
Discovery of Guanfacine as a Novel TAAR1 Agonist: A Combination Strategy through Molecular Modeling Studies and Biological Assays


 ,
, 
 , , ,
, , , 

Abstract
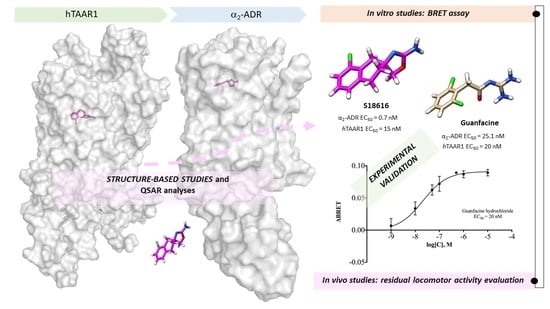
:
1. Introduction
2. Results and Discussion
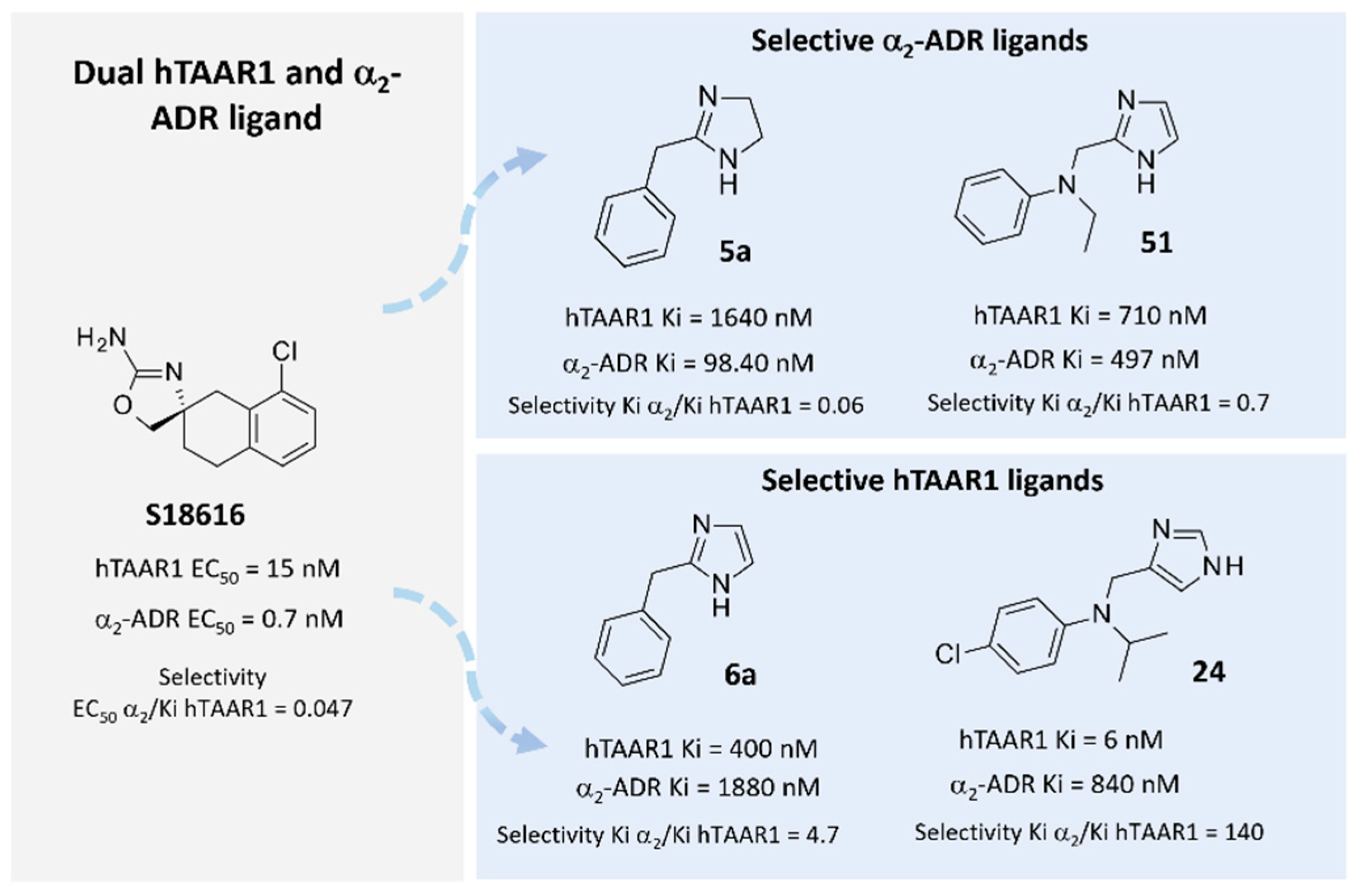
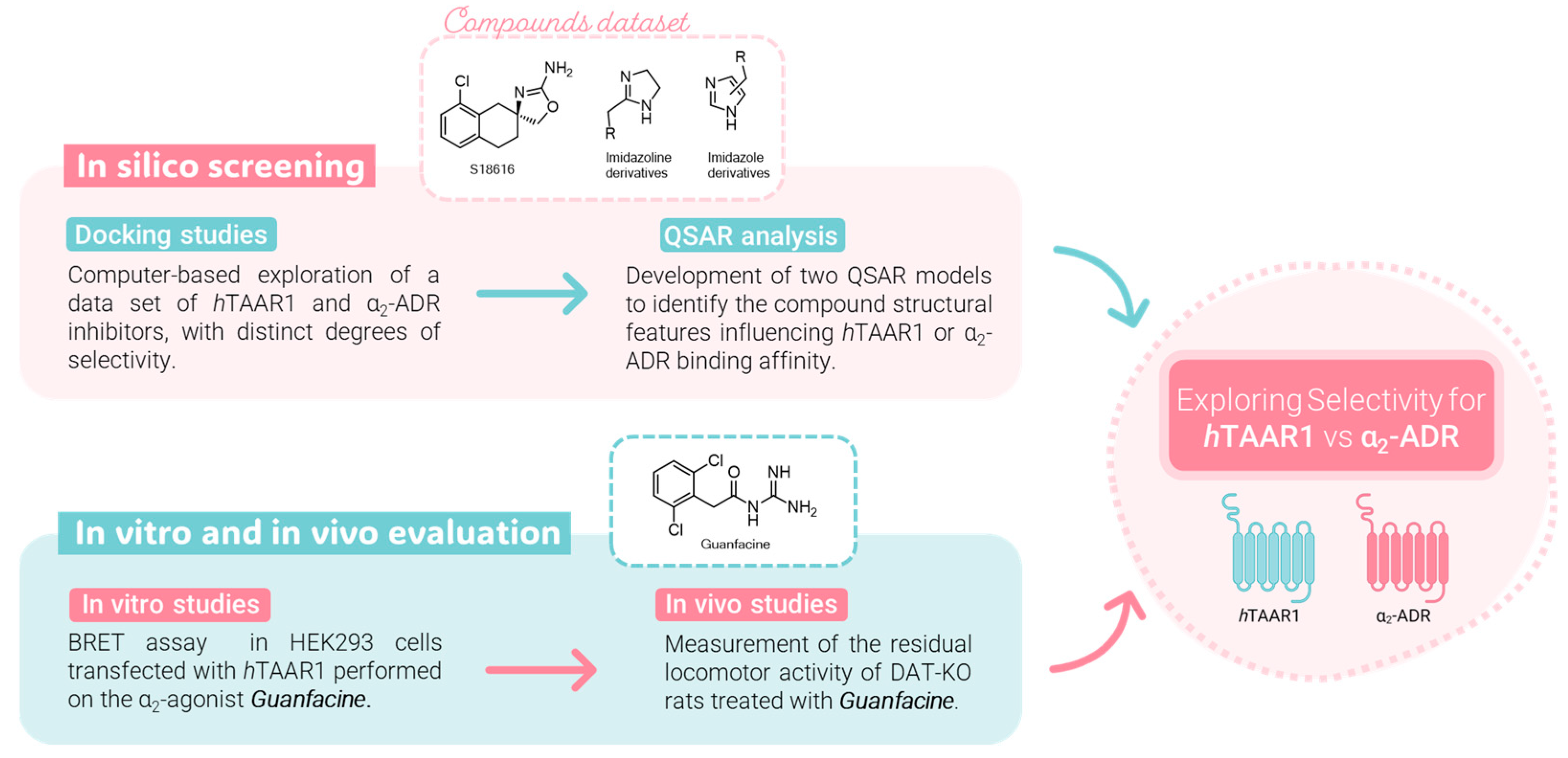
2.1. Probing In Silico S18616 as Dual TAAR1 and α2-ADR Ligands
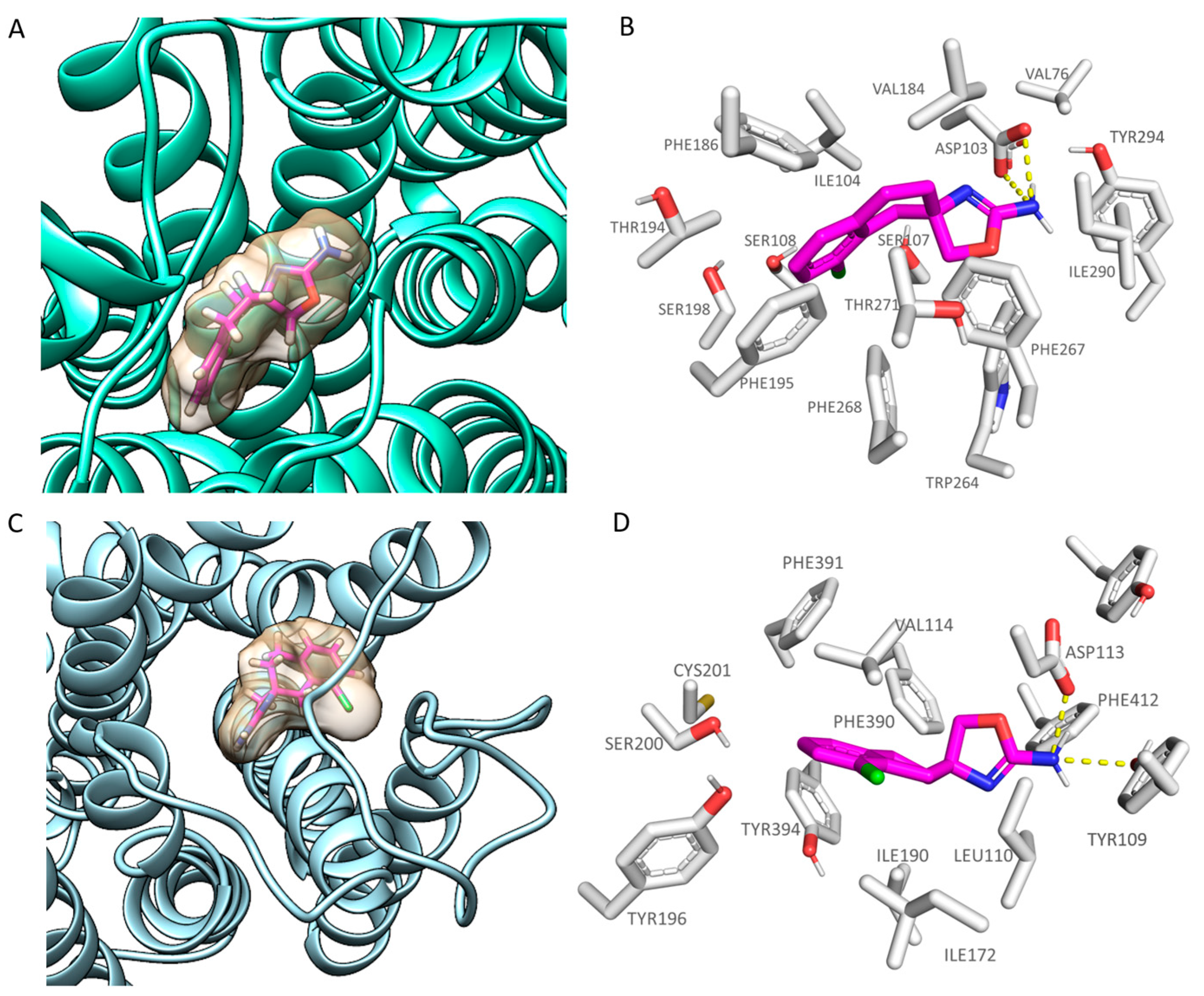
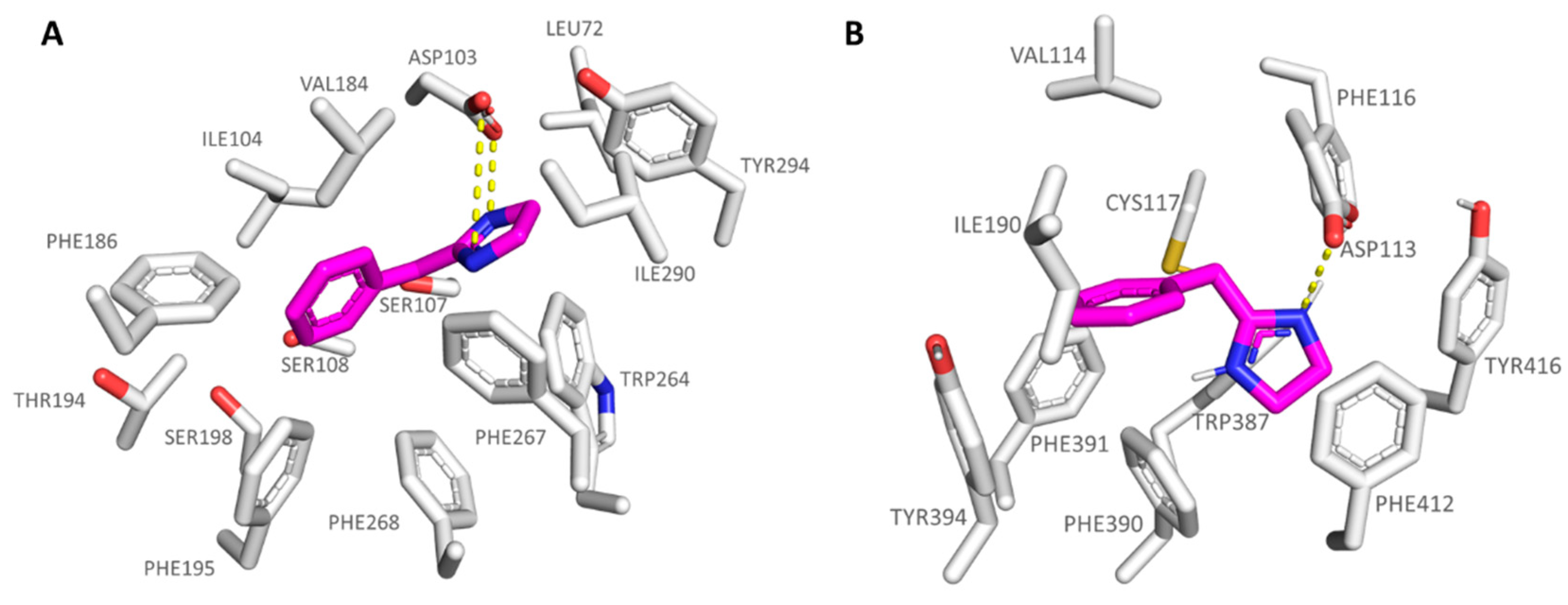
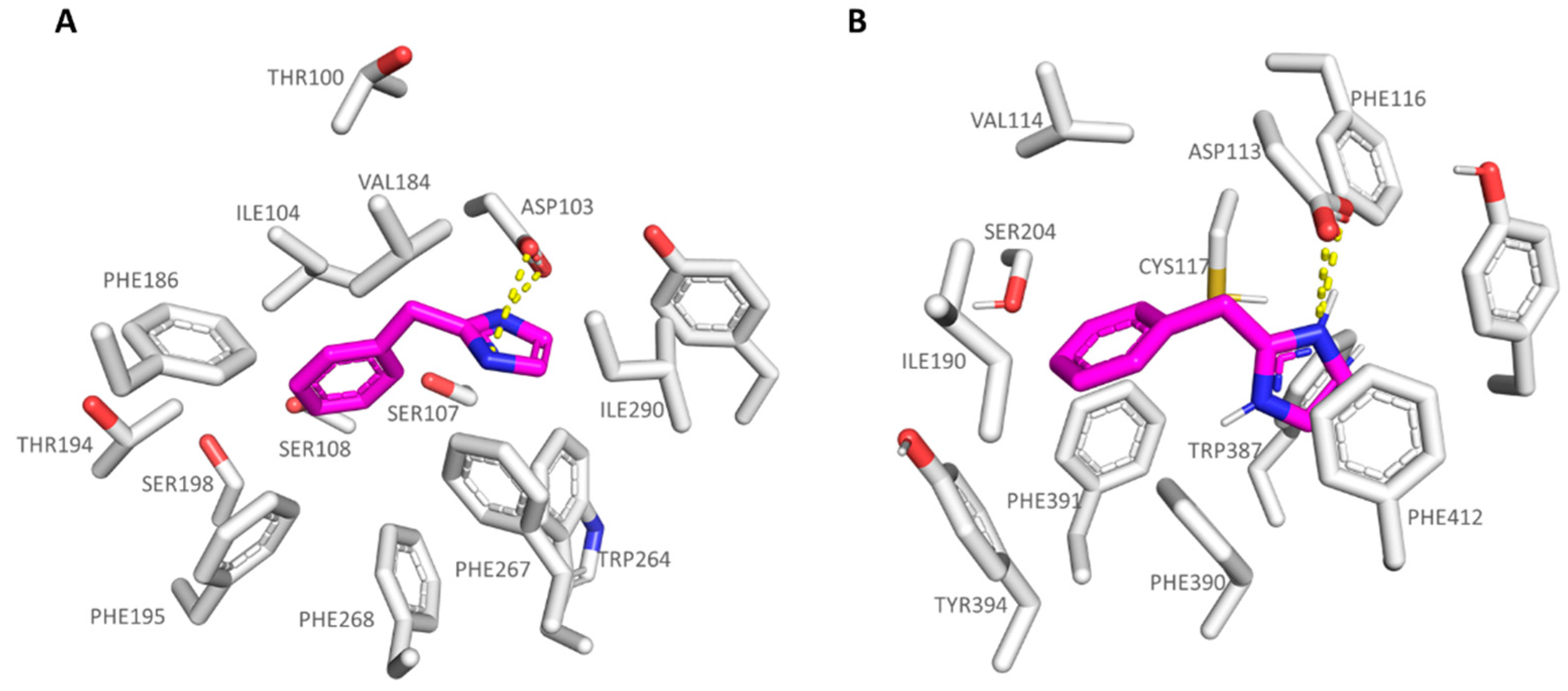
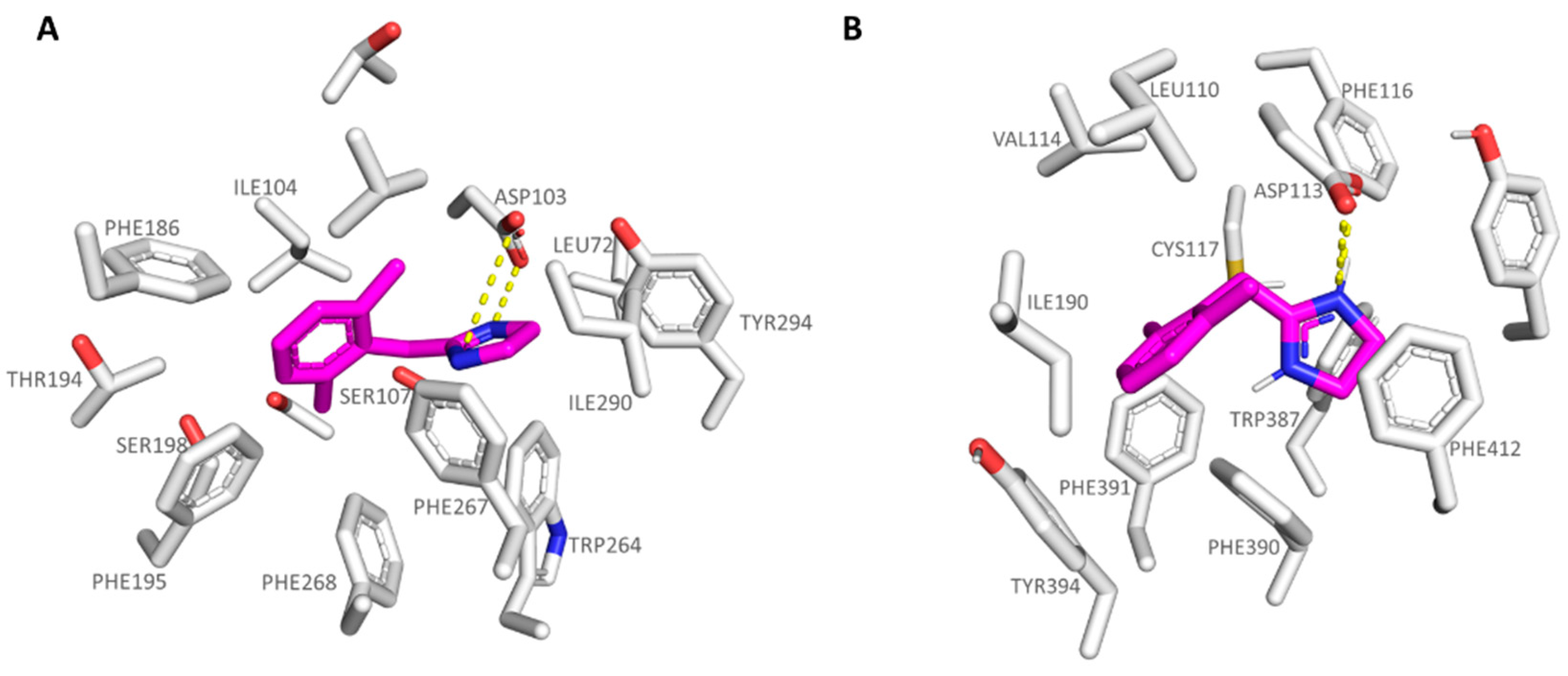
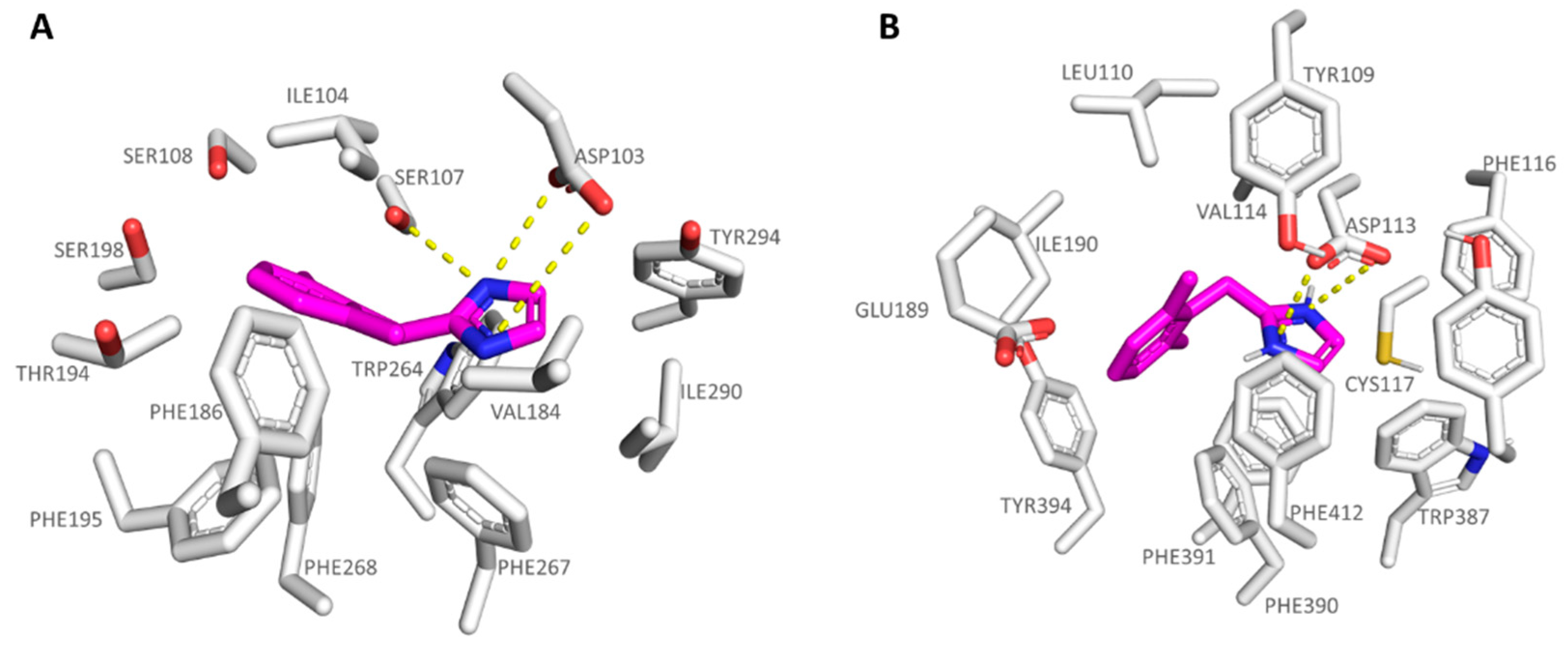
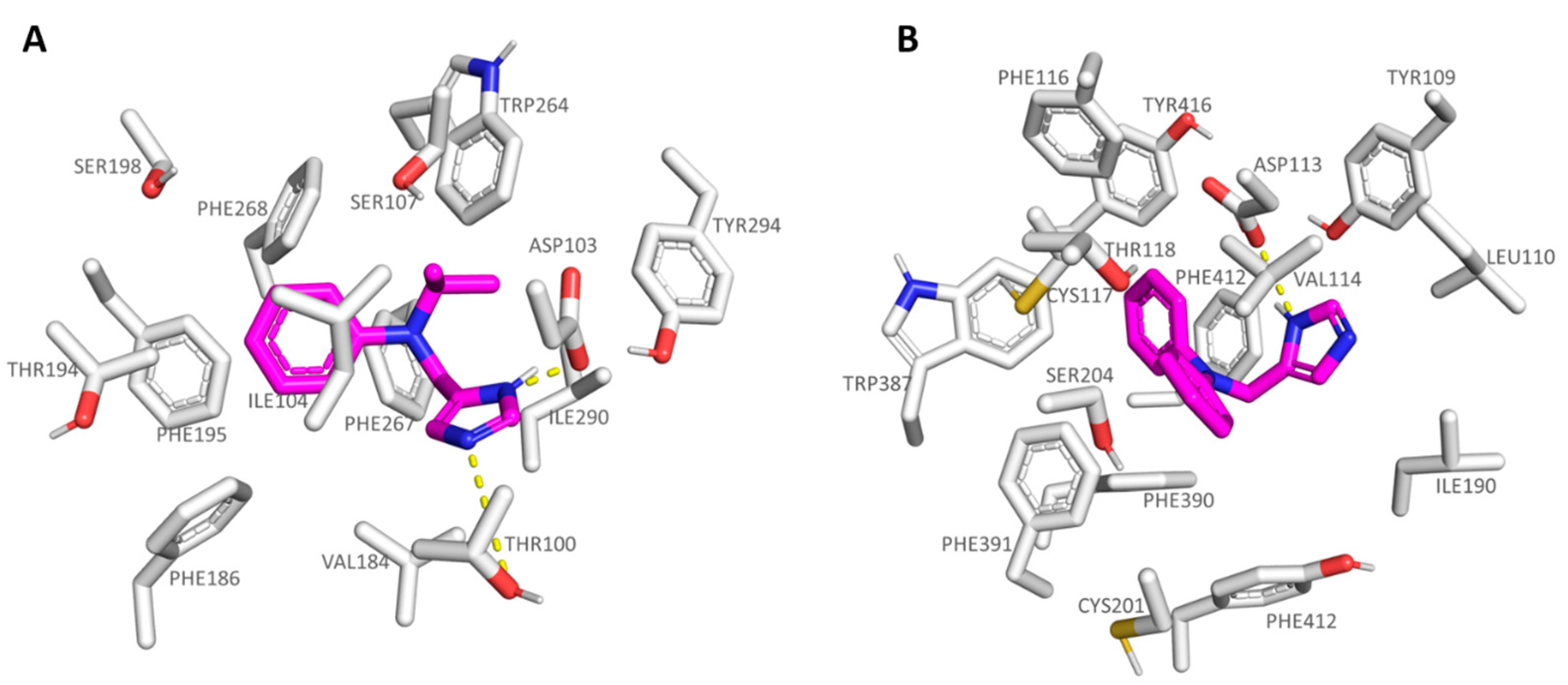
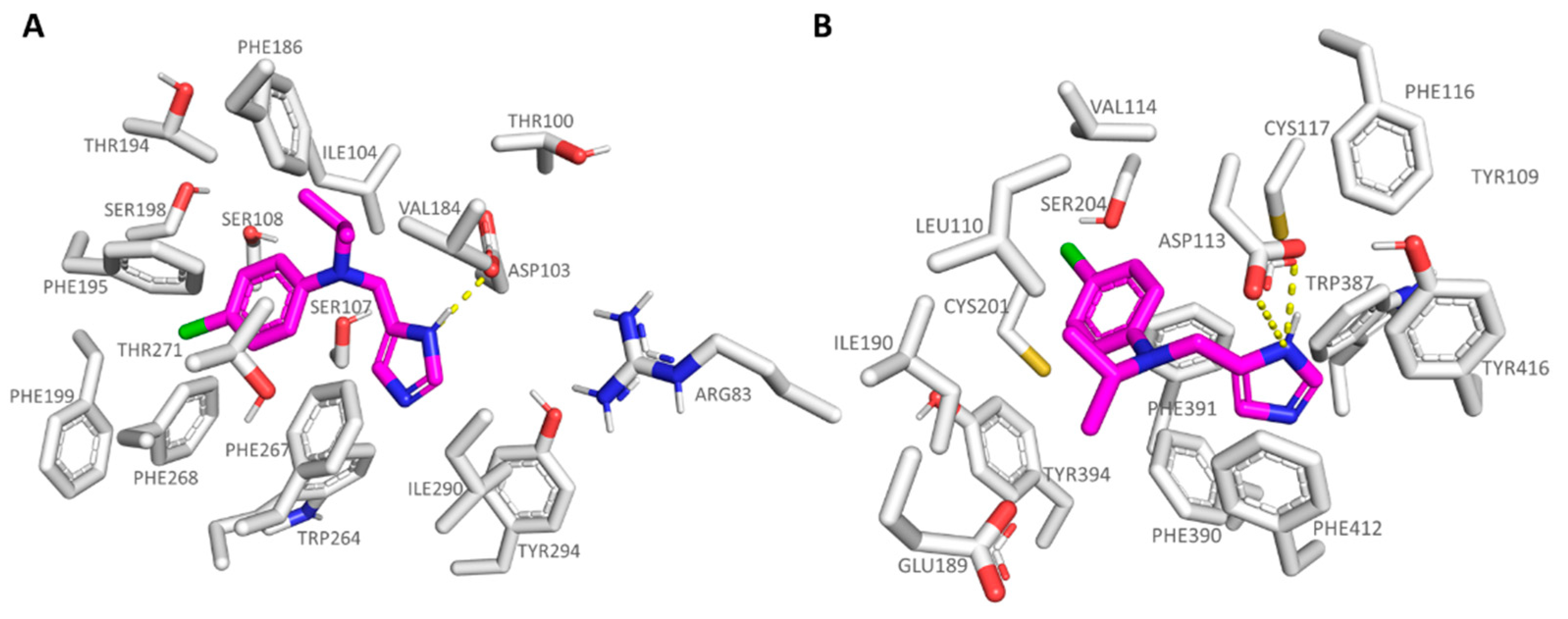
2.2. Comparative Molecular Docking of Dual Acting Compounds
2.3. QSAR Analyses and Pharmacophore Modeling
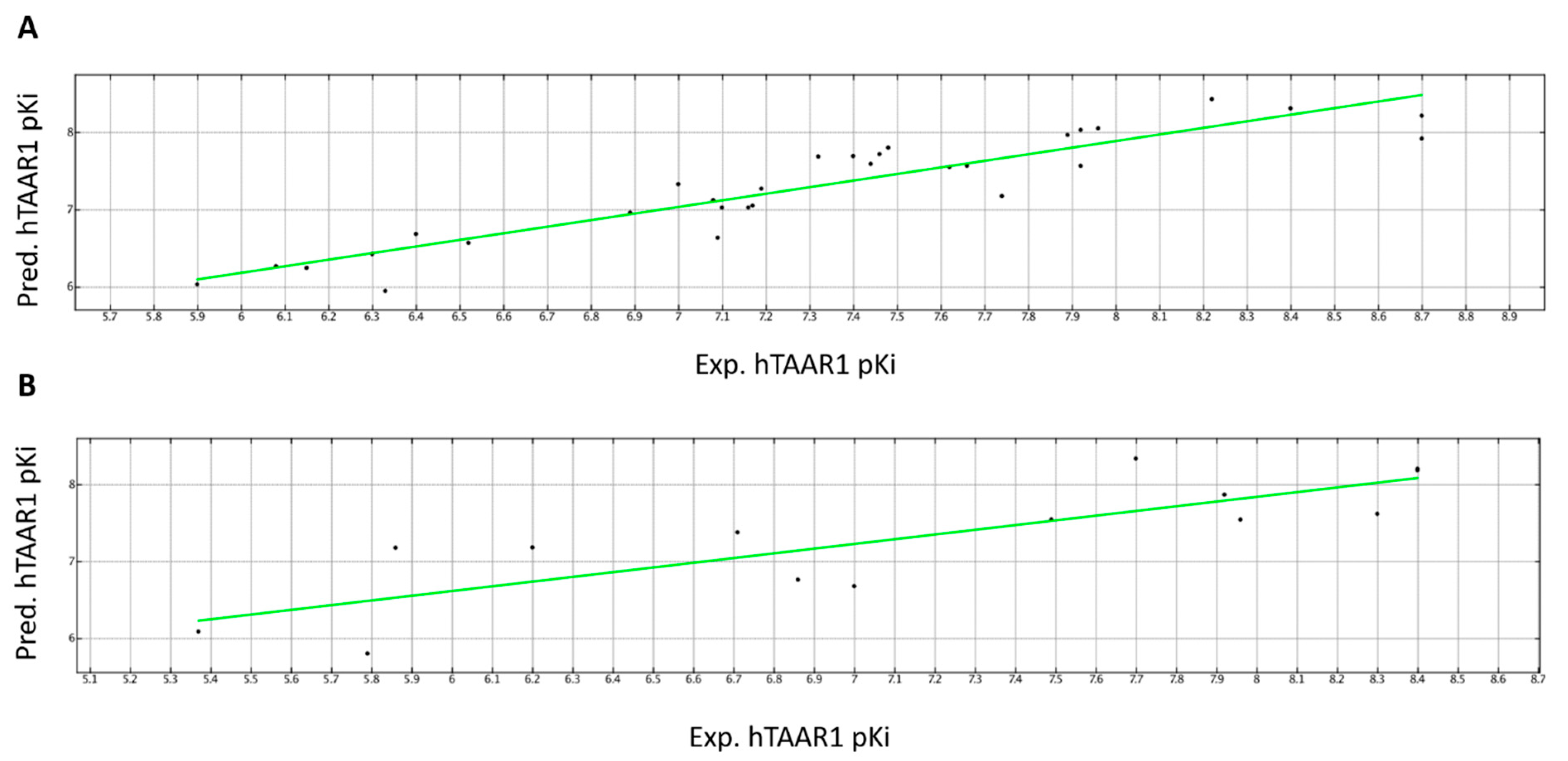
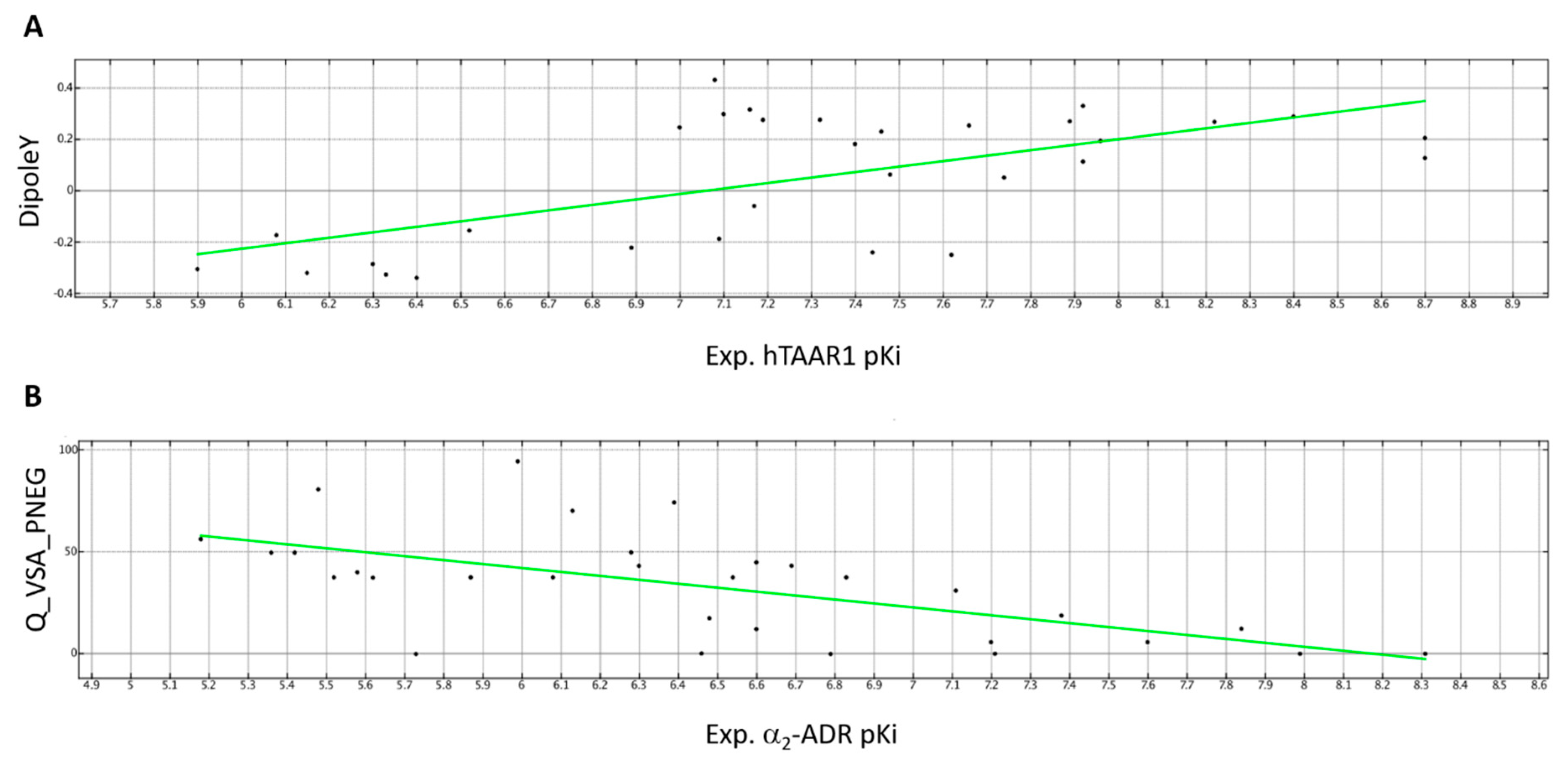
2.3.1. QSAR Model A—hTAAR1 Binding Affinity
2.3.2. QSAR Model B—α2-ADR Binding Affinity
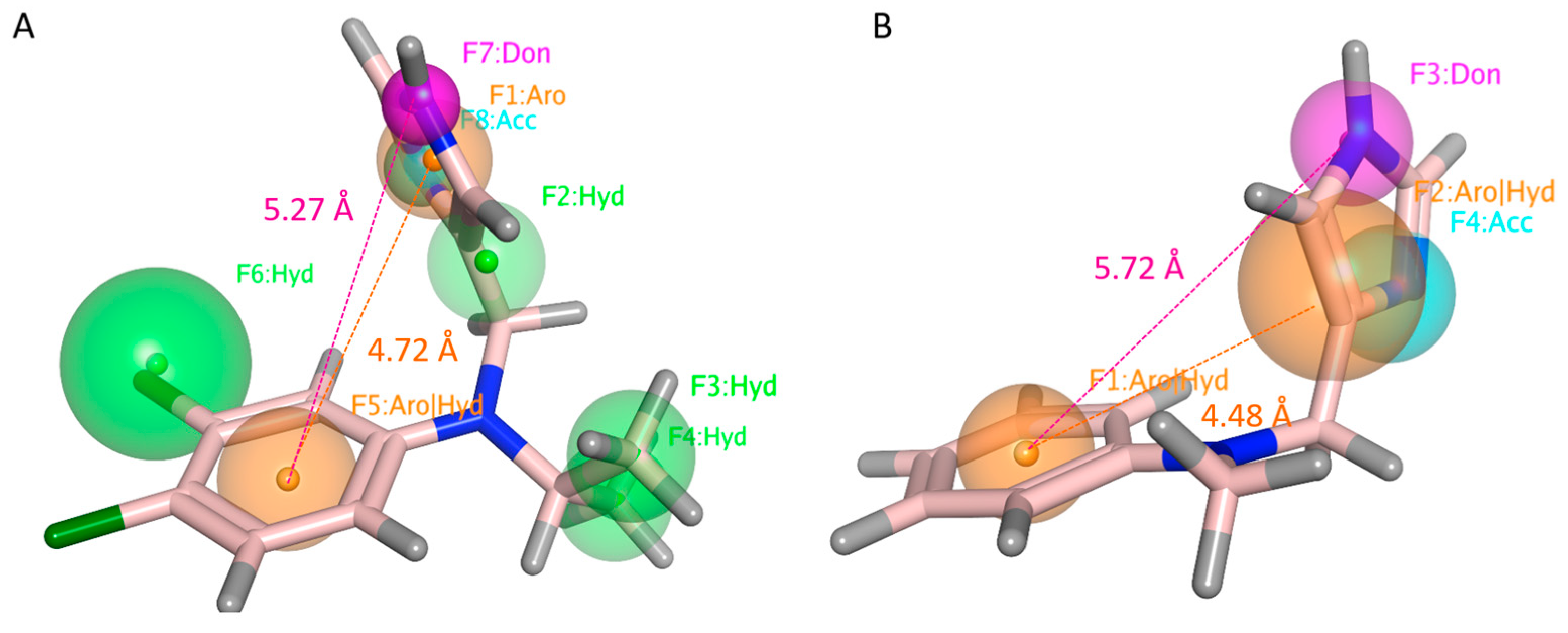
2.3.3. Pharmacophore Modeling
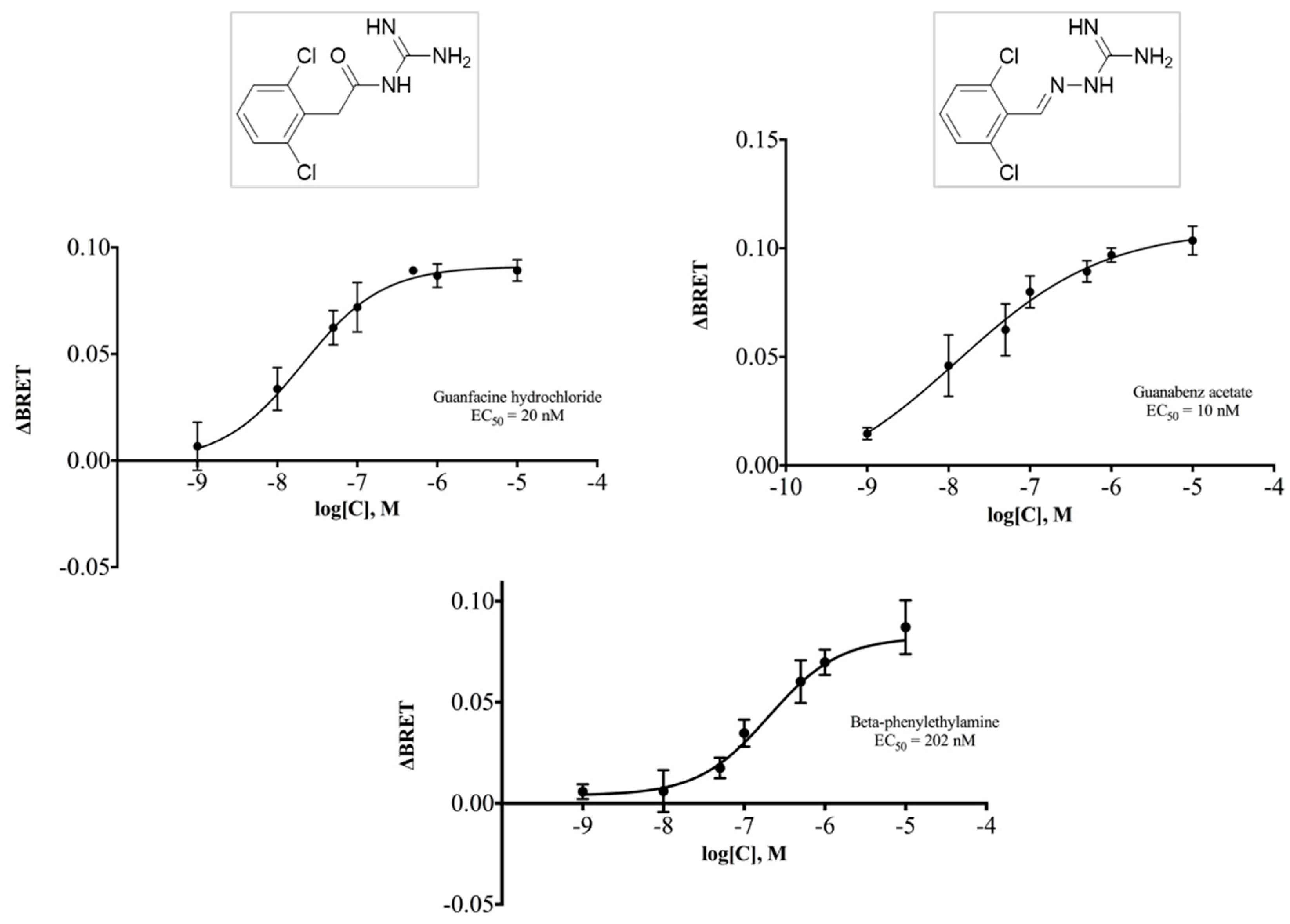
2.4. Guanfacine and Guanabenz Are TAAR1 Agonists
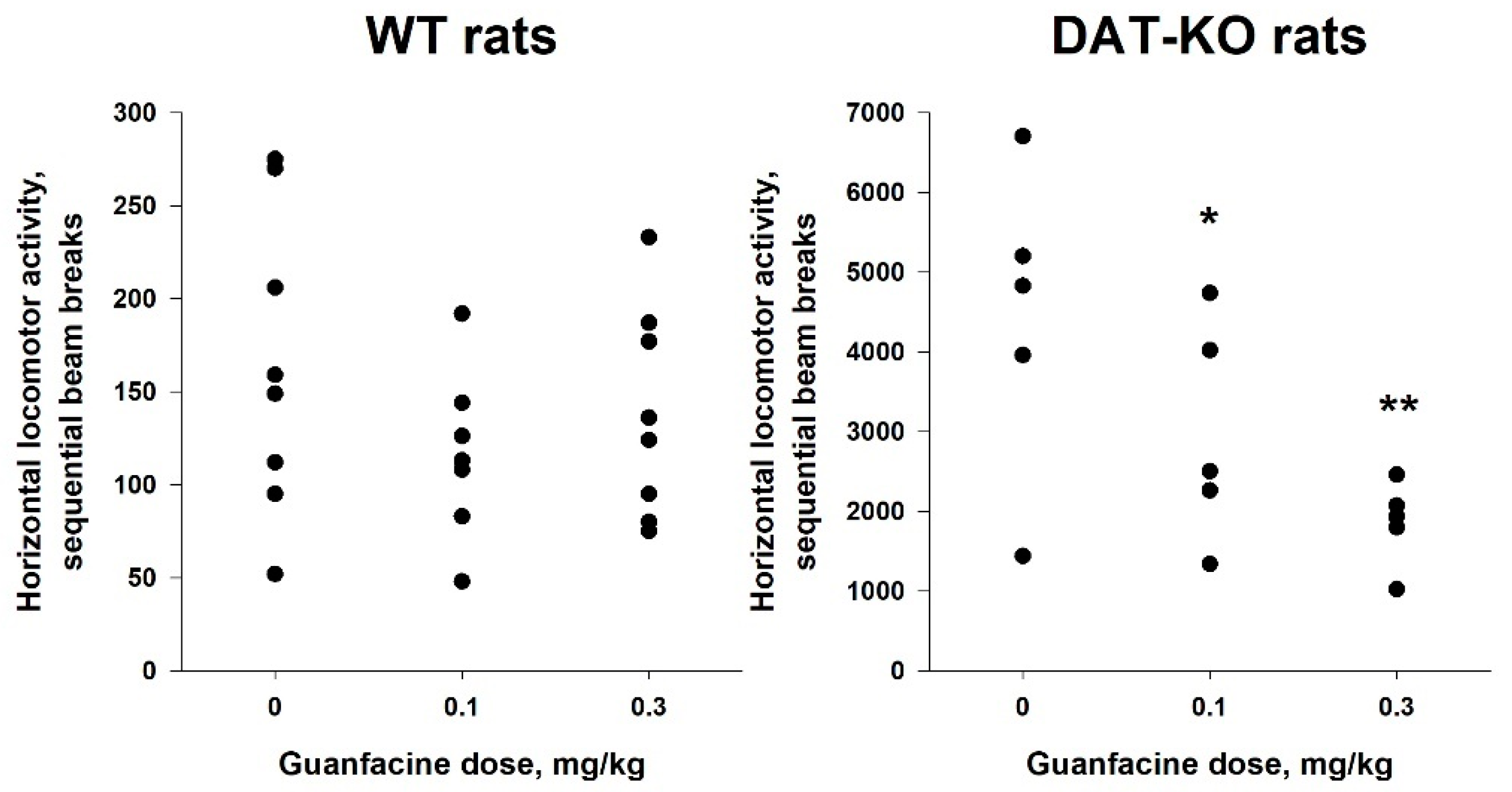
2.5. Administration of Guanfacine Resulted in Decrease in Locomotor Activity of DAT-KO Rats
3. Materials and Methods
3.1. Ligand and Protein Preparation
3.2. Molecular Docking Studies
3.3. QSAR Analyses
3.4. Reagents
3.5. Cell Culture and BRET Experiment
3.6. Subjects
3.7. Locomotor Activity Measurement
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rutigliano, G.; Accorroni, A.; Zucchi, R. The Case for TAAR1 as a Modulator of Central Nervous System Function. Front. Pharmacol. 2017, 8, 987. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.A.; Hoener, M.C.; Moore, C.S.; Berry, M.D. TAAR1 Regulates Purinergic-Induced TNF Secretion from Peripheral, But Not CNS-Resident, Macrophages. J. Neuroimmune Pharmacol. 2023, 18, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Halff, E.F.; Rutigliano, G.; Garcia-Hidalgo, A.; Howes, O.D. Trace Amine-Associated Receptor 1 (TAAR1) Agonism as a New Treatment Strategy for Schizophrenia and Related Disorders. Trends Neurosci. 2023, 46, 60–74. [Google Scholar] [CrossRef]
- Zhai, L.; Xiao, H.; Lin, C.; Wong, H.L.X.; Lam, Y.Y.; Gong, M.; Wu, G.; Ning, Z.; Huang, C.; Zhang, Y.; et al. Gut Microbiota-Derived Tryptamine and Phenethylamine Impair Insulin Sensitivity in Metabolic Syndrome and Irritable Bowel Syndrome. Nat. Commun. 2023, 14, 4986. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, Y.; Wang, Y.; Wang, Y.; He, X.; Xu, P.; Huang, S.; Yuan, Q.; Zhang, X.; Wang, L.; et al. Recognition of Methamphetamine and Other Amines by Trace Amine Receptor TAAR1. Nature 2023. