

Construction of an Exudative Age-Related Macular Degeneration Diagnostic and Therapeutic Molecular Network Using Multi-Layer Network Analysis, a Fuzzy Logic Model, and Deep Learning Techniques: Are Retinal and Brain Neurodegenerative Disorders Related?
 , ,
, ,  , , , , ,
, , , , ,
Abstract
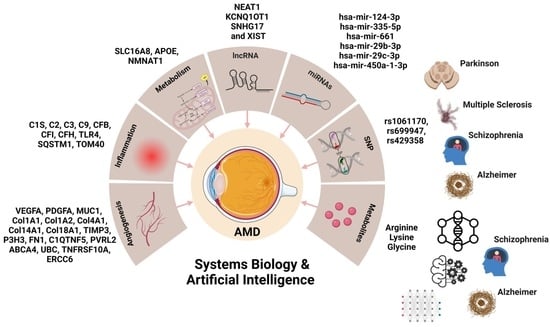
1. Introduction
2. Results
2.1. The NeDRex Plugin’s Network for Identifying Disease Modules
2.2. Gene Regulatory Network Analysis
2.3. Enrichment Analysis
2.4. Metabolite Pathway Analysis
2.5. Joint Pathway Analysis
2.6. Metabolite–Gene–Disease Interaction Network
2.7. Identification of Genes Related to nAMD–Single Nucleotide Polymorphisms (SNPs)
2.8. Results of the Developed Binary-GA Search Method for Maximization of the Model in the 56-Dimensional Space
2.9. Summary of All Results
3. Discussion
3.1. ECM Proteins, Complement System, and Pathogenesis of nAMD
3.2. ECM Remodeling by Cytoskeleton-Related Proteins and Angioinflammatory Factors
3.3. Aging and Pathogenesis of nAMD
3.4. AMD and Other Neurodegenerative Diseases
3.5. Autophagy and AMD
3.6. Non-Coding RNAs and AMD
3.7. Metabolic Activity in nAMD and Neurodegeneration
4. Materials and Methods
4.1. First Data Sources
4.2. Network Construction
4.3. Topological Network Analysis
4.4. Gene Regulatory Network Construction
4.5. Second Data Sources: nAMD-Related Metabolites and SNPs
4.6. Enrichment Analysis
4.7. Data Pre-Processing for the First Fuzzy Logic Model
4.8. Metabolites Merit Calculation Using Fuzzy Logic Model
4.9. Data Pre-Processing for the Second Fuzzy Logic Model
4.10. Calculating the Output Merit of the Metabolite Route Using the Fuzzy Logic Model
4.11. Metabolic Route Model Development Using Long Short-Term Memory (LSTM) Network
- -
- The training was performed using the Adam optimizer.
- -
- The network was trained for 100,000 epochs. For larger data sets, a lower number of epochs may suffice for achieving a good fit.
- -
- The sequences and responses used for validation were specified.
- -
- The learning rate was set at 0.0005.
- -
- The network that gave the best validation loss, i.e., the lowest validation loss, was outputted.
4.12. Utilizing AI and Genetic Algorithms to Identify Key Metabolites in the Metabolic Route
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R.; Mahajan, V.B. Retinal and choroidal angiogenesis: A review of new targets. Int. J. Retin. Vitr. 2017, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.B. Introduction: Understanding the role of angiogenesis and antiangiogenic agents in age-related macular degeneration. Ophthalmology 2009, 116, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, J.L.; Giordano, R.J.; Regatieri, C.V. Ocular Angiogenesis; Hindawi: London, UK, 2015; Volume 2015. [Google Scholar]
- Papadopoulos, Z. Recent developments in the treatment of wet age-related macular degeneration. Curr. Med. Sci. 2020, 40, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell. Biochem. 2022, 123, 1938–1965. [Google Scholar] [CrossRef]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Langer, R.; Ferrara, N. Targeting angiogenesis in oncology, ophthalmology and beyond. Nat. Rev. Drug Discov. 2023, 22, 476–495. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Gacche, R. Compensatory angiogenesis and tumor refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef]
- Montemagno, C.; Pagès, G. Resistance to anti-angiogenic therapies: A mechanism depending on the time of exposure to the drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef]
- Saravanan, S.; Vimalraj, S.; Pavani, K.; Nikarika, R.; Sumantran, V.N. Intussusceptive angiogenesis as a key therapeutic target for cancer therapy. Life Sci. 2020, 252, 117670. [Google Scholar] [CrossRef]
- Cannell, I.G.; Sawicka, K.; Pearsall, I.; Wild, S.A.; Deighton, L.; Pearsall, S.M.; Lerda, G.; Joud, F.; Khan, S.; Bruna, A. FOXC2 promotes vasculogenic mimicry and resistance to anti-angiogenic therapy. Cell Rep. 2023, 42, 112791. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular mimicry: A novel neovascularization mechanism driving anti-angiogenic therapy (AAT) resistance in glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ji, Y.R.; Lee, Y.M. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch. Pharmacal Res. 2022, 45, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Kardideh, B.; Samimi, Z.; Norooznezhad, F.; Kiani, S.; Mansouri, K. Autophagy, cancer and angiogenesis: Where is the link? Cell Biosci. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Uebelhoer, M.; Iruela-Arispe, M.L. Cross-talk between signaling and metabolism in the vasculature. Vasc. Pharmacol. 2016, 83, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Zolfaghari, N.; Soheili, Z.-S.; Samiei, S.; Latifi-Navid, H.; Hafezi-Moghadam, A.; Ahmadieh, H.; Rezaei-Kanavi, M. microRNA-96 targets the INS/AKT/GLUT4 signaling axis: Association with and effect on diabetic retinopathy. Heliyon 2023, 9, e15539. [Google Scholar] [CrossRef]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef] [PubMed]
- Davari, M.; Soheili, Z.-S.; Latifi-Navid, H.; Samiee, S. Potential involvement of miR-183/96/182 cluster-gene target interactions in transdifferentiation of human retinal pigment epithelial cells into retinal neurons. Biochem. Biophys. Res. Commun. 2023, 663, 87–95. [Google Scholar] [CrossRef]
- Latifi-Navid, H.; Soheili, Z.S.; Samiei, S.; Sadeghi, M.; Taghizadeh, S.; Pirmardan, E.R.; Ahmadieh, H. Network analysis and the impact of Aflibercept on specific mediators of angiogenesis in HUVEC cells. J. Cell. Mol. Med. 2021, 25, 8285–8299. [Google Scholar] [CrossRef]
- Sadegh, S.; Skelton, J.; Anastasi, E.; Bernett, J.; Blumenthal, D.B.; Galindez, G.; Salgado-Albarrán, M.; Lazareva, O.; Flanagan, K.; Cockell, S. Network medicine for disease module identification and drug repurposing with the NeDRex platform. Nat. Commun. 2021, 12, 6848. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.-H.; Rivera, C.G.; Popel, A.S.; Bader, J.S. Constructing the angiome: A global angiogenesis protein interaction network. Physiol. Genom. 2012, 44, 915–924. [Google Scholar] [CrossRef]
- Pool, F.M.; Kiel, C.; Serrano, L.; Luthert, P.J. Repository of proposed pathways and protein–protein interaction networks in age-related macular degeneration. NPJ Aging Mech. Dis. 2020, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. BioMed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [PubMed]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M. The collagen suprafamily: From biosynthesis to advanced biomaterial development. Adv. Mater. 2019, 31, 1801651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Zhang, H.; Wang, J.; Hua, H.; Jiang, Y. The role of network-forming collagens in cancer progression. Int. J. Cancer 2022, 151, 833–842. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Y.; Wu, Y.; Gao, Y.; Li, Q.; Abdulrahman, A.A.; Liu, X.F.; Ji, G.Q.; Gao, J.; Li, L. Identification of COL1A1 as an invasion-related gene in malignant astrocytoma. Int. J. Oncol. 2018, 53, 2542–2554. [Google Scholar] [CrossRef]
- Ma, Z.; Mao, C.; Jia, Y.; Fu, Y.; Kong, W. Extracellular matrix dynamics in vascular remodeling. Am. J. Physiol.-Cell Physiol. 2020, 319, C481–C499. [Google Scholar] [CrossRef]
- Kubota, K.; Okazaki, J.; Louie, O.; Kent, K.C.; Liu, B. TGF-β stimulates collagen (I) in vascular smooth muscle cells via a short element in the proximal collagen promoter. J. Surg. Res. 2003, 109, 43–50. [Google Scholar] [CrossRef]
- Guerrero, P.A.; McCarty, J.H. TGF-β activation and signaling in angiogenesis. In Physiologic and Pathologic Angiogenesis—Signaling Mechanisms and Targeted Therapy; InTechOpen: London, UK, 2017. [Google Scholar]
- Zou, J.; Liu, K.-C.; Wang, W.-P.; Xu, Y. Circular RNA COL1A2 promotes angiogenesis via regulating miR-29b/VEGF axis in diabetic retinopathy. Life Sci. 2020, 256, 117888. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Yang, H.; Li, C.; Fang, C. Identification of potential genes related to breast cancer brain metastasis in breast cancer patients. Biosci. Rep. 2021, 41, BSR20211615. [Google Scholar] [CrossRef] [PubMed]
