
Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma
 , ,
, ,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
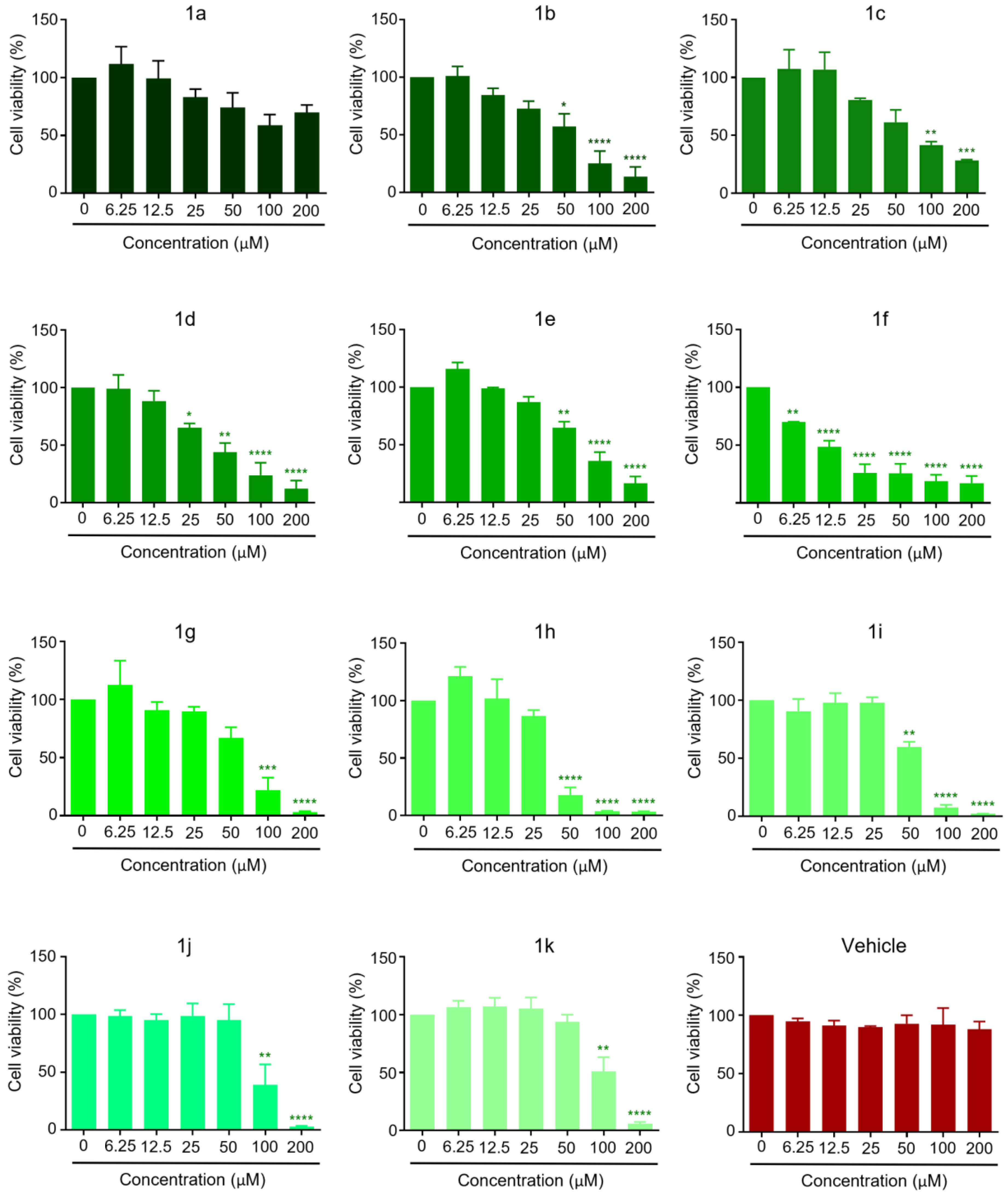
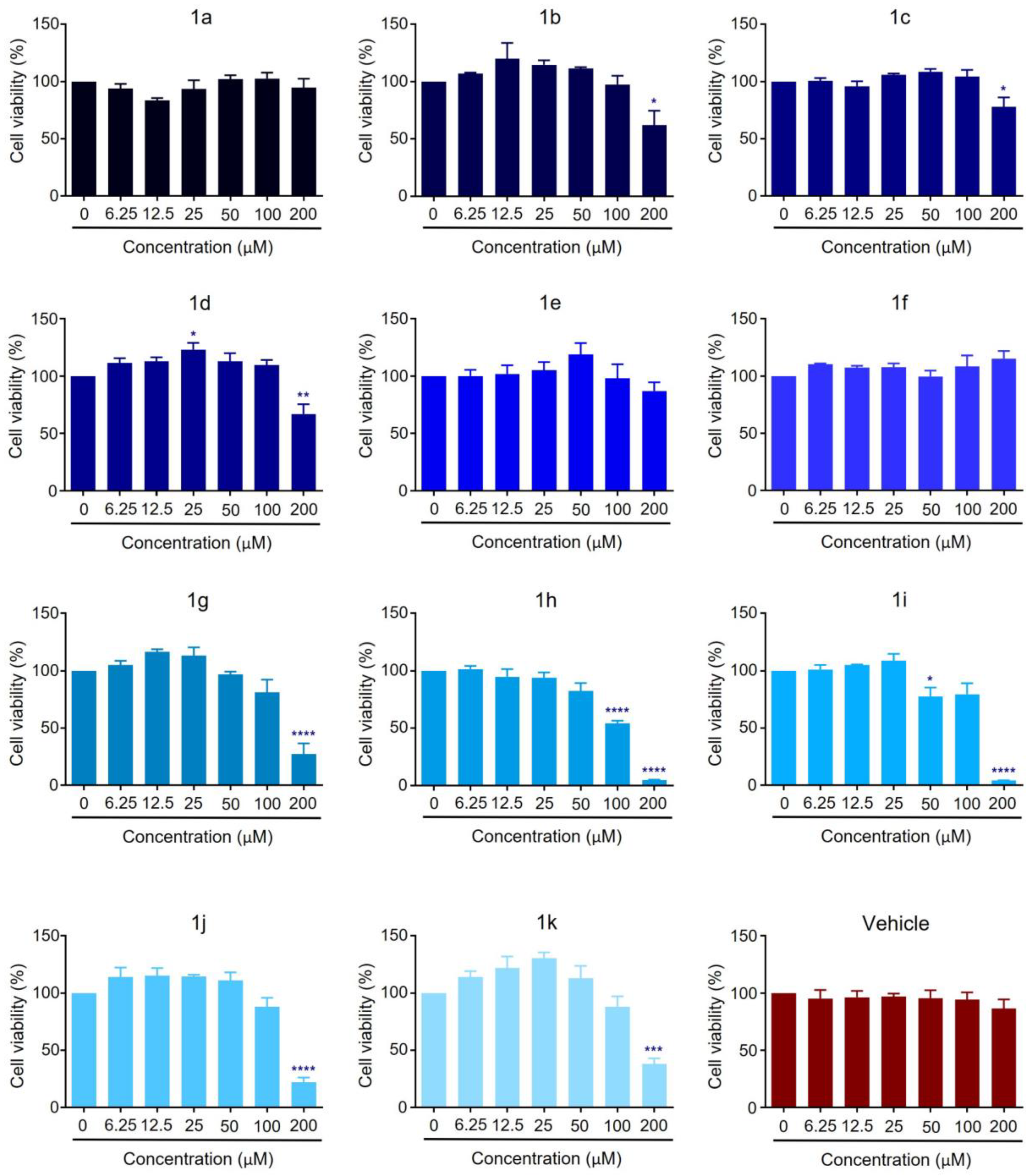
2.2. Anticancer Activity
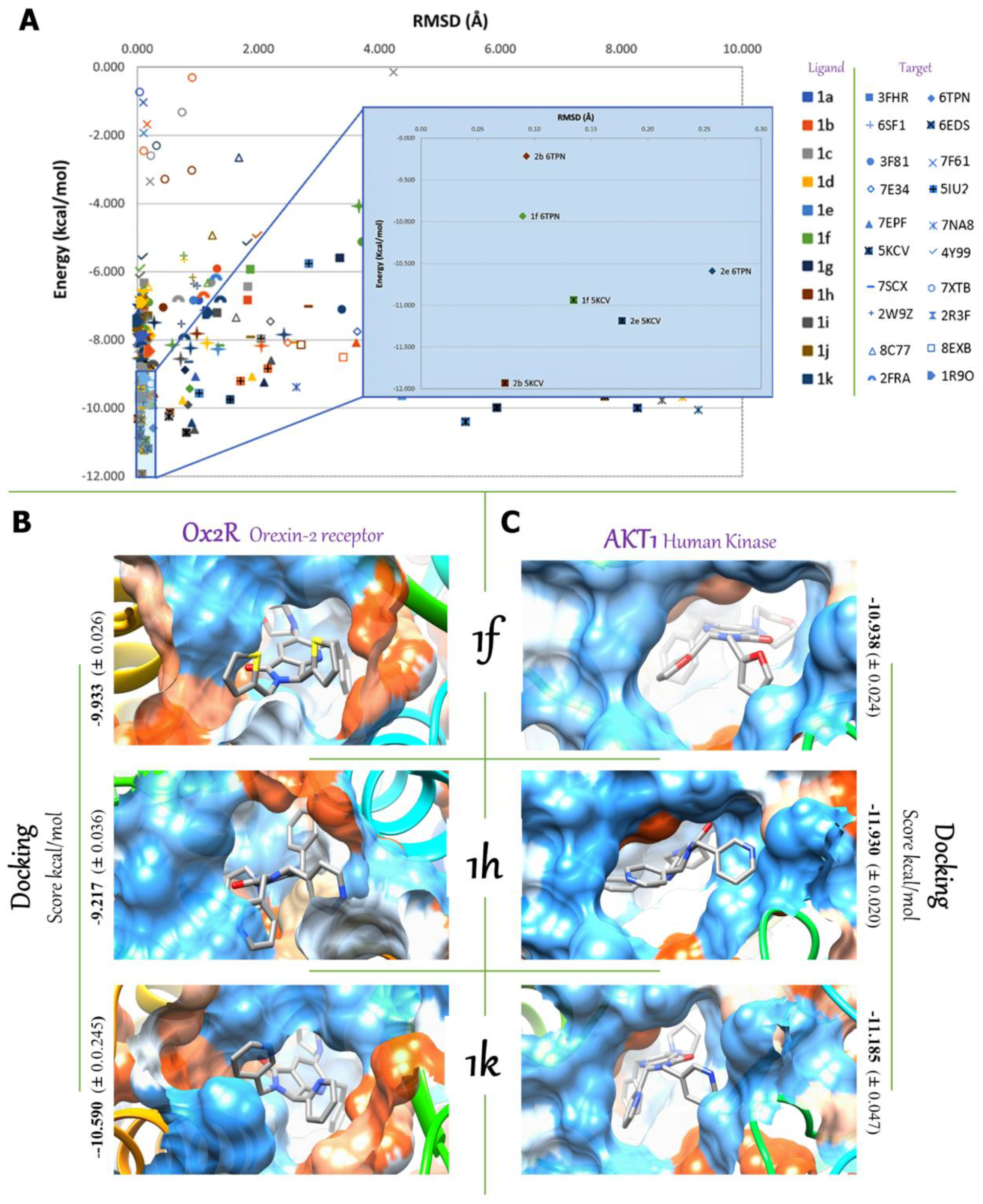
2.3. In Silico Studies
2.3.1. Multi-Target Molecular Docking
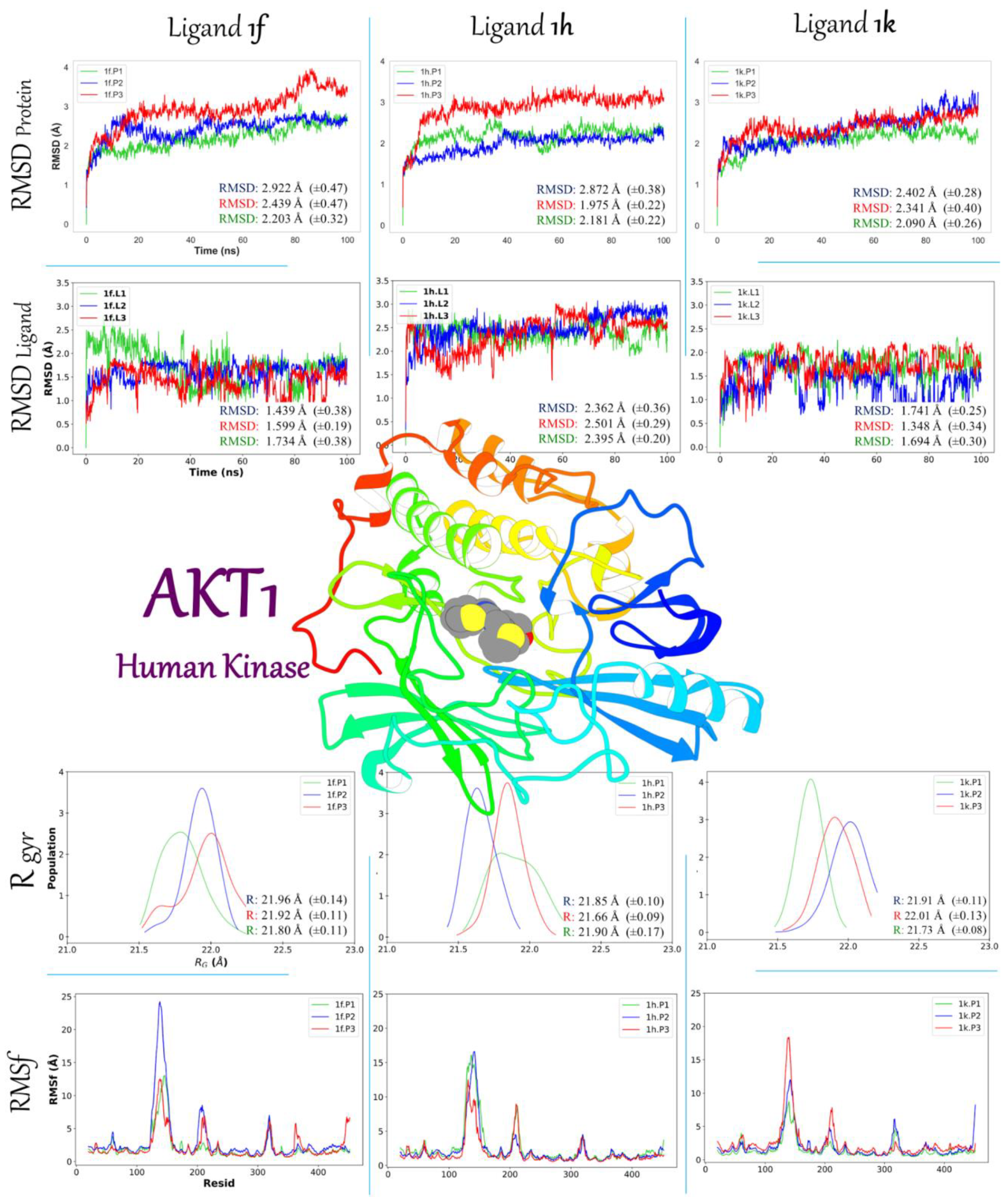
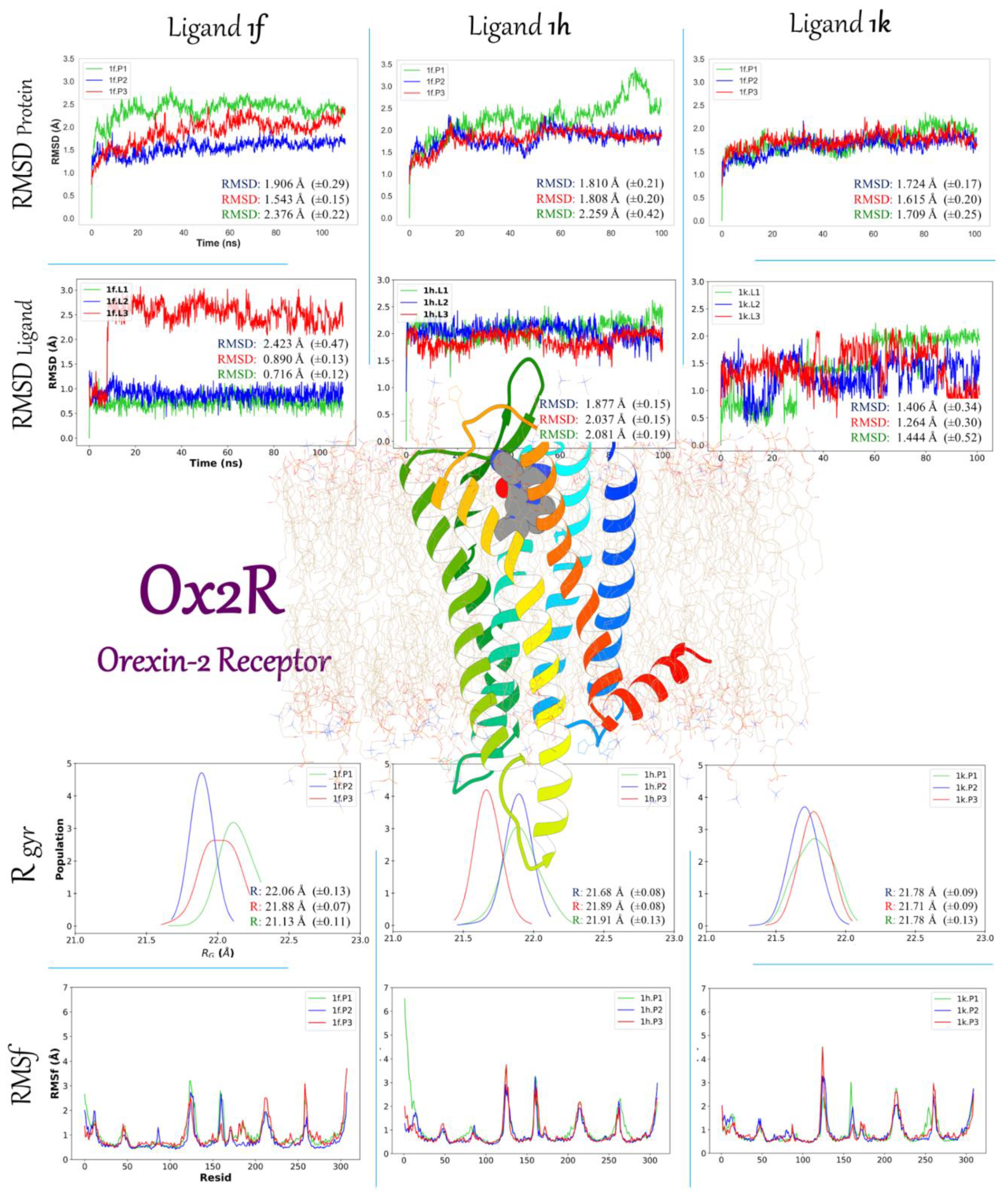
2.3.2. Molecular Dynamic Simulation Studies of 1f, 1h, and 1k on AKT1 and Ox2R