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, J.-X. Potential of Ligands for Trace Amine-Associated Receptor 1 (TAAR1) in the Management of Substance Use Disorders. CNS Drugs 2021, 35, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Alnefeesi, Y.; Tamura, J.K.; Lui, L.M.W.; Jawad, M.Y.; Ceban, F.; Ling, S.; Nasri, F.; Rosenblat, J.D.; McIntyre, R.S. Trace Amine-Associated Receptor 1 (TAAR1): Potential Application in Mood Disorders: A Systematic Review. Neurosci. Biobehav. Rev. 2021, 131, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Raony, Í.; Domith, I.; Lourenco, M.V.; Paes-de-Carvalho, R.; Pandolfo, P. Trace Amine-Associated Receptor 1 Modulates Motor Hyperactivity, Cognition, and Anxiety-like Behavior in an Animal Model of ADHD. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 117, 110555. [Google Scholar] [CrossRef]
- Sakanoue, W.; Yokoyama, T.; Hirakawa, M.; Maesawa, S.; Sato, K.; Saino, T. 3-Iodothyronamine, a Trace Amine-Associated Receptor Agonist, Regulates Intracellular Ca2+ Increases via CaMK II through Epac2 in Rat Cerebral Arterioles. Biomed. Res. 2023, 44, 219–232. [Google Scholar] [CrossRef]
- Decker, A.M.; Brackeen, M.F.; Mohammadkhani, A.; Kormos, C.M.; Hesk, D.; Borgland, S.L.; Blough, B.E. Identification of a Potent Human Trace Amine-Associated Receptor 1 Antagonist. ACS Chem. Neurosci. 2022, 13, 1082–1095. [Google Scholar] [CrossRef]
- Bugda Gwilt, K.; González, D.P.; Olliffe, N.; Oller, H.; Hoffing, R.; Puzan, M.; El Aidy, S.; Miller, G.M. Actions of Trace Amines in the Brain-Gut-Microbiome Axis via Trace Amine-Associated Receptor-1 (TAAR1). Cell. Mol. Neurobiol. 2020, 40, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Nazimek, J.M.; Mckie, S.; Capitão, L.P.; Scaife, J.; Pal, D.; Browning, M.; Dawson, G.R.; Nishikawa, H.; Campbell, U.; et al. Effects of Ulotaront on Brain Circuits of Reward, Working Memory, and Emotion Processing in Healthy Volunteers with High or Low Schizotypy. Schizophrenia 2023, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Ågren, R.; Betari, N.; Saarinen, M.; Zeberg, H.; Svenningsson, P.; Sahlholm, K. In Vitro Comparison of Ulotaront (SEP-363856) and Ralmitaront (RO6889450): Two TAAR1 Agonist Candidate Antipsychotics. Int. J. Neuropsychopharmacol. 2023, 26, 599–606. [Google Scholar] [CrossRef]
- Tonelli, M.; Cichero, E. Trace Amine Associated Receptor 1 (TAAR1) Modulators: A Patent Review (2010-Present). Expert. Opin. Ther. Pat. 2020, 30, 137–145. [Google Scholar] [CrossRef]