- Sertié, A.L.; Sossi, V.; Camargo, A.A.; Zatz, M.; Brahe, C.; Passos-Bueno, M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 2000, 9, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Aikio, M.; Hurskainen, M.; Brideau, G.; Hägg, P.; Sormunen, R.; Heljasvaara, R.; Gould, D.B.; Pihlajaniemi, T. Collagen XVIII short isoform is critical for retinal vascularization, and overexpression of the Tsp-1 domain affects eye growth and cataract formation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7450–7462. [Google Scholar] [CrossRef] [PubMed]
- Pokidysheva, E.; Boudko, S.; Vranka, J.; Zientek, K.; Maddox, K.; Moser, M.; Fässler, R.; Ware, J.; Bächinger, H.P. Biological role of prolyl 3-hydroxylation in type IV collagen. Proc. Natl. Acad. Sci. USA 2014, 111, 161–166. [Google Scholar] [CrossRef]
- Shah, R.; Smith, P.; Purdie, C.; Quinlan, P.; Baker, L.; Aman, P.; Thompson, A.; Crook, T. The prolyl 3-hydroxylases P3H2 and P3H3 are novel targets for epigenetic silencing in breast cancer. Br. J. Cancer 2009, 100, 1687–1696. [Google Scholar] [CrossRef]
- Labelle, M.; Hynes, R.O. The Initial Hours of Metastasis: The Importance of Cooperative Host–Tumor Cell Interactions during Hematogenous DisseminationHost–Tumor Cell Interactions during Metastatic Dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef]
- Buergy, D.; Wenz, F.; Groden, C.; Brockmann, M.A. Tumor–platelet interaction in solid tumors. Int. J. Cancer 2012, 130, 2747–2760. [Google Scholar] [CrossRef]
- Clark, S.J.; Schmidt, C.Q.; White, A.M.; Hakobyan, S.; Morgan, B.P.; Bishop, P.N. Identification of factor H–like protein 1 as the predominant complement regulator in Bruch’s membrane: Implications for age-related macular degeneration. J. Immunol. 2014, 193, 4962–4970. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Schwartz, S.G.; Flynn, H.W., Jr.; Emerson, G.G.; Choudhry, N.; Ferrone, P.J.; Goldberg, R.A.; Lim, J.I.; Witkin, A.J. Distinguishing between infectious endophthalmitis and noninfectious inflammation following intravitreal anti-VEGF injection. J. Vitr. Dis. 2019, 3, 42–44. [Google Scholar] [CrossRef]
- Anderson, W.J.; da Cruz, N.F.S.; Lima, L.H.; Emerson, G.G.; Rodrigues, E.B.; Melo, G.B. Mechanisms of sterile inflammation after intravitreal injection of antiangiogenic drugs: A narrative review. Int. J. Retin. Vitr. 2021, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Degn, S.E.; Jensenius, J.C.; Bjerre, M. The lectin pathway and its implications in coagulation, infections and auto-immunity. Curr. Opin. Organ Transplant. 2011, 16, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.R.; Boer, M.A.d.; Strasser, J.; Zwarthoff, S.A.; Beurskens, F.J.; de Haas, C.J.; Aerts, P.C.; Wang, G.; de Jong, R.N.; Bagnoli, F. Staphylococcal protein A inhibits complement activation by interfering with IgG hexamer formation. Proc. Natl. Acad. Sci. USA 2021, 118, e2016772118. [Google Scholar] [CrossRef]
- O’Callaghan, R.J. The pathogenesis of Staphylococcus aureus eye infections. Pathogens 2018, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Whiston, E.A.; Sugi, N.; Kamradt, M.C.; Sack, C.; Heimer, S.R.; Engelbert, M.; Wawrousek, E.F.; Gilmore, M.S.; Ksander, B.R.; Gregory, M.S. αB-crystallin protects retinal tissue during Staphylococcus aureus-induced endophthalmitis. Infect. Immun. 2008, 76, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Budoff, G.; Prenner, J.L.; Schwarzbauer, J.E. Minireview: Fibronectin in retinal disease. Exp. Biol. Med. 2017, 242, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.G.; Bader, J.S.; Popel, A.S. Angiogenesis-associated crosstalk between collagens, CXC chemokines, and thrombospondin domain-containing proteins. Ann. Biomed. Eng. 2011, 39, 2213–2222. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.J.; Sobel, M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: Enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef]
- Xiang, L.; Xie, G.; Ou, J.; Wei, X.; Pan, F.; Liang, H. The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PLoS ONE 2012, 7, e35378. [Google Scholar] [CrossRef]
- Wang, K.; Seo, B.R.; Fischbach, C.; Gourdon, D. Fibronectin mechanobiology regulates tumorigenesis. Cell. Mol. Bioeng. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Farjood, F.; Ahmadpour, A.; Ostvar, S.; Vargis, E. Acute mechanical stress in primary porcine RPE cells induces angiogenic factor expression and in vitro angiogenesis. J. Biol. Eng. 2020, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.C.; Shlamkovitch, T.; Papo, N.; Barton, W.A. Constitutive association of Tie1 and Tie2 with endothelial integrins is functionally modulated by angiopoietin-1 and fibronectin. PLoS ONE 2016, 11, e0163732. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dong, J.; Zhou, H.; Deng, X.; Li, R.; Chen, N.; Luo, M.; Li, Y.; Wu, J.; Wang, L. Glycation of fibronectin inhibits VEGF-induced angiogenesis by uncoupling VEGF receptor-2-c-Src crosstalk. J. Cell. Mol. Med. 2020, 24, 9154–9164. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum. Mol. Genet. 2009, 18, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2020, 25, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef]
- Smookler, D.S.; Mohammed, F.F.; Kassiri, Z.; Duncan, G.S.; Mak, T.W.; Khokha, R. Cutting edge: Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J. Immunol. 2006, 176, 721–725. [Google Scholar] [CrossRef]
- Mohammed, F.F.; Smookler, D.S.; Taylor, S.E.; Fingleton, B.; Kassiri, Z.; Sanchez, O.H.; English, J.L.; Matrisian, L.M.; Au, B.; Yeh, W.-C. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat. Genet. 2004, 36, 969–977. [Google Scholar] [CrossRef]
- Kassiri, Z.; Oudit, G.Y.; Sanchez, O.; Dawood, F.; Mohammed, F.F.; Nuttall, R.K.; Edwards, D.R.; Liu, P.P.; Backx, P.H.; Khokha, R. Combination of tumor necrosis factor-α ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ. Res. 2005, 97, 380–390. [Google Scholar] [CrossRef]
- Janssen, A.; Hoellenriegel, J.; Fogarasi, M.; Schrewe, H.; Seeliger, M.; Tamm, E.; Ohlmann, A.; May, C.A.; Weber, B.H.; Stöhr, H. Abnormal vessel formation in the choroid of mice lacking tissue inhibitor of metalloprotease-3. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Dewing, J.M.; Carare, R.O.; Lotery, A.J.; Ratnayaka, J.A. The Diverse Roles of TIMP-3: Insights into degenerative diseases of the senescent retina and brain. Cells 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Ishikawa, K.; Fukuda, Y.; Ji, R.; Wada, I.; Kubo, Y.; Akiyama, M.; Notomi, S.; Murakami, Y.; Nakao, S. TNFRSF10A downregulation induces retinal pigment epithelium degeneration during the pathogenesis of age-related macular degeneration and central serous chorioretinopathy. Hum. Mol. Genet. 2022, 31, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhsh, F.; Merikhian, P.; Eisavand, M.R.; Farahmand, L. Crosstalk between MUC1 and VEGF in angiogenesis and metastasis: A review highlighting roles of the MUC1 with an emphasis on metastatic and angiogenic signaling. Cancer Cell Int. 2021, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Yokoyama, S.; Higashi, M.; Yamada, N.; Takao, S.; Yonezawa, S. MUC1 enhances hypoxia-driven angiogenesis through the regulation of multiple proangiogenic factors. Oncogene 2013, 32, 4614–4621. [Google Scholar] [CrossRef]
- Aubert, S.; Fauquette, V.; Hémon, B.; Lepoivre, R.; Briez, N.; Bernard, D.; Van Seuningen, I.; Leroy, X.; Perrais, M. MUC1, a new hypoxia inducible factor target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer Res. 2009, 69, 5707–5715. [Google Scholar] [CrossRef]
- Stanton, C.M.; Borooah, S.; Drake, C.; Marsh, J.A.; Campbell, S.; Lennon, A.; Soares, D.C.; Vallabh, N.A.; Sahni, J.; Cideciyan, A.V. Novel pathogenic mutations in C1QTNF5 support a dominant negative disease mechanism in late-onset retinal degeneration. Sci. Rep. 2017, 7, 12147. [Google Scholar] [CrossRef]