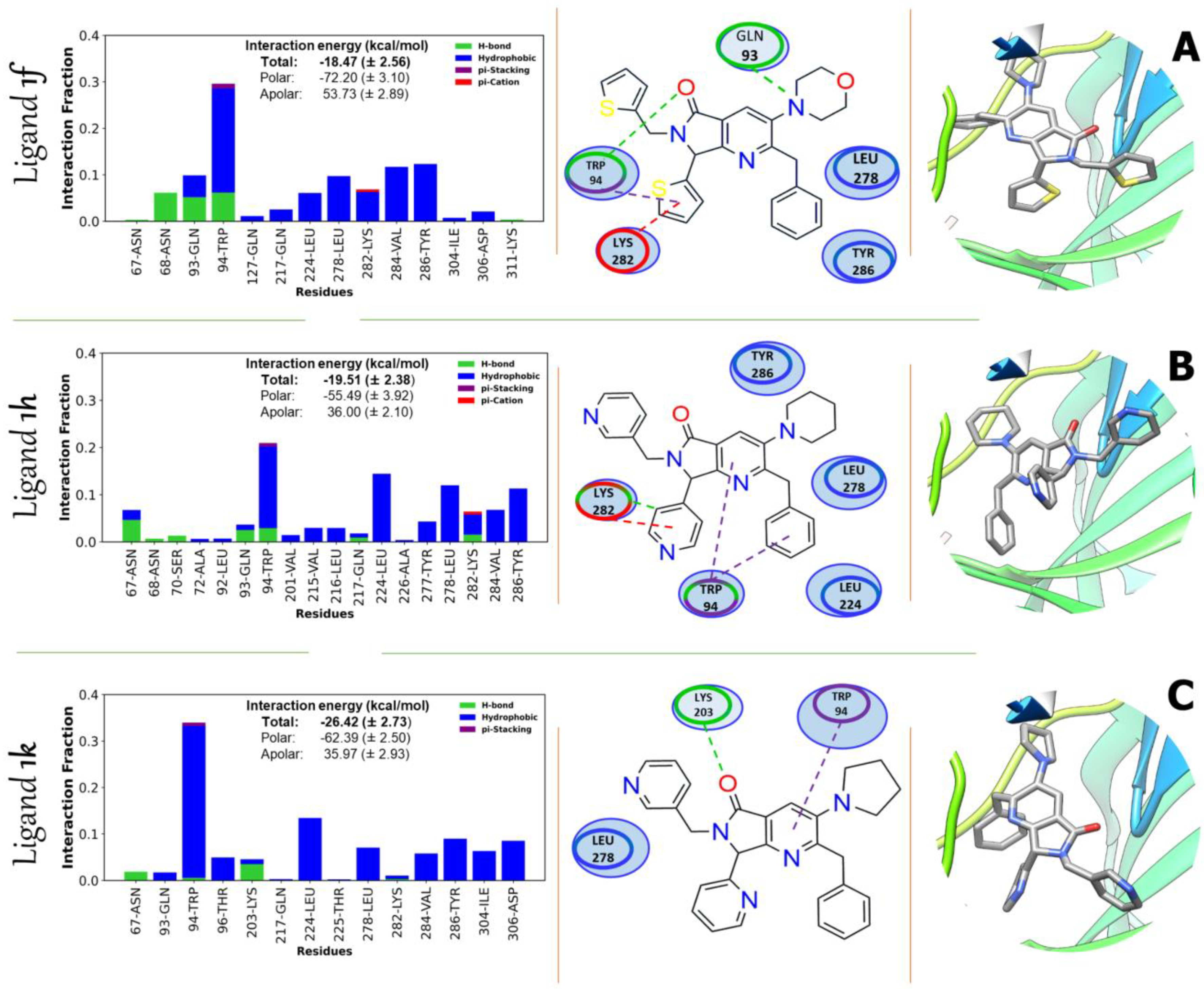
2.3.3. Binding Free Energy 1f, 1h, and 1k
3. Materials and Methods
3.1. Synthesis
3.1.1. General Information, Instrumentation, Software, and Chemicals
3.1.2. Synthesis and Characterization of the Pyrrolo[3,4-b]pyridin-5-ones 1g–1k
- 2-benzyl-3-(diethylamino)-7-(pyridin-3-yl)-6-(pyridin-3-ylmethyl)-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one (1g)
- 2-benzyl-3-(piperidin-1-yl)-6-(pyridin-3-ylmethyl)-7-(pyridin-4-yl)-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one (1h)
- 2-benzyl-3-(piperidin-1-yl)-7-(pyridin-2-yl)-6-(pyridin-3-ylmethyl)-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one (1i)
- 2-benzyl-3-(diethylamino)-7-(pyridin-2-yl)-6-(pyridin-3-ylmethyl)-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one (1j)
- 2-benzyl-7-(pyridin-2-yl)-6-(pyridin-3-ylmethyl)-3-(pyrrolidin-1-yl)-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one (1k)
3.2. Anticancer Activity
3.2.1. Reagents
3.2.2. Cell Culture
3.2.3. Cell Viability Assays
3.2.4. Statistical Analysis
3.3. In Silico Studies
3.3.1. ADME and Tox Properties
3.3.2. Target Selection and Active Pocket Determination
3.3.3. Ligand Optimization
3.3.4. Homology Modeling and Docking Simulations
3.3.5. Molecular Dynamics Simulations
3.3.6. Binding Free Energy
- : represents the binding free energy.
