, S.; Di Tommaso, M.; De Simone, C.; Rapino, C.; Pasquariello, N.; Dainese, E.; Finazzi-Agrò, A.; Maccarrone, M. Characterization of Biotin-Anandamide, a Novel Tool for the Visualization of Anandamide Accumulation. J. Lipid Res. 2008, 49, 1216–1223. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Chemical Structures | Indications |
|---|---|---|
| Aminopterin (APGA) | 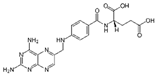 | Pediatric leukemia |
| Avanafil | 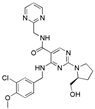 | Erectile dysfunction |
| Ceftriaxone |  | Bacterial infections, such as endocarditis, meningitis, pneumonia, skin infections, urinary tract infections |
| Methotrexate |  | Cancer, Autoimmune diseases, Ectopic pregnancies |
| Miltefosine | 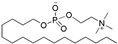 | Leishmaniasis, Breast cancer (topical treatment) |
| PGE-1 | 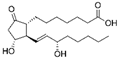 | Erectile dysfunction, Neonatal congenital heart defects |
| Raloxifene |  | Osteoporosis, Breast cancer prevention |
| Raltegravir | 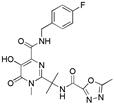 | HIV |
| Riociguat | 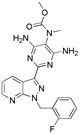 | Pulmonary hypertension |
| Valsartan | 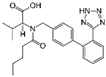 | Hypertension, Heart failure, Diabetes, Kidney disease |
| Drugs | Commercial Name | Dose | Features |
|---|---|---|---|
| Methotrexate | Trexall, Otrexup, Rasuvo, Xatmep, RediTrex, Jylamvo | Dosage depends on pathology | Methotrexate is a folate derivative that inhibits several enzymes responsible for nucleotide synthesis. It is used to treat inflammation caused by arthritis or to control cell division in neoplastic diseases. |
| Miltefosine | Impavido | 50 mg BID/TID | Miltefosine is a broad spectrum antimicrobial, anti-leishmanial, phospholipid drug developed in the 1980s as an anti-cancer agent. |
| Raltegravir | Isentress | 600 mg BID | Raltegravir is an antiretroviral agent used for the treatment of HIV infections. It is the first of a new class of HIV drugs. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criscuolo, E.; De Sciscio, M.L.; De Cristofaro, A.; Nicoara, C.; Maccarrone, M.; Fezza, F. Computational and Experimental Drug Repurposing of FDA-Approved Compounds Targeting the Cannabinoid Receptor CB1. Pharmaceuticals 2023, 16, 1678. https://doi.org/10.3390/ph16121678
Criscuolo E, De Sciscio ML, De Cristofaro A, Nicoara C, Maccarrone M, Fezza F. Computational and Experimental Drug Repurposing of FDA-Approved Compounds Targeting the Cannabinoid Receptor CB1. Pharmaceuticals. 2023; 16(12):1678. https://doi.org/10.3390/ph16121678
Chicago/Turabian StyleCriscuolo, Emanuele, Maria Laura De Sciscio, Angela De Cristofaro, Catalin Nicoara, Mauro Maccarrone, and Filomena Fezza. 2023. "Computational and Experimental Drug Repurposing of FDA-Approved Compounds Targeting the Cannabinoid Receptor CB1" Pharmaceuticals 16, no. 12: 1678. https://doi.org/10.3390/ph16121678
APA StyleCriscuolo, E., De Sciscio, M. L., De Cristofaro, A., Nicoara, C., Maccarrone, M., & Fezza, F. (2023). Computational and Experimental Drug Repurposing of FDA-Approved Compounds Targeting the Cannabinoid Receptor CB1. Pharmaceuticals, 16(12), 1678. https://doi.org/10.3390/ph16121678