- Seldin, M.M.; Tan, S.Y.; Wong, G.W. Metabolic function of the CTRP family of hormones. Rev. Endocr. Metab. Disord. 2014, 15, 111–123. [Google Scholar] [CrossRef]
- Schäffler, A.; Buechler, C. CTRP family: Linking immunity to metabolism. Trends Endocrinol. Metab. 2012, 23, 194–204. [Google Scholar] [CrossRef]
- Shang, M.; Zhang, Y.; Zhang, T. IFI44L and C1QTNF5 as promising biomarkers of proliferative diabetic retinopathy. Medicine 2022, 101, e31961. [Google Scholar] [CrossRef]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Runge, P.; Haghayegh Jahromi, N.; Naboth, H.; Landtwing, A.; Montecchi, R.; Leicht, N.; Hunter, M.C.; Takai, Y.; Halin, C. CD112 regulates angiogenesis and T cell entry into the spleen. Cells 2021, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, Y.; Shinojima, A.; Kawamura, A.; Yuzawa, M. Correlation of aging and segmental choroidal thickness measurement using swept source optical coherence tomography in healthy eyes. PLoS ONE 2015, 10, e0144156. [Google Scholar] [CrossRef] [PubMed]
- Chirco, K.; Sohn, E.; Stone, E.; Tucker, B.; Mullins, R. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye 2017, 31, 10–25. [Google Scholar] [CrossRef]
- Jiménez-Gómez, Y.; Alba-Molina, D.; Blanco-Blanco, M.; Pérez-Fajardo, L.; Reyes-Ortega, F.; Ortega-Llamas, L.; Villalba-González, M.; Fernández-Choquet de Isla, I.; Pugliese, F.; Stoikow, I. Novel Treatments for Age-Related Macular Degeneration: A Review of Clinical Advances in Sustained Drug Delivery Systems. Pharmaceutics 2022, 14, 1473. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, W.; Thomas, M.; Hanke, P.; Sam, J.; Osl, F.; Weininger, D.; Baehner, M.; Seeber, S.; Kettenberger, H.; Schanzer, J. Anti-tumoral, anti-angiogenic and anti-metastatic efficacy of a tetravalent bispecific antibody (TAvi6) targeting VEGF-A and angiopoietin-2. MAbs 2016, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Lindsley, K.; Vedula, S.S.; Krzystolik, M.G.; Hawkins, B.S. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2014, 8. [Google Scholar] [CrossRef]
- Koh, G.Y.; Augustin, H.G.; Campochiaro, P.A. Viewpoints: Dual-blocking antibody against VEGF-A and angiopoietin-2 for treating vascular diseases of the eye. Trends Mol. Med. 2022, 28, 347–349. [Google Scholar] [CrossRef]
- Arepalli, S.; Kaiser, P.K. Pipeline therapies for neovascular age related macular degeneration. Int. J. Retin. Vitr. 2021, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Manosalva, C.; Quiroga, J.; Hidalgo, A.I.; Alarcón, P.; Anseoleaga, N.; Hidalgo, M.A.; Burgos, R.A. Role of lactate in inflammatory processes: Friend or foe. Front. Immunol. 2022, 12, 808799. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family—From monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflügers Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Thounaojam, M.C.; Bartoli, M.; Martin, P.M. Implications of NAD(+) Metabolism in the Aging Retina and Retinal Degeneration. Oxid. Med. Cell Longev. 2020, 2020, 2692794. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K. NAMPT-mediated NAD+ biosynthesis is essential for vision in mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef]
- Kuribayashi, H.; Baba, Y.; Iwagawa, T.; Arai, E.; Murakami, A.; Watanabe, S. Roles of Nmnat1 in the survival of retinal progenitors through the regulation of pro-apoptotic gene expression via histone acetylation. Cell Death Dis. 2018, 9, 891. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, K.-k.; Tong, Y.; Zhou, Y.-l.; Wang, Y.-x.; Zhao, P.-q.; Wang, Z.-y. Exogenous NAD+ decreases oxidative stress and protects H2O2-treated RPE cells against necrotic death through the up-regulation of autophagy. Sci. Rep. 2016, 6, 26322. [Google Scholar] [CrossRef]
- Bergen, A.A. Nicotinamide, iRPE-in-a dish, and age-related macular degeneration therapy development. Stem Cell Investig. 2017, 4, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Green, C.F.; Hui, Y.-H.; Prieto, L.; Shepard, R.; Dong, S.; Wang, T.; Tan, B.; Gong, X.; Kays, L. Discovery of a highly selective NAMPT inhibitor that demonstrates robust efficacy and improved retinal toxicity with nicotinic acid coadministration. Mol. Cancer Ther. 2017, 16, 2677–2688. [Google Scholar] [CrossRef] [PubMed]
- Zabka, T.S.; Singh, J.; Dhawan, P.; Liederer, B.M.; Oeh, J.; Kauss, M.A.; Xiao, Y.; Zak, M.; Lin, T.; McCray, B. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol. Sci. 2015, 144, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.-H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.-Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The impact of oxidative stress on blood-retinal barrier physiology in age-related macular degeneration. Cells 2021, 10, 64. [Google Scholar] [CrossRef]
- Tuo, J.; Ning, B.; Bojanowski, C.M.; Lin, Z.-N.; Ross, R.J.; Reed, G.F.; Shen, D.; Jiao, X.; Zhou, M.; Chew, E.Y. Synergic effect of polymorphisms in ERCC6 5′ flanking region and complement factor H on age-related macular degeneration predisposition. Proc. Natl. Acad. Sci. USA 2006, 103, 9256–9261. [Google Scholar] [CrossRef]
- Shughoury, A.; Sevgi, D.D.; Ciulla, T.A. Molecular Genetic Mechanisms in Age-Related Macular Degeneration. Genes 2022, 13, 1233. [Google Scholar] [CrossRef]
- Baas, D.C.; Despriet, D.D.; Gorgels, T.G.; Bergeron-Sawitzke, J.; Uitterlinden, A.G.; Hofman, A.; van Duijn, C.M.; Merriam, J.E.; Smith, R.T.; Barile, G.R. The ERCC6 gene and age-related macular degeneration. PLoS ONE 2010, 5, e13786. [Google Scholar] [CrossRef]
- Liutkeviciene, R.; Vilkeviciute, A.; Gedvilaite, G.; Kaikaryte, K.; Kriauciuniene, L. Haplotypes of HTRA1 rs1120638, TIMP3 rs9621532, VEGFA rs833068, CFI rs10033900, ERCC6 rs3793784, and KCTD10 rs56209061 gene polymorphisms in age-related macular degeneration. Dis. Markers 2019, 2019, 9602949. [Google Scholar] [CrossRef]
- Liu, M.M.; Agrón, E.; Chew, E.; Meyerle, C.; Ferris, F.L.; Chan, C.-C.; Tuo, J. Copy number variations in candidate genes in neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3129–3135. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Synowiec, E.; Salminen, A.; Kaarniranta, K. Genetic variability in DNA repair proteins in age-related macular degeneration. Int. J. Mol. Sci. 2012, 13, 13378–13397. [Google Scholar] [CrossRef] [PubMed]
- Zweifel, S.A.; Spaide, R.F.; Curcio, C.A.; Malek, G.; Imamura, Y. Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology 2010, 117, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Mrejen, S.; Sato, T.; Curcio, C.A.; Spaide, R.F. Assessing the cone photoreceptor mosaic in eyes with pseudodrusen and soft drusen in vivo using adaptive optics imaging. Ophthalmology 2014, 121, 545–551. [Google Scholar] [CrossRef]
- Finger, R.P.; Wu, Z.; Luu, C.D.; Kearney, F.; Ayton, L.N.; Lucci, L.M.; Hubbard, W.C.; Hageman, J.L.; Hageman, G.S.; Guymer, R.H. Reticular pseudodrusen: A risk factor for geographic atrophy in fellow eyes of individuals with unilateral choroidal neovascularization. Ophthalmology 2014, 121, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Domalpally, A.; Agrón, E.; Pak, J.W.; Keenan, T.D.; Ferris III, F.L.; Clemons, T.E.; Chew, E.Y. Prevalence, risk, and genetic association of reticular pseudodrusen in age-related macular degeneration: Age-related eye disease study 2 report 21. Ophthalmology 2019, 126, 1659–1666. [Google Scholar] [CrossRef]
- Hu, M.L.; Quinn, J.; Xue, K. Interactions between apolipoprotein E metabolism and retinal inflammation in age-related macular degeneration. Life 2021, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Ishida, B.Y.; Bailey, K.R.; Duncan, K.G.; Chalkley, R.J.; Burlingame, A.; Kane, J.P.; Schwartz, D.M. Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J. Lipid Res. 2004, 45, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Ozaki, S.; Nealon, M.; Neitz, J.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: Implications for the process of drusen formation. Am. J. Ophthalmol. 2001, 131, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.M.; Zorapapel, N.C.; Rich, K.A.; Wagstaff, R.E.; Lambert, R.W.; Rosenberg, S.E.; Moghaddas, F.; Pirouzmanesh, A.; Aoki, A.M.; Kenney, M.C. Effects of cholesterol and apolipoprotein E on retinal abnormalities in ApoE-deficient mice. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1891–1900. [Google Scholar]