- Frycz, B.A.; Nowicka, K.; Konopka, A.; Hoener, M.C.; Bulska, E.; Kaczmarek, L.; Stefaniuk, M. Activation of Trace Amine-Associated Receptor 1 (TAAR1) Transiently Reduces Alcohol Drinking in Socially Housed Mice. Addict. Biol. 2023, 28, e13285. [Google Scholar] [CrossRef] [PubMed]
- Grinchii, D.; Hoener, M.C.; Khoury, T.; Dekhtiarenko, R.; Nejati Bervanlou, R.; Jezova, D.; Dremencov, E. Effects of Acute and Chronic Administration of Trace Amine-Associated Receptor 1 (TAAR1) Ligands on in Vivo Excitability of Central Monoamine-Secreting Neurons in Rats. Mol. Psychiatry 2022, 27, 4861–4868. [Google Scholar] [CrossRef] [PubMed]
- Dorotenko, A.; Tur, M.; Dolgorukova, A.; Bortnikov, N.; Belozertseva, I.V.; Zvartau, E.E.; Gainetdinov, R.R.; Sukhanov, I. The Action of TAAR1 Agonist RO5263397 on Executive Functions in Rats. Cell. Mol. Neurobiol. 2020, 40, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, J.; Wang, K.; Huang, Y.; Zhang, Y.; Li, J.-X. Effects of a Trace Amine-Associated Receptor 1 Agonist RO 5263397 on Ethanol-Induced Behavioral Sensitization. Behav. Brain Res. 2020, 390, 112641. [Google Scholar] [CrossRef]
- Polini, B.; Ricardi, C.; Bertolini, A.; Carnicelli, V.; Rutigliano, G.; Saponaro, F.; Zucchi, R.; Chiellini, G. T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line. Int. J. Mol. Sci. 2023, 24, 11569. [Google Scholar] [CrossRef]
- Kane, J.M. A New Treatment Paradigm: Targeting Trace Amine-Associated Receptor 1 (TAAR1) in Schizophrenia. J. Clin. Psychopharmacol. 2022, 42, S1–S13. [Google Scholar] [CrossRef]
- Dedic, N.; Dworak, H.; Zeni, C.; Rutigliano, G.; Howes, O.D. Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 13185. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T. Trace Amine-Associated Receptor 1 as a Target for the Development of New Antipsychotics: Current Status of Research and Future Directions. CNS Drugs 2021, 35, 1153–1161. [Google Scholar] [CrossRef]
- Dedic, N.; Jones, P.G.; Hopkins, S.C.; Lew, R.; Shao, L.; Campbell, J.E.; Spear, K.L.; Large, T.H.; Campbell, U.C.; Hanania, T.; et al. SEP-363856, a Novel Psychotropic Agent with a Unique, Non-D2 Receptor Mechanism of Action. J. Pharmacol. Exp. Ther. 2019, 371, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.; Carvalho, A.F.; Puri, B.K.; Maes, M.; Bortolasci, C.C.; Morris, G.; Berk, M. Trace Amine-Associated Receptor 1 (TAAR1): A New Drug Target for Psychiatry? Neurosci. Biobehav. Rev. 2021, 120, 537–541. [Google Scholar] [CrossRef]