- : is the energy of the complex.
- : in complex is the energy of the protein in the complex.
- : in complex is the energy of the ligand in the complex.
- denotes the calculated free energy value.
- stands for the molecular mechanics energy.
- is the solvation energy.
- represents the temperature.
- refers to the average molecular mechanics entropy.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The Lingering Mysteries of Metastatic Recurrence in Breast Cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Duggento, A.; Indovina, I.; Guerrisi, M.; Toschi, N. Radiomics in Breast Cancer Classification and Prediction. Semin. Cancer Biol. 2021, 72, 238–250. [Google Scholar] [CrossRef]
- Boix-Montesinos, P.; Soriano-Teruel, P.M.; Armiñán, A.; Orzáez, M.; Vicent, M.J. The Past, Present, and Future of Breast Cancer Models for Nanomedicine Development. Adv. Drug Deliv. Rev. 2021, 173, 306–330. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT inhibitors: New weapons in the fight against breast cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- George, B.; Gui, B.; Raguraman, R.; Paul, A.M.; Nakshatri, H.; Pillai, M.R.; Kumar, R. AKT1 transcriptomic landscape in breast cancer cells. Cells 2022, 11, 2290. [Google Scholar] [CrossRef]
- Marcos, P.; Coveñas, R. Involvement of the Orexinergic System in Cancer: Antitumor Strategies and Future Perspectives. Appl. Sci. 2023, 13, 7596. [Google Scholar] [CrossRef]
- Kishida, M.; Ishige, K.; Horibe, T.; Tada, N.; Koibuchi, N.; Shoda, J.; Kita, K.; Kawakami, K. Orexin 2 receptor as a potential target for immunotoxin and antibody-drug conjugate cancer therapy. Oncol. Lett. 2012, 3, 525–529. [Google Scholar] [CrossRef]
- Ibarra, I.A.; Islas-Jácome, A.; González-Zamora, E. Synthesis of polyheterocycles via multicomponent reactions. Org. Biomol. Chem. 2018, 16, 1402. [Google Scholar] [CrossRef]
- Flores-Reyes, J.C.; Islas-Jácome, A.; González-Zamora, E. The Ugi three-component reaction and its variants. Org. Chem. Front. 2021, 8, 5460–5514. [Google Scholar] [CrossRef]
- Morales-Salazar, I.; Montes-Enríquez, F.P.; Garduño-Albino, C.E.; García-Sánchez, M.A.; Ibarra, I.A.; Rojas-Aguirre, Y.; García-Hernández, M.E.; Sarmiento-Silva, R.E.; Alcaraz-Estrada, S.L.; Díaz-Cervantes, E.; et al. Synthesis of bis-furyl-pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu reaction and in vitro activity assays against human SARS-CoV-2 and in silico studies on its main proteins. RSC Med. Chem. 2023, 14, 154–165. [Google Scholar] [CrossRef]
- Morales-Salazar, I.; Rincón-Guevara, M.A.; González-Zamora, E.; Islas-Jácome, A. 2-Benzyl-3-morpholino-7-(thiophen-2-yl)-6-(thiophen-2-ylmethyl)-6,7-dihydro-5H-pyrrolo [3,4-b] pyridin-5-one. Molbank 2022, 4, M1503. [Google Scholar] [CrossRef]
- Fayol, A.; Housseman, C.; Sun, X.; Janvier, P.; Bienaymé, H.; Zhu, J. Synthesis of α-Isocyano-α-alkyl(aryl)acetamides and their use in the multicomponent synthesis of 5-aminooxazole, pyrrolo[3,4-b]pyridin-5-one and 4,5,6,7-tetrahydrofuro[2,3-c]pyridine. Synthesis 2005, 1, 161–165. [Google Scholar] [CrossRef]
- Zamudio-Medina, A.; García-González, A.N.; Herrera-Carrillo, G.K.; Zárate-Zárate, D.; Benavides-Macías, A.; Tamariz, J.; Ibarra, I.A.; Islas-Jácome, A.; González-Zamora, E. Synthesis of polyheterocyclic pyrrolo[3,4-b]pyridin-5-ones via a one-pot (Ugi-3CR/aza diels-alder/N-acylation/aromatization/SN2) process. A suitable alternative towards novel aza-analogues of falipamil. Molecules 2018, 23, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Speck, K.; Magauer, T. The chemistry of isoindole natural products. Beilstein J. Org. Chem. 2013, 9, 2048–2078. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Mansour, A.F.; Abdel-Hamid, H.; Abu-Serie, M.M.; Mohyeldin, S.M.; Teleb, M. Nature-inspired new isoindole-based Passerini adducts as efficient tumor-selective apoptotic inducers via caspase-3/7 activation. Eur. J. Med. Chem. 2023, 245, 114865. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spano, V.; Rocca, R.; Bivacqua, R.; Gualtieri, G.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Manfreda, L.; Bai, R.; et al. Identification of pyrrolo [3′,4′:3,4] cyclohepta [1,2-d][1,2] oxazoles as promising new candidates for the treatment of lymphomas. Eur. J. Med. Chem. 2023, 254, 115372. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Yaglioglu, A.S.; Kishali, N.H.; Sahin, E.; Kara, Y. Evaluation of cytotoxic potentials of some isoindole-1,3-dione derivatives on HeLa, C6 and A549 cancer cell lines. Med. Chem. 2020, 16, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef] [PubMed]