- Johnson, L.V.; Forest, D.L.; Banna, C.D.; Radeke, C.M.; Maloney, M.A.; Hu, J.; Spencer, C.N.; Walker, A.M.; Tsie, M.S.; Bok, D. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 18277–18282. [Google Scholar] [CrossRef] [PubMed]
- Löffler, K.U.; Edward, D.P.; Tso, M. Immunoreactivity against tau, amyloid precursor protein, and beta-amyloid in the human retina. Investig. Ophthalmol. Vis. Sci. 1995, 36, 24–31. [Google Scholar]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s Aβ-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef] [PubMed]
- Dentchev, T.; Milam, A.H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Dunaief, J.L. Amyloid-β is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Am. J. Ophthalmol. 2003, 136, 787. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, C.-W.; Zhou, Y.; Wong, W.Q.; Lee, L.C.; Ong, W.Y.; Yoon, S.O.; Hong, W.; Fu, X.-Y.; Soong, T.W. APP upregulation contributes to retinal ganglion cell degeneration via JNK3. Cell Death Differ. 2018, 25, 663–678. [Google Scholar] [CrossRef]
- Cerf, E.; Gustot, A.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 2011, 25, 1585–1595. [Google Scholar] [CrossRef]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef]
- Petrlova, J.; Hong, H.S.; Bricarello, D.A.; Harishchandra, G.; Lorigan, G.A.; Jin, L.W.; Voss, J.C. A differential association of Apolipoprotein E isoforms with the amyloid-β oligomer in solution. Proteins: Struct. Funct. Bioinform. 2011, 79, 402–416. [Google Scholar] [CrossRef]
- LaDu, M.J.; Falduto, M.T.; Manelli, A.M.; Reardon, C.A.; Getz, G.S.; Frail, D.E. Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 1994, 269, 23403–23406. [Google Scholar] [CrossRef]
- Huang, Y.-W.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef] [PubMed]
- Thakkinstian, A.; Bowe, S.; McEvoy, M.; Smith, W.; Attia, J. Association between apolipoprotein E polymorphisms and age-related macular degeneration: A HuGE review and meta-analysis. Am. J. Epidemiol. 2006, 164, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.K.; Simpson, J.A.; Richardson, A.J.; English, D.R.; Aung, K.Z.; Makeyeva, G.A.; Guymer, R.H.; Giles, G.G.; Hopper, J.; Robman, L.D. Apolipoprotein E gene associations in age-related macular degeneration: The Melbourne Collaborative Cohort Study. Am. J. Epidemiol. 2012, 175, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Baird, P.N.; Richardson, A.J.; Robman, L.D.; Dimitrov, P.N.; Tikellis, G.; McCarty, C.A.; Guymer, R.H. Apolipoprotein (APOE) gene is associated with progression of age-related macular degeneration (AMD). Hum. Mutat. 2006, 27, 337–342. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.J.; Patterson, C.C.; Chakravarthy, U.; Dasari, S.; Klaver, C.C.; Vingerling, J.R.; Ho, L.; de Jong, P.T.; Fletcher, A.E.; Young, I.S. Evidence of association of APOE with age-related macular degeneration-a pooled analysis of 15 studies. Hum. Mutat. 2011, 32, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Lim, A.; Liu, X.; Snellingen, T.; Wang, N.; Liu, N. Apolipoprotein E gene and age-related macular degeneration in a Chinese population. Mol. Vis. 2011, 17, 997. [Google Scholar]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Shimada, N.; Ichinose, S.; Sato, T.; Mochizuki, M.; Morita, I. Amyloid-β up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J. Cell. Physiol. 2009, 220, 119–128. [Google Scholar] [CrossRef]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef]
- Bradt, B.M.; Kolb, W.P.; Cooper, N.R. Complement-dependent proinflammatory properties of the Alzheimer’s disease β-peptide. J. Exp. Med. 1998, 188, 431–438. [Google Scholar] [CrossRef]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P. Complement activation by beta-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Kojima, A.; Shimada, N.; Nakahama, K.-i.; Safranova, O.; Iwata, N.; Saido, T.C.; Mochizuki, M. Altered function of factor I caused by amyloid β: Implication for pathogenesis of age-related macular degeneration from drusen. J. Immunol. 2008, 181, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ohno-Matsui, K.; Ichinose, S.; Sato, T.; Iwata, N.; Saido, T.C.; Hisatomi, T.; Mochizuki, M.; Morita, I. The potential role of amyloid β in the pathogenesis of age-related macular degeneration. J. Clin. Investig. 2005, 115, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Lee, S.E.; Guo, J.; Qu, W.; Hudson, B.I.; Schmidt, A.M.; Barile, G.R. RAGE ligand upregulation of VEGF secretion in ARPE-19 cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Wang, A.; To, E.; Gao, J.; Cao, S.; Cui, J.Z.; Matsubara, J.A. Vinpocetine inhibits amyloid-beta induced activation of NF-κB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp. Eye Res. 2014, 127, 49–58. [Google Scholar] [CrossRef]
- Jo, D.H.; Cho, C.S.; Kim, J.H.; Kim, J.H. Intracellular amyloid-β disrupts tight junctions of the retinal pigment epithelium via NF-κB activation. Neurobiol. Aging 2020, 95, 115–122. [Google Scholar] [CrossRef]
- Chen, L.; Bai, Y.; Zhao, M.; Jiang, Y. TLR4 inhibitor attenuates amyloid-β-induced angiogenic and inflammatory factors in ARPE-19 cells: Implications for age-related macular degeneration. Mol. Med. Rep. 2016, 13, 3249–3256. [Google Scholar] [CrossRef]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. BioMed Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- O’Neill, L.A. When signaling pathways collide: Positive and negative regulation of toll-like receptor signal transduction. Immunity 2008, 29, 12–20. [Google Scholar] [CrossRef]
- Lin, Q.; Li, M.; Fang, D.; Fang, J.; Su, S.B. The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int. Immunopharmacol. 2011, 11, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMP s and DAMP s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, M.; Bai, Y.; Huang, L.; Yu, W.; Bian, Z.; Zhao, M.; Li, X. Retinal ischemia/reperfusion injury is mediated by Toll-like receptor 4 activation of NLRP3 inflammasomes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5466–5475. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Shamsuddin, N. Retinal Muller glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS ONE 2012, 7, e29830. [Google Scholar]
- Tu, Z.; Portillo, J.-A.C.; Howell, S.; Bu, H.; Subauste, C.S.; Al-Ubaidi, M.R.; Pearlman, E.; Lin, F. Photoreceptor cells constitutively express functional TLR4. J. Neuroimmunol. 2011, 230, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Redfern, R.L.; McDermott, A.M. Toll-like receptors in ocular surface disease. Exp. Eye Res. 2010, 90, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Abedin, A.; Tsintzas, K.; Abedin, S.A.; Otri, A.M.; Hopkinson, A.; Mathew, M.; Dua, H.S. Increased expression of hepcidin and toll-like receptors 8 and 10 in viral keratitis. Cornea 2011, 30, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Said, D.G.; Dua, H.S. Human antimicrobial peptides in ocular surface defense. Prog. Retin. Eye Res. 2017, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A.; Wei, R.; Branch, M.J.; Sidney, L.E.; Amoaku, W.M. Expression of Toll-like receptors in human retinal and choroidal vascular endothelial cells. Exp. Eye Res. 2015, 138, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.V.; Nagineni, C.N.; Chin, M.S.; Hooks, J.J.; Detrick, B. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J. Neuroimmunol. 2004, 153, 7–15. [Google Scholar] [CrossRef]
- Ambati, J. Age-related macular degeneration and the other double helix the cogan lecture. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2166–2169. [Google Scholar] [CrossRef]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef]