- Achtyes, E.D.; Hopkins, S.C.; Dedic, N.; Dworak, H.; Zeni, C.; Koblan, K. Ulotaront: Review of Preliminary Evidence for the Efficacy and Safety of a TAAR1 Agonist in Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2023, 273, 1543–1556. [Google Scholar] [CrossRef]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A Non-D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Højlund, M.; Correll, C.U. Ulotaront: A TAAR1/5-HT1A Agonist in Clinical Development for the Treatment of Schizophrenia. Expert Opin. Investig. Drugs 2022, 31, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.E.; Suchland, K.L.; Miyakawa, M.; Bunzow, J.R.; Grandy, D.K.; Scanlan, T.S. Trace Amine-Associated Receptor Agonists: Synthesis and Evaluation of Thyronamines and Related Analogues. J. Med. Chem. 2006, 49, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel Biguanide-Based Derivatives Scouted as TAAR1 Agonists: Synthesis, Biological Evaluation, ADME Prediction and Molecular Docking Studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational Design, Chemical Synthesis and Biological Evaluation of Novel Biguanides Exploring Species-Specificity Responsiveness of TAAR1 Agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef]
- Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2020, 13, 391. [Google Scholar] [CrossRef]
- Cichero, E.; Tonelli, M. Targeting Species-Specific Trace Amine-Associated Receptor 1 Ligands: To Date Perspective of the Rational Drug Design Process. Future Med. Chem. 2017, 9, 1507–1527. [Google Scholar] [CrossRef]
- Xu, Z.; Li, Q. TAAR Agonists. Cell. Mol. Neurobiol. 2020, 40, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J.; Dekeyne, A.; Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Milligan, G.; Duqueyroix, D.; Girardon, S.; Mullot, J.; Boutin, J.A.; et al. S18616, a Highly Potent, Spiroimidazoline Agonist at Alpha(2)-Adrenoceptors: I. Receptor Profile, Antinociceptive and Hypothermic Actions in Comparison with Dexmedetomidine and Clonidine. J. Pharmacol. Exp. Ther. 2000, 295, 1192–1205. [Google Scholar] [PubMed]
- Galley, G.; Stalder, H.; Goergler, A.; Hoener, M.C.; Norcross, R.D. Optimisation of Imidazole Compounds as Selective TAAR1 Agonists: Discovery of RO5073012. Bioorganic Med. Chem. Lett. 2012, 22, 5244–5248. [Google Scholar] [CrossRef]
- Galley, G.; Beurier, A.; Décoret, G.; Goergler, A.; Hutter, R.; Mohr, S.; Pähler, A.; Schmid, P.; Türck, D.; Unger, R.; et al. Discovery and Characterization of 2-Aminooxazolines as Highly Potent, Selective, and Orally Active TAAR1 Agonists. ACS Med. Chem. Lett. 2016, 7, 192–197. [Google Scholar] [CrossRef]