- Krishnappa, S.; Naganna, C.M.; Rajan, H.K.; Rajashekarappa, S.; Gowdru, H.B. Cytotoxic and apoptotic effects of chemogenic and biogenic nano-sulfur on human carcinoma cells: A comparative study. ACS Omega 2021, 6, 32548–32562. [Google Scholar] [CrossRef]
- Shoaib, S.; Ansari, M.A.; Ghazwani, M.; Hani, U.; Jamous, Y.F.; Alali, Z.; Ahmad, W.; Weir, S.A.; Alomary, M.N.; Yusuf, N.; et al. Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers 2023, 15, 697. [Google Scholar] [CrossRef] [PubMed]
- Fuso, P.; Muratore, M.; D’angelo, T.; Paris, I.; Carbognin, L.; Tiberi, G.; Pavese, F.; Duranti, S.; Orlandi, A.; Tortora, G.; et al. PI3K Inhibitors in Advanced Breast Cancer: The Past, The Present, New Challenges and Future Perspectives. Cancers 2022, 14, 2161. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, X.; Zhang, C.; Xue, L.; Yang, L. Expression and clinical significance of MAPK and EGFR in triple–negative breast cancer. Oncol. Lett. 2020, 19, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Collin, L.J.; Maliniak, M.L.; Cronin-Fenton, D.P.; Ahern, T.P.; Christensen, K.B.; Ulrichsen, S.P.; Lash, T.L. Hypoxia-inducible factor-1α expression and breast cancer recurrence in a Danish population-based case control study. Breast Cancer Res. 2021, 23, 103. [Google Scholar] [CrossRef]
- 29. Adelusi, T.I.; Oyedele AQ, K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Abdul-Hammed, M. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Receptor-Based Virtual Screening of Large Libraries in a Multi-Level In Silico Approach. In Advanced Methods in Structural Biology, 1st ed.; Springer: New York, NY, USA, 2023; pp. 261–267. [Google Scholar]
- Sivakumar, K.C.; Haixiao, J.; Naman, C.B.; Sajeevan, T.P. Prospects of multitarget drug designing strategies by linking molecular docking and molecular dynamics to explore the protein–ligand recognition process. Drug Dev. Res. 2020, 81, 685–699. [Google Scholar] [CrossRef]
- Mayr, F.; Möller, G.; Garscha, U.; Fischer, J.; Rodríguez Castaño, P.; Inderbinen, S.G.; Temml, V.; Waltenberger, B.; Schwaiger, S.; Hartmann, R.W.; et al. Finding new molecular targets of familiar natural products using in silico target prediction. Int. J. Mol. Sci. 2020, 21, 7102. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Zhang, L.X.; Ge, Y.J.; Zhang, J.W.; Hu, J.X.; Shen, C.Y.; Lu, A.P.; Hou, T.J.; Cao, D.S. In-silico target prediction by ensemble chemogenomic model based on multi-scale information of chemical structures and protein sequences. J. Cheminform. 2023, 15, 48. [Google Scholar] [CrossRef]
- Karasev, D.A.; Sobolev, B.N.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. The method predicting interaction between protein targets and small-molecular ligands with the wide applicability domain. Comput. Biol. Chem. 2022, 98, 107674. [Google Scholar] [CrossRef]
- Madanagopal, P.; Ramprabhu, N.; Jagadeesan, R. In silico prediction and structure-based multitargeted molecular docking analysis of selected bioactive compounds against mucormycosis. Bull. Natl. Res. Cent. 2022, 46, 24. [Google Scholar] [CrossRef] [PubMed]
- El-Bindary, A.A.; Mohamed, G.G.; El-Sonbati, A.Z.; Diab, M.A.; Hassan, W.M.I.; Morgan, S.M.; Elkholy, A.K. Geometrical structure, potentiometric, molecular docking and thermodynamic studies of azo dye ligand and its metal complexes. J. Mol. Liq. 2016, 218, 138–149. [Google Scholar] [CrossRef]
- Boittier, E.D.; Tang, Y.Y.; Buckley, M.E.; Schuurs, Z.P.; Richard, D.J.; Gandhi, N.S. Assessing molecular docking tools to guide targeted drug discovery of CD38 inhibitors. Int. J. Mol. Sci. 2020, 21, 5183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Meng, J.; Jiang, K.; Lan, H.; Wang, Z.; Lin, M.; Li, W.; Guo, H.; Wei, Y.; Mu, Y. Improving protein–ligand docking and screening accuracies by incorporating a scoring function correction term. Brief. Bioinform. 2022, 23, bbac051. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Kode, J.; Kovvuri, J.; Nagaraju, B.; Jadhav, S.; Barkume, M.; Sen, S.; Kasinathan, N.K.; Chaudhari, P.; Mohanty, B.S.; Gour, J.; et al. Synthesis, biological evaluation, and molecular docking analysis of phenstatin based indole linked chalcones as anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 105, 104447. [Google Scholar] [CrossRef]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Millán-Pacheco, C.; Rios-Soto, L.; Corral-Rodríguez, N.; Sierra-Campos, E.; Valdez-Solana, M.; Téllez-Valencia, A.; Avitia-Domínguez, C. Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design. Pharmaceuticals 2023, 16, 1148. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef]