- Landström, M. The TAK1–TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Titi-Lartey, O.; Mohammed, I.; Amoaku, W.M. Toll-like Receptor Signalling Pathways and the Pathogenesis of Retinal Diseases. Front. Ophthalmol. 2022, 2, 13. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.-F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-D.; Amaral, J.; Lee, J.W.; Rodriguez, I.R. 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS ONE 2014, 9, e100985. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.; Kwon, M.-Y.; Woo, J.M.; Chung, S.W. Oxidative stress-induced pentraxin 3 expression human retinal pigment epithelial cells is involved in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci. 2019, 20, 6028. [Google Scholar] [CrossRef]
- Mulfaul, K.; Ozaki, E.; Fernando, N.; Brennan, K.; Chirco, K.R.; Connolly, E.; Greene, C.; Maminishkis, A.; Salomon, R.G.; Linetsky, M. Toll-like receptor 2 facilitates oxidative damage-induced retinal degeneration. Cell Rep. 2020, 30, 2209–2224. [Google Scholar] [CrossRef]
- Zareparsi, S.; Buraczynska, M.; Branham, K.E.; Shah, S.; Eng, D.; Li, M.; Pawar, H.; Yashar, B.M.; Moroi, S.E.; Lichter, P.R. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2005, 14, 1449–1455. [Google Scholar] [CrossRef]
- Kaur, I.; Hussain, A.; Hussain, N.; Das, T.; Pathangay, A.; Mathai, A.; Hussain, A.; Nutheti, R.; Nirmalan, P.K.; Chakrabarti, S. Analysis of CFH, TLR4, and APOE polymorphism in India suggests the Tyr402His variant of CFH to be a global marker for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3729–3735. [Google Scholar] [CrossRef]
- Despriet, D.D.; Bergen, A.A.; Merriam, J.E.; Zernant, J.; Barile, G.R.; Smith, R.T.; Barbazetto, I.A.; van Soest, S.; Bakker, A.; de Jong, P.T. Comprehensive analysis of the candidate genes CCL2, CCR2, and TLR4 in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 364–371. [Google Scholar] [CrossRef]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [PubMed]
- Viiri, J.; Amadio, M.; Marchesi, N.; Hyttinen, J.M.; Kivinen, N.; Sironen, R.; Rilla, K.; Akhtar, S.; Provenzani, A.; D’Agostino, V.G. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS ONE 2013, 8, e69563. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between autophagy and the ubiquitin-proteasome system and its role in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [PubMed]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2023, 19, 388–400. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Surgucheva, I.; Ninkina, N.; Buchman, V.L.; Grasing, K.; Surguchov, A. Protein aggregation in retinal cells and approaches to cell protection. Cell. Mol. Neurobiol. 2005, 25, 1051–1066. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Maruszczak, K.K.; Jung, M.; Rasool, S.; Trempe, J.-F.; Rapaport, D. The role of the individual TOM subunits in the association of PINK1 with depolarized mitochondria. J. Mol. Med. 2022, 100, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Takahashi, Y.; He, H.; Hattori, T.; Chen, C.; Liang, X.; Chen, H.; Young, M.M.; Wang, H.-G. TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 2019, 28, 1744–1757. [Google Scholar] [CrossRef] [PubMed]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, C.E.; Wooldredge, A.C.; Fong, B.; Kennedy, B.K.; Zhou, C. Tom70-based transcriptional regulation of mitochondrial biogenesis and aging. Elife 2022, 11, e75658. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9, 1122. [Google Scholar] [CrossRef]
- Roses, A.; Sundseth, S.; Saunders, A.; Gottschalk, W.; Burns, D.; Lutz, M. Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 687–694. [Google Scholar] [CrossRef]
- Tang, W.; Guo, J.; Gu, R.; Lei, B.; Ding, X.; Ma, J.; Xu, G. MicroRNA-29b-3p inhibits cell proliferation and angiogenesis by targeting VEGFA and PDGFB in retinal microvascular endothelial cells. Mol. Vis. 2020, 26, 64–75. [Google Scholar]
- Hu, Y. Evaluation of miR-29c inhibits endotheliocyte migration and angiogenesis of human endothelial cells by suppressing the insulin like growth factor 1. Am. J. Transl. Res. 2015, 7, 866–877. [Google Scholar]
- Smyth, A.; Callaghan, B.; Willoughby, C.E.; O’Brien, C. The Role of miR-29 Family in TGF-β Driven Fibrosis in Glaucomatous Optic Neuropathy. Int. J. Mol. Sci. 2022, 23, 10216. [Google Scholar] [PubMed]
- Toyono, T.; Usui, T.; Villarreal, G., Jr.; Kallay, L.; Matthaei, M.; Vianna, L.M.M.; Zhu, A.Y.; Kuroda, M.; Amano, S.; Jun, A.S. MicroRNA-29b Overexpression Decreases Extracellular Matrix mRNA and Protein Production in Human Corneal Endothelial Cells. Cornea 2016, 35, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cai, Y.; Rong, X.; Chen, J.; Zheng, D.; Chen, L.; Zhang, J.; Luo, R.; Zhao, P.; Ruan, J. MiR-661 promotes tumor invasion and metastasis by directly inhibiting RB1 in non small cell lung cancer. Mol. Cancer 2017, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P.V. MicroRNAs as diagnostic and prognostic biomarkers of age-related macular degeneration: Advances and limitations. Neural Regen. Res. 2021, 16, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Pawlick, J.S.; Zuzic, M.; Pasquini, G.; Swiersy, A.; Busskamp, V. MiRNA Regulatory Functions in Photoreceptors. Front. Cell Dev. Biol. 2021, 8, 620249. [Google Scholar] [CrossRef]
- Chu-Tan, J.A.; Rutar, M.; Saxena, K.; Aggio-Bruce, R.; Essex, R.W.; Valter, K.; Jiao, H.; Fernando, N.; Wooff, Y.; Madigan, M.C.; et al. MicroRNA-124 Dysregulation is Associated With Retinal Inflammation and Photoreceptor Death in the Degenerating Retina. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4094–4105. [Google Scholar] [CrossRef]
- Tomesz, A.; Szabo, L.; Molnar, R.; Deutsch, A.; Darago, R.; Raposa, B.L.; Ghodratollah, N.; Varjas, T.; Nemeth, B.; Orsos, Z.; et al. Changes in miR-124-1, miR-212, miR-132, miR-134, and miR-155 Expression Patterns after 7,12-Dimethylbenz(a)anthracene Treatment in CBA/Ca Mice. Cells 2022, 11, 1020. [Google Scholar] [CrossRef]
- Tomé, M.; López-Romero, P.; Albo, C.; Sepúlveda, J.C.; Fernández-Gutiérrez, B.; Dopazo, A.; Bernad, A.; González, M.A. miR-335 orchestrates cell proliferation, migration and differentiation in human mesenchymal stem cells. Cell Death Differ. 2011, 18, 985–995. [Google Scholar] [CrossRef]
- Lu, W.; Ma, Y.Y.; Shao, Q.Q.; Liang, J.; Qi, T.T.; Huang, Y.; Wang, Q.J. ROS/p53/miR-335-5p/Sp1 axis modulates the migration and epithelial to mesenchymal transition of JEG-3 cells. Mol. Med. Rep. 2020, 21, 1208–1216. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Z.; Zhang, S.; Zhu, P.; Ko, J.K.; Yung, K.K. MUC1: Structure, Function, and Clinic Application in Epithelial Cancers. Int. J. Mol. Sci. 2021, 22, 6567. [Google Scholar] [CrossRef]
- Salo, A.M.; Myllyharju, J. Prolyl and lysyl hydroxylases in collagen synthesis. Exp. Dermatol. 2021, 30, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Ashok, G.; Miryala, S.K.; Saju, M.T.; Anbarasu, A.; Ramaiah, S. FN1 encoding fibronectin as a pivotal signaling gene for therapeutic intervention against pancreatic cancer. Mol. Genet. Genom. 2022, 297, 1565–1580. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.; Jomary, C.; Mullins, R.; Cree, A.; Chen, X.; MacLeod, A.; Jones, S.; Collins, A.; Stone, E.; Lotery, A. Association between the SERPING1 gene and age-related macular degeneration: A two-stage case–control study. Lancet 2008, 372, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Zauhar, R.; Biber, J.; Jabri, Y.; Kim, M.; Hu, J.; Kaplan, L.; Pfaller, A.; Schäfer, N.; Enzmann, V.; Schlötzer-Schrehardt, U.; et al. As in Real Estate, Location Matters: Cellular Expression of Complement Varies Between Macular and Peripheral Regions of the Retina and Supporting Tissues. Front. Immunol. 2022, 13, 895519. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, X.; Sun, M.; Zhang, Z.; Sun, D. Potential epigenetic molecular regulatory networks in ocular neovascularization. Front. Genet. 2022, 13, 970224. [Google Scholar] [CrossRef]