- Childress, A.; Hoo-Cardiel, A.; Lang, P. Evaluation of the Current Data on Guanfacine Extended Release for the Treatment of ADHD in Children and Adolescents. Expert Opin. Pharmacother. 2020, 21, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Yamamuro, K.; Okazaki, K.; Kishimoto, T. Evaluating Guanfacine Hydrochloride in the Treatment of Attention Deficit Hyperactivity Disorder (ADHD) in Adult Patients: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2021, 15, 1965–1969. [Google Scholar] [CrossRef]
- Galvez-Contreras, A.Y.; Vargas-de la Cruz, I.; Beltran-Navarro, B.; Gonzalez-Castaneda, R.E.; Gonzalez-Perez, O. Therapeutic Approaches for ADHD by Developmental Stage and Clinical Presentation. Int. J. Environ. Res. Public Health 2022, 19, 12880. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Martino, D. Guanfacine for the Treatment of Attention Deficit Hyperactivity Disorder in Children and Adolescents. Expert Rev. Neurother. 2015, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Black, B.T.; Soden, S.E.; Kearns, G.L.; Jones, B.L. Clinical and Pharmacologic Considerations for Guanfacine Use in Very Young Children. J. Child Adolesc. Psychopharmacol. 2016, 26, 498–504. [Google Scholar] [CrossRef]
- Alamo, C.; López-Muñoz, F.; Sánchez-García, J. Mechanism of Action of Guanfacine: A Postsynaptic Differential Approach to the Treatment of Attention Deficit Hyperactivity Disorder (Adhd). Actas Esp. Psiquiatr. 2016, 44, 107–112. [Google Scholar]
- Ramos, B.P.; Arnsten, A.F.T. Adrenergic Pharmacology and Cognition: Focus on the Prefrontal Cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Jin, L.E. Guanfacine for the Treatment of Cognitive Disorders: A Century of Discoveries at Yale. Yale J. Biol. Med. 2012, 85, 45–58, PMCID:PMC3313539. [Google Scholar] [PubMed]
- Arnsten, A.F.T. Guanfacine’s Mechanism of Action in Treating Prefrontal Cortical Disorders: Successful Translation across Species. Neurobiol. Learn. Mem. 2020, 176, 107327. [Google Scholar] [CrossRef]
- Fox, H.; Sofuoglu, M.; Sinha, R. Guanfacine Enhances Inhibitory Control and Attentional Shifting in Early Abstinent Cocaine-Dependent Individuals. J. Psychopharmacol. 2015, 29, 312–323. [Google Scholar] [CrossRef]
- Franowicz, J.S.; Kessler, L.E.; Borja, C.M.D.; Kobilka, B.K.; Limbird, L.E.; Arnsten, A.F.T. Mutation of the alpha2A-Adrenoceptor Impairs Working Memory Performance and Annuls Cognitive Enhancement by Guanfacine. J. Neurosci. 2002, 22, 8771–8777. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Qu, L.; Zhou, Q.T.; Wu, D.; Zhao, S.W. Crystal Structure of the alpha2A Adrenergic Receptor in Complex with a Partial Agonist. Available online: https://www.rcsb.org/structure/6KUY (accessed on 4 December 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Version 2.5.2. Schrödinger, LLC: New York, NY, USA, 2020.