- Lapierre, J.M.; Eathiraj, S.; Vensel, D.; Liu, Y.; Bull, C.O.; Cornell-Kennon, S.; Iimura, S.; Kelleher, E.W.; Kizer, D.E.; Koerner, S.; et al. Discovery of 3-(3-(4-(1-Aminocyclobutyl) phenyl)-5-phenyl-3 H-imidazo [4,5-b] pyridin-2-yl) pyridin-2-amine (ARQ 092): An orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem. 2016, 59, 6455–6469. [Google Scholar] [CrossRef]
- Asada, H.; Im, D.; Hotta, Y.; Yasuda, S.; Murata, T.; Suno, R.; Iwata, S. Molecular basis for anti-insomnia drug design from structure of lemborexant-bound orexin 2 receptor. Structure 2022, 30, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Couvineau, A.; Nicole, P.; Gratio, V.; Voisin, T. The Orexin receptors: Structural and anti-tumoral properties. Front. Endocrinol. 2022, 13, 931970. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Zhu, M.; Yan, H. Computationally predicting binding affinity in protein–ligand complexes: Free energy-based simulations and machine learning-based scoring functions. Brief. Bioinform. 2021, 22, bbaa107. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Z.; Wang, L.; Tian, S.; Hou, T.; Sun, H. Predicting the mutation effects of protein–ligand interactions via end-point binding free energy calculations: Strategies and analyses. J. Cheminform. 2022, 14, 56. [Google Scholar] [CrossRef]
- Muegge, I.; Hu, Y. Recent Advances in Alchemical Binding Free Energy Calculations for Drug Discovery. ACS Med. Chem. Lett. 2023, 14, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, X.; Hou, T.; Cai, L.; Man, V.H.; Wang, J. Development and test of highly accurate endpoint free energy methods. 1: Evaluation of ABCG2 charge model on solvation free energy prediction and optimization of atom radii suitable for more accurate solvation free energy prediction by the PBSA method. J. Comput. Chem. 2023, 44, 1334–1346. [Google Scholar] [CrossRef]
- Yang, M.; Bo, Z.; Xu, T.; Xu, B.; Wang, D.; Zheng, H. Uni-GBSA: An open-source and web-based automatic workflow to perform MM/GB (PB) SA calculations for virtual screening. Brief. Bioinform. 2023, 24, bbad218. [Google Scholar] [CrossRef] [PubMed]
- Molani, F.; Webb, S.; Cho, A.E. Combining QM/MM Calculations with Classical Mining Minima to Predict Protein–Ligand Binding Free Energy. J. Chem. Inf. Model. 2023, 63, 2728–2734. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Saitoh, T.; Kutsumura, N.; Irukayama-Tomobe, Y.; Ogawa, Y.; Kuroda, D.; Gouda, H.; Kumagai, H.; Fujii, H.; Yanagisawa, M.; et al. Design and synthesis of non-peptide, selective orexin receptor 2 agonists. J. Med. Chem. 2015, 58, 7931–7937. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, A.; Barker, O.; Morris, G.B.; Law, R.J.; Slack, M.; Biggin, P.C. Toward an understanding of agonist binding to human Orexin-1 and Orexin-2 receptors with G-protein-coupled receptor modeling and site-directed mutagenesis. Biochemistry 2013, 52, 8246–8260. [Google Scholar] [CrossRef] [PubMed]
- Janockova, J.; Dolezal, R.; Nepovimova, E.; Kobrlova, T.; Benkova, M.; Kuca, K.; Konecny, J.; Mezeiova, E.; Melikova, M.; Hepnarova, V.; et al. Investigation of new orexin 2 receptor modulators using in silico and in vitro methods. Molecules 2018, 23, 2926. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Byrne, N.J.; Zamlynny, B.; Tummala, S.; Xiao, L.; Shipman, J.M.; Partridge, A.T.; Minnick, C.; Breslin, M.J.; Rudd, M.T.; et al. Structures of active-state orexin receptor 2 rationalize peptide and small-molecule agonist recognition and receptor activation. Nat. Commun. 2021, 12, 815. [Google Scholar] [CrossRef]
- Karhu, L.; Magarkar, A.; Bunker, A.; Xhaard, H. Determinants of Orexin Receptor Binding and Activation—A Molecular Dynamics Study. J. Phys. Chem. B. 2019, 123, 2609–2622. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Pogodin, P.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. PASS Targets: Ligand-based multi-target computational system based on a public data and naïve Bayes approach. SAR QSAR Environ. Res. 2015, 26, 783–793. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Guterres, H.; Park, S.J.; Cao, Y.; Im, W. CHARMM-GUI ligand designer for template-based virtual ligand design in a binding site. J. Chem. Inf. Model 2021, 61, 5336–5342. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Softw Inc.: San Diego, CA, USA, 2015. [Google Scholar]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 277, pp. 396–404. [Google Scholar]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Genet. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Jofily, P.; Pascutti, P.G.; Torres, P.H. Improving blind docking in DOCK6 through an automated preliminary fragment probing strategy. Molecules 2021, 26, 1224. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Zhang, Y. BSP-SLIM: A blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins Struct. Funct. Genet. 