- Duan, M.-Y.; Li, M.; Tian, H.; Tang, G.; Yang, Y.-C.; Peng, N.-C. Down-regulation of lncRNA NEAT1 regulated by miR-194-5p/DNMT3A facilitates acute myeloid leukemia. Blood Cells Mol. Dis. 2020, 82, 102417. [Google Scholar] [CrossRef]
- Chen, Q.; Cai, J.; Wang, Q.; Wang, Y.; Liu, M.; Yang, J.; Zhou, J.; Kang, C.; Li, M.; Jiang, C. Long Noncoding RNA NEAT1, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/β-Catenin Pathway by Scaffolding EZH2Oncogenic Function of NEAT1 in Glioblastoma. Clin. Cancer Res. 2018, 24, 684–695. [Google Scholar] [CrossRef]
- Zhou, Z.-W.; Zheng, L.-J.; Ren, X.; Li, A.-P.; Zhou, W.-S. LncRNA NEAT1 facilitates survival and angiogenesis in oxygen-glucose deprivation (OGD)-induced brain microvascular endothelial cells (BMECs) via targeting miR-377 and upregulating SIRT1, VEGFA, and BCL-XL. Brain Res. 2019, 1707, 90–98. [Google Scholar] [CrossRef]
- Hu, X.; Li, F.; He, J.; Yang, J.; Jiang, Y.; Jiang, M.; Wei, D.; Chang, L.; Hejtmancik, J.F.; Hou, L. LncRNA NEAT1 recruits SFPQ to regulate MITF splicing and control RPE cell proliferation. Investig. Ophthalmol. Vis. Sci. 2021, 62, 18. [Google Scholar] [CrossRef]
- Yang, M.; So, K.-F.; Lo, A.C.Y.; Lam, W.C. The effect of Lycium barbarum polysaccharides on pyroptosis-associated amyloid β1-40 oligomers-induced adult retinal pigment epithelium 19 cell damage. Int. J. Mol. Sci. 2020, 21, 4658. [Google Scholar] [CrossRef]
- Chen, H.; Deng, Y.; Gan, X.; Li, Y.; Huang, W.; Lu, L.; Wei, L.; Su, L.; Luo, J.; Zou, B. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol. Neurodegener. 2020, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gan, X.; Li, Y.; Gu, J.; Liu, Y.; Deng, Y.; Wang, X.; Hong, Y.; Hu, Y.; Su, L. NLRP12-and NLRC4-mediated corneal epithelial pyroptosis is driven by GSDMD cleavage accompanied by IL-33 processing in dry eye. Ocul. Surf. 2020, 18, 783–794. [Google Scholar] [CrossRef]
- Zha, X.; Xi, X.; Fan, X.; Ma, M.; Zhang, Y.; Yang, Y. Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging 2020, 12, 8137. [Google Scholar] [CrossRef] [PubMed]
- Shome, A.; Mugisho, O.O.; Niederer, R.L.; Rupenthal, I.D. Blocking the inflammasome: A novel approach to treat uveitis. Drug Discov. Today 2021, 26, 2839–2857. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Rong, R.; Xia, X. Spotlight on pyroptosis: Role in pathogenesis and therapeutic potential of ocular diseases. J. Neuroinflammation 2022, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jin, X.; Wang, J.; Li, X.; Zhang, H. Dexamethasone attenuates dry eye-induced pyroptosis by regulating the KCNQ1OT1/miR-214 cascade. Steroids 2022, 186, 109073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gong, Y.; Li, N.; Liu, X.; Zhang, Y.; Ye, F.; Guo, Q.; Zheng, J. Long noncoding RNA Kcnq1ot1 promotes sC5b-9-induced podocyte pyroptosis by inhibiting miR-486a-3p and upregulating NLRP3. Am. J. Physiol.-Cell Physiol. 2021, 320, C355–C364. [Google Scholar] [CrossRef]
- Li, Z.; Ren, R.; Wang, L.; Wang, Z.; Zong, X.; Sun, P.; Zhu, C.; Guo, M.; Guo, G.; Hu, G. lncRNA KCNQ1OT1 promotes emt, angiogenesis, and stemness of pituitary adenoma by upregulation of RAB11A. J. Oncol. 2022, 2022, 4474476. [Google Scholar] [CrossRef]
- Ma, L.; Gao, J.; Zhang, N.; Wang, J.; Xu, T.; Lei, T.; Zou, X.; Wei, C.; Wang, Z. Long noncoding RNA SNHG17: A novel molecule in human cancers. Cancer Cell Int. 2022, 22, 104. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Tang, D.; Li, M.; Zhao, P.; Yang, W.; Shu, L.; Wang, J.; He, Z.; Li, Y. SNHG17 promotes the proliferation and migration of colorectal adenocarcinoma cells by modulating CXCL12-mediated angiogenesis. Cancer Cell Int. 2020, 20, 566. [Google Scholar] [CrossRef]
- Wang, W.; Min, L.; Qiu, X.; Wu, X.; Liu, C.; Ma, J.; Zhang, D.; Zhu, L. Biological function of long non-coding RNA (LncRNA) Xist. Front. Cell Dev. Biol. 2021, 9, 645647. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wan, G.; Peng, G.; Yan, P.; Qian, C.; Li, F. Long non-coding RNA XIST regulates hyperglycemia-associated apoptosis and migration in human retinal pigment epithelial cells. Biomed. Pharmacother. 2020, 125, 109959. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.R.; Jarrar, Z.; Wormald, R.; Cook, D.G.; Fletcher, A.; Owen, C.G. Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: A meta-analysis. Ophthalmology 2012, 119, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Bai, X.; Liu, C.; Hu, Z. Long noncoding RNA XIST participates hypoxia-induced angiogenesis in human brain microvascular endothelial cells through regulating miR-485/SOX7 axis. Am. J. Transl. Res. 2019, 11, 6487. [Google Scholar] [PubMed]
- Wang, C.; Dong, J.; Sun, J.; Huang, S.; Wu, F.; Zhang, X.; Pang, D.; Fu, Y.; Li, L. Silencing of lncRNA XIST impairs angiogenesis and exacerbates cerebral vascular injury after ischemic stroke. Mol. Ther.-Nucleic Acids 2021, 26, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 32, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Kleppisch, T. NO/cGMP-dependent modulation of synaptic transmission. In Pharmacology of Neurotransmitter Release; Springer: Berlin/Heidelberg, Germany, 2008; pp. 529–560. [Google Scholar]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.; Higgs, E. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Giuffrida Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Baba, H.; Suzuki, T.; Arai, H.; Emson, P.C. Expression of nNOS and soluble guanylate cyclase in schizophrenic brain. Neuroreport 2004, 15, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Djordjević, V.V.; Stojanović, I.; Stanković-Ferlež, D.; Ristić, T.; Lazarević, D.; Ćosić, V.; Djordjević, V.B. Plasma nitrite/nitrate concentrations in patients with schizophrenia. Clin. Chem. Lab. Med. 2010, 48, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.K.; Leonard, S.; Reddy, R.D. Increased nitric oxide radicals in postmortem brain from patients with schizophrenia. Schizophr. Bull. 2004, 30, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Yanik, M.; Vural, H.; Kocyigit, A.; Tutkun, H.; Zoroglu, S.S.; Herken, H.; Savaş, H.A.; Köylü, A.; Akyol, Ö. Is the arginine-nitric oxide pathway involved in the pathogenesis of schizophrenia? Neuropsychobiology 2003, 47, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Uzbay, T.; Goktalay, G.; Kayir, H.; Eker, S.S.; Sarandol, A.; Oral, S.; Buyukuysal, L.; Ulusoy, G.; Kirli, S. Increased plasma agmatine levels in patients with schizophrenia. J. Psychiatr. Res. 2013, 47, 1054–1060. [Google Scholar] [CrossRef]
- Zeinoddini, A.; Ahadi, M.; Farokhnia, M.; Rezaei, F.; Tabrizi, M.; Akhondzadeh, S. L-lysine as an adjunct to risperidone in patients with chronic schizophrenia: A double-blind, placebo-controlled, randomized trial. J. Psychiatr. Res. 2014, 59, 125–131. [Google Scholar] [CrossRef]
- Wass, C.; Klamer, D.; Katsarogiannis, E.; Pålsson, E.; Svensson, L.; Fejgin, K.; Bogren, I.-B.; Engel, J.A.; Rembeck, B. L-lysine as adjunctive treatment in patients with schizophrenia: A single-blinded, randomized, cross-over pilot study. BMC Med. 2011, 9, 40. [Google Scholar] [CrossRef]
- Ohnishi, T.; Balan, S.; Toyoshima, M.; Maekawa, M.; Ohba, H.; Watanabe, A.; Iwayama, Y.; Fujita, Y.; Tan, Y.; Hisano, Y. Investigation of betaine as a novel psychotherapeutic for schizophrenia. EBioMedicine 2019, 45, 432–446. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef]
- Jiang, H.; Wu, M.; Liu, Y.; Song, L.; Li, S.; Wang, X.; Zhang, Y.f.; Fang, J.; Wu, S. Serine racemase deficiency attenuates choroidal neovascularization and reduces nitric oxide and VEGF levels by retinal pigment epithelial cells. J. Neurochem. 2017, 143, 375–388. [Google Scholar] [CrossRef]
- Ando, A.; Yang, A.; Nambu, H.; Campochiaro, P.A. Blockade of nitric-oxide synthase reduces choroidal neovascularization. Mol. Pharmacol. 2002, 62, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Pecháňová, O.; Šimko, F. The role of nitric oxide in the maintenance of vasoactive balance. Physiol. Res. 2007, 56, S7–S16. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.L.; Uppal, K.; Williamson, S.M.; Liu, K.; Burgess, L.G.; Tran, V.; Umfress, A.C.; Jarrell, K.L.; Bailey, J.N.C.; Agarwal, A. The carnitine shuttle pathway is altered in patients with neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4978–4985. [Google Scholar] [CrossRef]