- Wang, Y.-L.; Li, J.-Y.; Shi, X.-X.; Wang, Z.; Hao, G.-F.; Yang, G.-F. Web-Based Quantitative Structure—Activity Relationship Resources Facilitate Effective Drug Discovery. Top. Curr. Chem. 2021, 379, 37. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Righetti, G.; Casale, M.; Liessi, N.; Tasso, B.; Salis, A.; Tonelli, M.; Millo, E.; Pedemonte, N.; Fossa, P.; Cichero, E. Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors. Int. J. Mol. Sci. 2020, 21, 8084. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Forina, M.; Boggia, R.; Mosti, L.; Fossa, P. Zupan’s Descriptors in QSAR Applied to the Study of a New Class of Cardiotonic Agents. Farmaco 1997, 52, 411–419. [Google Scholar] [PubMed]
- Stanton, D.T.; Jurs, P.C. Development and Use of Charged Partial Surface Area Structural Descriptors in Computer-Assisted Quantitative Structure-Property Relationship Studies. Anal. Chem. 1990, 62, 2323–2329. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Balaban, A.T. Highly Discriminating Distance-Based Topological Index. Chem. Phys. Lett. 1982, 89, 399–404. [Google Scholar] [CrossRef]
- Moro, S.; Bacilieri, M.; Cacciari, B.; Bolcato, C.; Cusan, C.; Pastorin, G.; Klotz, K.-N.; Spalluto, G. The Application of a 3D-QSAR (autoMEP/PLS) Approach as an Efficient Pharmacodynamic-Driven Filtering Method for Small-Sized Virtual Library: Application to a Lead Optimization of a Human A3 Adenosine Receptor Antagonist. Bioorganic Med. Chem. 2006, 14, 4923–4932. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Knutelska, J.; Bednarski, M.; Nowiński, L.; Zygmunt, M.; Mordyl, B.; Głuch-Lutwin, M.; Kazek, G.; Sapa, J.; Pytka, K. A Comparison of the Anorectic Effect and Safety of the Alpha2-Adrenoceptor Ligands Guanfacine and Yohimbine in Rats with Diet-Induced Obesity. PLoS ONE 2015, 10, e0141327. [Google Scholar] [CrossRef]
- Kotańska, M.; Marcinkowska, M.; Kuder, K.J.; Walczak, M.; Bednarski, M.; Siwek, A.; Kołaczkowski, M. Metabolic and Cardiovascular Benefits and Risks of 4-Hydroxy Guanabenz Hydrochloride: A2-Adrenoceptor and Trace Amine-Associated Receptor 1 Ligand. Pharmacol. Rep. 2023, 75, 1211–1229. [Google Scholar] [CrossRef]
- Lam, V.M.; Rodríguez, D.; Zhang, T.; Koh, E.J.; Carlsson, J.; Salahpour, A. Discovery of Trace Amine-Associated Receptor 1 Ligands by Molecular Docking Screening against a Homology Model. MedChemComm 2015, 6, 2216–2223. [Google Scholar] [CrossRef]