2012, 80, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Smith, J.C. Speed vs accuracy: Effect on ligand pose accuracy of varying box size and exhaustiveness in AutoDock vina. Mol. Inform. 2023, 42, 2200188. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks III, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Balusek, C.; Hwang, H.; Lau, C.H.; Lundquist, K.; Hazel, A.; Pavlova, A.; Lynch, D.L.; Reggio, P.H.; Wang, Y.; Gumbart, J.C. Accelerating membrane simulations with hydrogen mass repartitioning. J. Chem. Theory Comput. 2019, 15, 4673–4686. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Melo, M.N.; Van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Pogozheva, I.D.; Armstrong, G.A.; Kong, L.; Hartnagel, T.J.; Carpino, C.A.; Gee, S.E.; Picarello, D.M.; Rubin, D.S.; Lee, J.; Park, S.; et al. Comparative molecular dynamics simulation studies of realistic eukaryotic, prokaryotic, and archaeal membranes. J. Chem. Inf. Model. 2022, 62, 1036–1051. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Forouzesh, N.; Mishra, N. An effective MM/GBSA protocol for absolute binding free energy calculations: A case study on SARS-CoV-2 spike protein and the human ACE2 receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef]
- Dasmahapatra, U.; Kumar, C.K.; Das, S.; Subramanian, P.T.; Murali, P.; Isaac, A.E.; Ramanathan, K.; MM, B.; Chanda, K. In-silico molecular modelling, MM/GBSA binding free energy and molecular dynamics simulation study of novel pyrido fused imidazo [4, 5-c] quinolines as potential anti-tumor agents. Front. Chem. 2022, 10, 991369. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 857, pp. 231–257. [Google Scholar]
- Carugo, O. How root-mean-square distance (rmsd) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Atanda, H.; Balogun, T.A.; Alshehri, M.M.; Olivos-Ramirez, G.; Vilca-Quispe, J.; Chenet-Zuta, M.; Cárdenas-Cárdenas, R.; Delgado Wong, H.; Ropón-Palacios, G.; Umar, H.I. In silico study revealed the inhibitory activity of selected phytomolecules of C. rotundus against VacA implicated in gastric ulcer. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MCF-7 IC50 (µM) | MDA-MB-231 IC50 (µM) |
|---|---|---|
| 1a | −2432.33696 | 114.998827 |
| 1b | 277.07712 | 89.7144132 |
| 1c | 554.323499 | 117.833333 |
| 1d | 331.476877 | 67.8459821 |
| 1e | 753.2345013 | 77.5989269 |
| 1f | 753.234501 | 14.8518557 |
| 1g | 158.183608 | 68.6064836 |
| 1h | 109.624029 | 75.5411255 |
| 1i | 123.0212854 | 59.4034271 |
| 1j | 159.288991 | 96.820554 |
| 1k | 187.18081 | 117.33474 |
| Contributions of Binding Interaction Energy (kcal/mol) | ||||||||
| Inhibitor | EvdW | EELE | EGB | ESURF | GApolar | Gpolar | Total | |
| AKT1 | 1f | −51.85 (±1.92) | −20.35 (±1.96) | 60.17 (±3.17) | −6.44 (±0.46) | −72.20 (±3.04) | 53.73 (±2.89) | −18.48 (±1.56) |
| 1h | −45.38 (±1.83) | −10.11 (±1.32) | 42.04 (±2.37) | −6.05 (±0.50) | −55.49 (±3.92) | 36.00 (±3.10) | −19.5 (±0.38) | |
| 1k | −54.5 (±2.31) | −7.88 (±0.66) | 42.70 (±2.30) | −6.74 (±0.50) | −62.39 (±2.50) | 35.97 (±3.93) | −26.42 (±0.73) | |
| Ox2R | 1f | −46.71 (±1.99) | −15.97 (±1.3) | 54.37 (±2.36) | −5.95 (±0.40) | −62.68 (±3.82) | 48.41 (±4.33) | −14.26 (±0.26) |
| 1h | −44.55 (±2.09) | −26.08 (±1.77) | 53.22 (±3.84) | −6.21 (±0.370) | −70.63 (±3.15) | 47.01 (±4.90) | −23.62 (±1.20) | |
| 1k | −48.87 (±2.71) | −24.26 (±1.22) | 56.38 (±3.08) | −6.31 (±0.56) | −73.13 (±2.01) | 50.07 (±2.78) | −23.06 (±1.12) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Salazar, I.; Garduño-Albino, C.E.; Montes-Enríquez, F.P.; Nava-Tapia, D.A.; Navarro-Tito, N.; Herrera-Zúñiga, L.D.; González-Zamora, E.; Islas-Jácome, A. Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma. Pharmaceuticals 2023, 16, 1562. https://doi.org/10.3390/ph16111562
Morales-Salazar I, Garduño-Albino CE, Montes-Enríquez FP, Nava-Tapia DA, Navarro-Tito N, Herrera-Zúñiga LD, González-Zamora E, Islas-Jácome A. Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma. Pharmaceuticals. 2023; 16(11):1562. https://doi.org/10.3390/ph16111562
Chicago/Turabian StyleMorales-Salazar, Ivette, Carlos E. Garduño-Albino, Flora P. Montes-Enríquez, Dania A. Nava-Tapia, Napoleón Navarro-Tito, Leonardo David Herrera-Zúñiga, Eduardo González-Zamora, and Alejandro Islas-Jácome. 2023. "Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma" Pharmaceuticals 16, no. 11: 1562. https://doi.org/10.3390/ph16111562
APA StyleMorales-Salazar, I., Garduño-Albino, C. E., Montes-Enríquez, F. P., Nava-Tapia, D. A., Navarro-Tito, N., Herrera-Zúñiga, L. D., González-Zamora, E., & Islas-Jácome, A. (2023). Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma. Pharmaceuticals, 16(11), 1562. https://doi.org/10.3390/ph16111562