- Elner, S.G.; Elner, V.M.; Field, M.G.; Park, S.; Heckenlively, J.R.; Petty, H.R. Retinal flavoprotein autofluorescence as a measure of retinal health. Trans. Am. Ophthalmol. Soc. 2008, 106, 215. [Google Scholar] [PubMed]
- Chao de la Barca, J.M.; Rondet-Courbis, B.; Ferré, M.; Muller, J.; Buisset, A.; Leruez, S.; Plubeau, G.; Macé, T.; Moureauzeau, L.; Chupin, S. A plasma metabolomic profiling of exudative age-related macular degeneration showing carnosine and mitochondrial deficiencies. J. Clin. Med. 2020, 9, 631. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Yun, J.; Yoon, Y.D.; Park, S.-I.; Seo, Y.-J.; Park, W.-S.; Chu, H.Y.; Park, K.H.; Lee, M.Y.; Lee, C.W. Blue light effect on retinal pigment epithelial cells by display devices. Integr. Biol. 2017, 9, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Elner, V.M.; Park, S.; Cornblath, W.; Hackel, R.; Petty, H.R. Flavoprotein autofluorescence detection of early ocular dysfunction. Arch. Ophthalmol. 2008, 126, 259–260. [Google Scholar] [CrossRef]
- Chen, A.X.; Conti, T.F.; Hom, G.L.; Greenlee, T.E.; Raimondi, R.; Briskin, I.N.; Rich, C.A.; Kampani, R.; Engel, R.; Sharma, S. Functional imaging of mitochondria in retinal diseases using flavoprotein fluorescence. Eye 2021, 35, 74–92. [Google Scholar] [CrossRef]
- Ahsanuddin, S.; Rios, H.A.; Otero-Marquez, O.A.; Macanian, J.; Zhou, D.; Rich, C.; Rosen, R. Flavoprotein Fluorescence (FPF) Elevation is a Marker of Mitochondrial Oxidative Stress in Patients with Retinal Disease. Front. Ophthalmol. 2023, 3, 1110501. [Google Scholar] [CrossRef]
- Field, M.G.; Comer, G.M.; Kawaji, T.; Petty, H.R.; Elner, V.M. Noninvasive imaging of mitochondrial dysfunction in dry age-related macular degeneration. Ophthalmic Surg. Lasers Imaging Retin. 2012, 43, 358–365. [Google Scholar] [CrossRef]
- Field, M.G.; Yang, D.; Bian, Z.-M.; Petty, H.R.; Elner, V.M. Retinal flavoprotein fluorescence correlates with mitochondrial stress, apoptosis, and chemokine expression. Exp. Eye Res. 2011, 93, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Andrade Romo, J.S.; Lynch, G.; Liu, K.; Kim, D.; Jansen, M.; Field, M.G.; Elner, V.M.; Rosen, R.B. Flavoprotein fluorescence correlation with visual acuity response in patients receiving anti-VEGF injection for diabetic macular edema. Oxidative Med. Cell. Longev. 2018, 2018, 3567306. [Google Scholar] [CrossRef] [PubMed]
- Geyman, L.S.; Suwan, Y.; Garg, R.; Field, M.G.; Krawitz, B.D.; Mo, S.; Pinhas, A.; Ritch, R.; Rosen, R.B. Noninvasive detection of mitochondrial dysfunction in ocular hypertension and primary open-angle glaucoma. J. Glaucoma 2018, 27, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Yamashina, S.; Ikejima, K.; Rusyn, I.; Sato, N. Glycine as a potent anti-angiogenic nutrient for tumor growth. J. Gastroenterol. Hepatol. 2007, 22, S62–S64. [Google Scholar] [CrossRef]
- Guo, D.; Murdoch, C.E.; Xu, H.; Shi, H.; Duan, D.D.; Ahmed, A.; Gu, Y. Vascular endothelial growth factor signaling requires glycine to promote angiogenesis. Sci. Rep. 2017, 7, 14749. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Tamura, K.; Sato, M.; Fujita, M.; Tamura, M. Glycine exerts dose-dependent biphasic effects on vascular development of zebrafish embryos. Biochem. Biophys. Res. Commun. 2020, 527, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Gholami, S.; Kamali, Y.; Rostamzad, M.R. Glycine supplementation ameliorates retinal neuronal damage in an experimental model of diabetes in rats: A light and electron microscopic study. J. Ophthalmic Vis. Res. 2019, 14, 448. [Google Scholar] [CrossRef]
- Xia, M.; Zhang, F. Amino Acids Metabolism in Retinopathy: From Clinical and Basic Research Perspective. Metabolites 2022, 12, 1244. [Google Scholar] [CrossRef]
- Deng, Y.; Shuai, P.; Wang, H.; Zhang, S.; Li, J.; Du, M.; Huang, P.; Qu, C.; Huang, L. Untargeted metabolomics for uncovering plasma biological markers of wet age-related macular degeneration. Aging 2021, 13, 13968. [Google Scholar] [CrossRef]
- Han, G.; Wei, P.; He, M.; Teng, H.; Chu, Y. Metabolomic Profiling of the Aqueous Humor in Patients with Wet Age-Related Macular Degeneration Using UHPLC–MS/MS. J. Proteome Res. 2020, 19, 2358–2366. [Google Scholar] [CrossRef]
- Luo, D.; Deng, T.; Yuan, W.; Deng, H.; Jin, M. Plasma metabolomic study in Chinese patients with wet age-related macular degeneration. BMC Ophthalmol. 2017, 17, 165. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Austin, S.A. Endothelial nitric oxide: Protector of a healthy mind. Eur. Heart J. 2014, 35, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Fleszar, M.G.; Wiśniewski, J.; Zboch, M.; Diakowska, D.; Gamian, A.; Krzystek-Korpacka, M. Targeted metabolomic analysis of nitric oxide/L-arginine pathway metabolites in dementia: Association with pathology, severity, and structural brain changes. Sci. Rep. 2019, 9, 13764. [Google Scholar] [CrossRef] [PubMed]
- Zaciragic, A. New insights into possible role of NOS-NO-ADMA pathway dysfunction in the development of cognitive decline and dementia: Exploring the vascular features of Alzheimer’s disease. Int. J. Neurol. Res. 2015, 1, 188–190. [Google Scholar] [CrossRef][Green Version]
- Sarı, İ.; Erşan, S.; Özmen, E.; Ayan, D.; Erşan, E.; Berisha, A.; Kaya, S. Changes in arginine metabolism in advanced Alzheimer’s patients: Experimental and theoretical analyses. J. Mol. Struct. 2023, 1282, 135254. [Google Scholar] [CrossRef]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against D-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflammation 2020, 17, 303. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Lo Surdo, P.; Calderone, A.; Iannuccelli, M.; Licata, L.; Peluso, D.; Castagnoli, L.; Cesareni, G.; Perfetto, L. DISNOR: A disease network open resource. Nucleic Acids Res. 2018, 46, D527–D534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. In ‘In Silico’ Simulation of Biological Processes: Novartis Foundation Symposium 247; Wiley: New York, NY, USA, 2002; pp. 91–103. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000Research 2015, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, S.; He, Z.; Reilly, J.; Zeng, Z.; Strang, N.; Shu, X. Metabolomics in Retinal Diseases: An Update. Biology 2021, 10, 944. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.N.; Green, B.D.; Thompson, R.B.; den Hollander, A.I.; Lengyel, I.; Eye-Risk Consortium. Metabolomics and Age-Related Macular Degeneration. Metabolites 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.W.; Wang, Y.; Pan, C.W. Metabolomics in Age-Related Macular Degeneration: A Systematic Review. Investig. Ophthalmol. Vis. Sci. 2020, 61, 13. [Google Scholar] [CrossRef] [PubMed]
- Lains, I.; Duarte, D.; Barros, A.S.; Martins, A.S.; Gil, J.; Miller, J.B.; Marques, M.; Mesquita, T.; Kim, I.K.; Cachulo, M.D.L.; et al. Human plasma metabolomics in age-related macular degeneration (AMD) using nuclear magnetic resonance spectroscopy. PLoS ONE 2017, 12, e0177749. [Google Scholar] [CrossRef]
- Han, X.; Gharahkhani, P.; Mitchell, P.; Liew, G.; Hewitt, A.W.; MacGregor, S. Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration. J. Hum. Genet. 2020, 65, 657–665. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- Maguire, M.G.; Ying, G.S.; Jaffe, G.J.; Toth, C.A.; Daniel, E.; Grunwald, J.; Martin, D.F.; Hagstrom, S.A.; Group, C.R. Single-Nucleotide Polymorphisms Associated With Age-Related Macular Degeneration and Lesion Phenotypes in the Comparison of Age-Related Macular Degeneration Treatments Trials. JAMA Ophthalmol. 2016, 134, 674–681. [Google Scholar] [CrossRef]
- Yan, Q.; Jiang, Y.; Huang, H.; Swaroop, A.; Chew, E.Y.; Weeks, D.E.; Chen, W.; Ding, Y. Genome-Wide Association Studies-Based Machine Learning for Prediction of Age-Related Macular Degeneration Risk. Transl. Vis. Sci. Technol. 2021, 10, 29. [Google Scholar] [CrossRef]