- Espinoza, S.; Masri, B.; Salahpour, A.; Gainetdinov, R.R. BRET Approaches to Characterize Dopamine and TAAR1 Receptor Pharmacology and Signaling. Methods Mol. Biol. 2013, 964, 107–122. [Google Scholar] [CrossRef]
- Hu, L.A.; Zhou, T.; Ahn, J.; Wang, S.; Zhou, J.; Hu, Y.; Liu, Q. Human and Mouse Trace Amine-Associated Receptor 1 Have Distinct Pharmacology towards Endogenous Monoamines and Imidazoline Receptor Ligands. Biochem. J. 2009, 424, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Iwasaki, Y.; Matsumoto, S.; Satoh, T.; Ozawa, A.; Yamada, E.; Kakizaki, S.; Trejo, J.A.O.; Uchiyama, Y.; Yamada, M.; et al. Administration of Small-Molecule Guanabenz Acetate Attenuates Fatty Liver and Hyperglycemia Associated with Obesity. Sci. Rep. 2020, 10, 13671. [Google Scholar] [CrossRef]
- Kotańska, M.; Knutelska, J.; Nicosia, N.; Mika, K.; Szafarz, M. Guanabenz-an Old Drug with a Potential to Decrease Obesity. Naunyn-Schmiedeberg–s Arch. Pharmacol. 2022, 395, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Michael, E.S.; Covic, L.; Kuliopulos, A. Trace Amine-Associated Receptor 1 (TAAR1) Promotes Anti-Diabetic Signaling in Insulin-Secreting Cells. J. Biol. Chem. 2019, 294, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Künnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like Effects of Small Molecule Trace Amine-Associated Receptor 1 Agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef]
- Leo, D.; Sukhanov, I.; Zoratto, F.; Illiano, P.; Caffino, L.; Sanna, F.; Messa, G.; Emanuele, M.; Esposito, A.; Dorofeikova, M.; et al. Pronounced Hyperactivity, Cognitive Dysfunctions, and BDNF Dysregulation in Dopamine Transporter Knock-out Rats. J. Neurosci. 2018, 38, 1959–1972. [Google Scholar] [CrossRef]
- Savchenko, A.; Targa, G.; Fesenko, Z.; Leo, D.; Gainetdinov, R.R.; Sukhanov, I. Dopamine Transporter Deficient Rodents: Perspectives and Limitations for Neuroscience. Biomolecules 2023, 13, 806. [Google Scholar] [CrossRef]
- Kurzina, N.; Belskaya, A.; Gromova, A.; Ignashchenkova, A.; Gainetdinov, R.R.; Volnova, A. Modulation of Spatial Memory Deficit and Hyperactivity in Dopamine Transporter Knockout Rats via α2A-Adrenoceptors. Front. Psychiatry 2022, 13, 851296. [Google Scholar] [CrossRef]
- V-PARVUS 2010. An Extendable Package of Programs for Explorative Data Analysis, Classification and Regression Analysis. Dip Chimica e Tecnologie Farmaceutiche, University of Genova. Available online: https://iris.unige.it/handle/11567/242687 (accessed on 23 September 2023).
- Boggia, R.; Forina, M.; Fossa, P.; Mosti, L. Chemometric Study and Validation Strategies in the Structure-Activity Relationships of New Cardiotonic Agents. Quant. Struct. Act. Relat. 1997, 16, 201–213. [Google Scholar] [CrossRef]
- Cruciani, G.; Crivori, P.; Carrupt, P.-A.; Testa, B. Molecular Fields in Quantitative Structure–Permeation Relationships: The VolSurf Approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Petitjean, M. Applications of the Radius-Diameter Diagram to the Classification of Topological and Geometrical Shapes of Chemical Compounds. J. Chem. Inf. Comput. Sci. 1992, 32, 331–337. [Google Scholar] [CrossRef]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-Pharmacophore Superpositioning and Pattern Matching in Computational Drug Design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Khalid, S.; Hanif, R.; Jabeen, I.; Mansoor, Q.; Ismail, M. Pharmacophore Modeling for Identification of Anti-IGF-1R Drugs and in-Vitro Validation of Fulvestrant as a Potential Inhibitor. PLoS ONE 2018, 13, e0196312. [Google Scholar] [CrossRef]
- Haidar, S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of Pharmacophore Model for Indeno[1,2-b]Indoles as Human Protein Kinase CK2 Inhibitors and Database Mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef]
- Sukhanov, I.; Dorofeikova, M.; Dolgorukova, A.; Dorotenko, A.; Gainetdinov, R.R. Trace Amine-Associated Receptor 1 Modulates the Locomotor and Sensitization Effects of Nicotine. Front. Pharmacol. 2018, 9, 329. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Corresponding Residues | ||||
|---|---|---|---|---|---|
| 6KUY | Asp113 | Val114 | Cys117 | Ile190 | Ser204 |
| hTAAR1 | Asp103 | Ile104 | Ser107 | Val184 | Ser198 |
| 6KUY | Trp387 | Phe390 | Phe391 | Phe412 | Tyr416 |
| hTAAR1 | Trp264 | Phe267 | Phe268 | Ile290 | Tyr294 |
| Molecular Descriptor Code | Descriptor Class | Description | Descriptor Type | Relative Importance (RI) |
|---|---|---|---|---|
| DipoleY | 3D-V | The y component of the dipole moment (external coordinates). | Conformation Dependent Charge Descriptors | 1.00000 |
| SlogP_VSA4 | 2D-II | Sum of vi such that Li is in (0.1, 0.15]. | Subdivided Surface Areas | 0.810513 |
| DCASA | 3D-V | Absolute value of the difference between CASA+ (Positive charge weighted surface area, ASA+ times max {qi > 0}) and CASA- (Negative charge weighted surface area, ASA- times max {qi < 0}). [58] | Conformation Dependent Charge Descriptors | 0.582248 |
| E | 3D-I | Value of the potential energy. The state of all term enable flags will be honored (in addition to the term weights). This means that the current potential setup accurately reflects what will be calculated. | Potential Energy Descriptors | 0.435867 |
| vsurf_IW5 | 3D-III | Hydrophilic integy moment (8 descriptors) | Surface Area, Volume and Shape Descriptors | 0.344589 |
| DipoleZ | 3D-V | The z component of the dipole moment (external coordinates). | Conformation Dependent Charge Descriptors | 0.325382 |
| Q_VSA_FHYD | 2D-V | Fractional hydrophobic van der Waals surface area. This is the sum of the vi such that |qi| is less than or equal to 0.2 divided by the total surface area. The vi are calculated using a connection table approximation. | Partial Charge Descriptors | 0.31853 |
| vsurf_EDmin1 | 3D-III | Lowest hydrophobic energy (3 descriptors) | Surface Area, Volume and Shape Descriptors | 0.29631 |
| Q_RPC- | 2D-V | Relative negative partial charge: the smallest negative qi divided by the sum of the negative qi. Q_RPC- is identical to RPC- which has been retained for compatibility. | Partial Charge Descriptors | 0.061043 |
| GCUT_SMR_0 | 2D-VII | The GCUT descriptors using atomic contribution to molar refractivity (using the Wildman and Crippen SMR method) [59] instead of partial charge. | Adjacency and Distance Matrix Descriptors | 0.027644 |
| Molecular Descriptor Code | Descriptor Class | Description | Descriptor Type | Relative Importance (RI) |
|---|---|---|---|---|
| Q_VSA_PNEG | 2D-V | Fractional negative van der Waals surface area. This is the sum of the vi such that qi is negative divided by the total surface area. The vi are calculated using a connection table approximation. | Partial Charge Descriptors | 1.000000 |
| Q_VSA_POL | 2D-V | Total positive van der Waals surface area. This is the sum of the vi such that qi is non-negative. The vi are calculated using a connection table approximation. | Partial Charge Descriptors | 0.795835 |
| SlogP_VSA3 | 2D-II | Sum of vi such that Li is in (0,0.1]. | Subdivided Surface Areas | 0.195358 |
| vsurf_ID7 | 3D-III | Hydrophobic integy moment (8 descriptors) | Surface Area, Volume and Shape Descriptors | 0.156047 |
| vsa_other | 2D-VI | Approximation to the sum of VDW surface areas (Å2) of atoms typed as “other”. | Pharmacophore Feature Descriptors | 0.102077 |
| vsurf_IW4 | 3D-III | Hydrophilic integy moment (8 descriptors) | Surface Area, Volume and Shape Descriptors | 0.1005 |
| E_tor | 3D-I | Torsion (proper and improper) potential energy. In the Potential Setup panel, the term enable (Bonded) flag is ignored, but the term weight is applied. | Potential Energy Descriptors | 0.098422 |
| balabanJ | 2D-VII | Balaban’s connectivity topological index [60]. | Adjacency and Distance Matrix Descriptors | 0.085904 |
| vsurf_ID1 | 3D-III | Hydrophobic integy moment (8 descriptors) | Surface Area, Volume and Shape Descriptors | 0.022599 |
| GCUT_SMR_1 | 2D-VII | The GCUT descriptors using atomic contribution to molar refractivity (using the Wildman and Crippen SMR method) instead of partial charge. | Adjacency and Distance Matrix Descriptors | 0.005108 |
| Q_VSA_FHYD | 2D-V | Fractional hydrophobic van der Waals surface area. This is the sum of the vi such that |qi| is less than or equal to 0.2 divided by the total surface area. The vi are calculated using a connection table approximation. | Partial Charge Descriptors | 0.001779 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cichero, E.; Francesconi, V.; Casini, B.; Casale, M.; Kanov, E.; Gerasimov, A.S.; Sukhanov, I.; Savchenko, A.; Espinoza, S.; Gainetdinov, R.R.; et al. Discovery of Guanfacine as a Novel TAAR1 Agonist: A Combination Strategy through Molecular Modeling Studies and Biological Assays. Pharmaceuticals 2023, 16, 1632. https://doi.org/10.3390/ph16111632
Cichero E, Francesconi V, Casini B, Casale M, Kanov E, Gerasimov AS, Sukhanov I, Savchenko A, Espinoza S, Gainetdinov RR, et al. Discovery of Guanfacine as a Novel TAAR1 Agonist: A Combination Strategy through Molecular Modeling Studies and Biological Assays. Pharmaceuticals. 2023; 16(11):1632. https://doi.org/10.3390/ph16111632
Chicago/Turabian StyleCichero, Elena, Valeria Francesconi, Beatrice Casini, Monica Casale, Evgeny Kanov, Andrey S. Gerasimov, Ilya Sukhanov, Artem Savchenko, Stefano Espinoza, Raul R. Gainetdinov, and et al. 2023. "Discovery of Guanfacine as a Novel TAAR1 Agonist: A Combination Strategy through Molecular Modeling Studies and Biological Assays" Pharmaceuticals 16, no. 11: 1632. https://doi.org/10.3390/ph16111632
APA StyleCichero, E., Francesconi, V., Casini, B., Casale, M., Kanov, E., Gerasimov, A. S., Sukhanov, I., Savchenko, A., Espinoza, S., Gainetdinov, R. R., & Tonelli, M. (2023). Discovery of Guanfacine as a Novel TAAR1 Agonist: A Combination Strategy through Molecular Modeling Studies and Biological Assays. Pharmaceuticals, 16(11), 1632. https://doi.org/10.3390/ph16111632