- Yan, Q.; Ding, Y.; Liu, Y.; Sun, T.; Fritsche, L.G.; Clemons, T.; Ratnapriya, R.; Klein, M.L.; Cook, R.J.; Liu, Y.; et al. Genome-wide analysis of disease progression in age-related macular degeneration. Hum. Mol. Genet. 2018, 27, 929–940. [Google Scholar] [CrossRef]
- Cipriani, V.; Tierney, A.; Griffiths, J.R.; Zuber, V.; Sergouniotis, P.I.; Yates, J.R.W.; Moore, A.T.; Bishop, P.N.; Clark, S.J.; Unwin, R.D. Beyond factor H: The impact of genetic-risk variants for age-related macular degeneration on circulating factor-H-like 1 and factor-H-related protein concentrations. Am. J. Hum. Genet. 2021, 108, 1385–1400. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; MacGregor, S.; Han, X.; Francis, J.; Harris, C.; Kavanagh, D.; Lotery, A.; Waheed, N.; SCOPE Study Group. Evaluating a Causal Relationship between Complement Factor I Protein Level and Advanced Age-Related Macular Degeneration Using Mendelian Randomization. Ophthalmol. Sci. 2022, 2, 100146. [Google Scholar] [CrossRef] [PubMed]
- Waksmunski, A.R.; Igo, R.P., Jr.; Song, Y.E.; Cooke Bailey, J.N.; Laux, R.; Fuzzell, D.; Fuzzell, S.; Adams, L.D.; Caywood, L.; Prough, M.; et al. Rare variants and loci for age-related macular degeneration in the Ohio and Indiana Amish. Hum. Genet. 2019, 138, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Strunz, T.; Pollmann, M.; Gamulescu, M.A.; Tamm, S.; Weber, B.H.F. Genetic Association Analysis of Anti-VEGF Treatment Response in Neovascular Age-Related Macular Degeneration. Int. J. Mol. Sci. 2022, 23, 6094. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395–7400. [Google Scholar] [CrossRef] [PubMed]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Holliday, E.G.; Smith, A.V.; Cornes, B.K.; Buitendijk, G.H.; Jensen, R.A.; Sim, X.; Aspelund, T.; Aung, T.; Baird, P.N.; Boerwinkle, E.; et al. Insights into the genetic architecture of early stage age-related macular degeneration: A genome-wide association study meta-analysis. PLoS ONE 2013, 8, e53830. [Google Scholar] [CrossRef]
- Black, J.R.; Clark, S.J. Age-related macular degeneration: Genome-wide association studies to translation. Genet. Med. 2016, 18, 283–289. [Google Scholar] [CrossRef]
- Xue, Z.; Yuan, J.; Chen, F.; Yao, Y.; Xing, S.; Yu, X.; Li, K.; Wang, C.; Bao, J.; Qu, J.; et al. Genome-wide association meta-analysis of 88,250 individuals highlights pleiotropic mechanisms of five ocular diseases in UK Biobank. EBioMedicine 2022, 82, 104161. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Backes, C.; Khaleeq, Q.T.; Meese, E.; Keller, A. miEAA: microRNA enrichment analysis and annotation. Nucleic Acids Res. 2016, 44, W110–W116. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, M.; Yasukawa, T. A fuzzy-logic-based approach to qualitative modeling. IEEE Trans. Fuzzy Syst. 1993, 1, 7. [Google Scholar] [CrossRef]
- Barro, S.; Marín, R. Fuzzy Logic in Medicine; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001; Volume 83. [Google Scholar]
- Raza, K. Fuzzy logic based approaches for gene regulatory network inference. Artif. Intell. Med. 2019, 97, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, J. Long Short-Term Memory Networks with Python: Develop Sequence Prediction Models with Deep Learning; Machine Learning Mastery: Vermont, Australia, 2017. [Google Scholar]
- Colin, R. Genetic algorithms. In Modern Heuristic Techniques for Combinatorial Problems; Wiley: New York, NY, USA, 2010; pp. 151–196. [Google Scholar]
- McCall, J. Genetic algorithms for modelling and optimisation. J. Comput. Appl. Math. 2005, 184, 205–222. [Google Scholar] [CrossRef]
- Al-Tashi, Q.; Kadir, S.J.A.; Rais, H.M.; Mirjalili, S.; Alhussian, H. Binary optimization using hybrid grey wolf optimization for feature selection. IEEE Access 2019, 7, 39496–39508. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L-Leucine | Inosine | L-Glutamic Acid | L-Aspartate |
|---|---|---|---|
| Zinc (II) ion | SM(d18:1/18:0) | L-Alanine | L-Cystine |
| cis-Aconitic acid | Dopaquinone | L-Serine | L-Lysine |
| L-Aspartic acid | L-Histidine | Betaine | Adenosine |
| Glutathione | Cytidine | L-Arginine | L-Valine |
| Urea | Glycerol | L-Glutamine | Glycine |
| Taurine |
| AMD-Related Results | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Gene Name | miRNAs | lncRNAs | Metabolites | ||||
| Disease Module | Gene Regulatory Network | AMD-SNP Data | Shared Metabolites between the MMIN and MGDIN Networks via Degree and Betweenness centralities | Fuzzy Logic Model + Deep Learning + Genetic Algorithm | ||||
| 1 | SLC16A8 | PDGFA | CFH | hsa-mir-450a-1-3p | NEAT1 | Pyruvic acid | L-Leucine | L-Glutamic acid |
| 2 | RPGR | COL1A1 | VEGFA | hsa-mir-661 | SNHG17 | Glycine | Zinc (II) ion | L-Alanine |
| 3 | ERCC6 | COL1A2 | APOE | hsa-mir-335-5p | KCNQ1QT1 | Citric acid | cis-Aconitic acid | L-Serine |
| 4 | NMNAT1 | COL4A1 | TOMM40 | hsa-mir-124-3p | XIST | L-Lysine | L-Aspartic acid | Betaine |
| 5 | VEGFA | COL14A1 | PVRL2 | hsa-mir-29b-3p | L-Alanine | Glutathione | L-Arginine | |
| 6 | TNFRSF10A | COL18A1 | ABCA1 | hsa-mir-29c-3p | L-Arginine | Urea | L-Glutamine | |
| 7 | SQSTM1 | UBC | L-Methionine | Taurine | L-Aspartate | |||
| 8 | C9 | C1S | Inosine | L-Cystine | ||||
| 9 | APOE | P3H3 | SM(d18:1/18:0) | L-Lysine | ||||
| 10 | TLR4 | FN1 | Dopaquinone | Adenosine | ||||
| 11 | ABCA4 | MUC1 | L-Histidine | L-Valine | ||||
| 12 | TIMP3 | BMP1 | Cytidine | Glycine | ||||
| 13 | C3 | SERPING1 | Glycerol | |||||
| 14 | CFH | SPDYE1 | ||||||
| 15 | C2 | |||||||
| 16 | CFI | |||||||
| 17 | CFB | |||||||
| 18 | C1QTNF5 | |||||||
| AMD-Related Results | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | Common Metabolites | AMD-SNPs | Common SNPs | |||||
| AMD and Schizophrenia | AMD and Alzheimer’s Disease | AMD and Multiple Sclerosis | AMD and Schizophrenia | AMD and Parkinson’s Disease | AMD and Alzheimer’s Disease | |||
| 1 | L-Lactic acid | Glycine | rs17576 | rs114254831 | rs3025039 | rs699947 | rs3025039 | rs800292 |
| 2 | Cortisol | L-Lysine | rs1061170 | rs116503776 | rs699947 | rs2230205 | rs429358 | rs9332739 |
| 3 | Cholesterol | L-Arginine | rs699947 | rs2740488 | rs11755724 | rs1061170 | rs699947 | rs1061170 |
| 4 | (R)-3-Hydroxybutyric acid | Calcium | rs429358 | rs12678919 | rs7412 | rs4151667 | ||
| 5 | Glycine | rs2043085 | rs7679 | rs2230199 | rs2075650 | |||
| 6 | L-Lysine | rs3764261 | rs3918242 | rs1061170 | rs2740488 | |||
| 7 | L-Arginine | rs4073 | rs800292 | rs429358 | rs7412 | |||
| 8 | Pyruvic acid | rs243865 | rs3025039 | rs699947 | ||||
| 9 | rs964184 | rs7412 | rs2736911 | |||||
| 10 | rs2075650 | rs2070895 | rs429358 | |||||
| 11 | rs174547 | rs1800775 | rs6857 | |||||
| 12 | rs2071559 | rs17577 | ||||||
| 13 | rs1800961 | rs1065489 | ||||||
| 14 | rs6857 | rs10468017 | ||||||
| 15 | rs1837253 | rs17231506 | ||||||
| Pathway Enrichment Analysis | ||||||
|---|---|---|---|---|---|---|
| NO | Genes | miRNAs | Metabolites | |||
| Enrichment Analysis | Metabolic Pathway Analysis | Joint Pathway Analysis | ||||
| KEGG | SMPDB | |||||
| 1 | Staphylococcus aureus infection (FDR = 0.000152) | Terpenoid backbone biosynthesis (FDR = 0.0022855) | Aminoacyl–tRNA biosynthesis (FDR = 7.02 × 10−12) | Urea cycle (FDR = 0.0308) | Aminoacyl–tRNA biosynthesis (FDR = 8.02 × 10−12) | Alanine, aspartate, and glutamate metabolism (FDR = 0.0005232) |
| 2 | Protein digestion and absorption (FDR = 0.00031) | Arachidonic acid metabolism (FDR = 0.0280283) | Glyoxylate and dicarboxylate metabolism (FDR = 0.000176) | Glycine and serine metabolism (FDR = 0.0458) | Alanine, aspartate, and glutamate metabolism (FDR = 0.021047) | Glycine, serine, and threonine metabolism (FDR = 0.000031504) |
| 3 | ECM–receptor interaction (FDR = 0.00427) | Hippo signaling pathway—multiple species (FDR = 0.0280283) | Arginine biosynthesis (FDR = 0.00562) | Glycine, Serine, and Threonine metabolism (FDR = 0.029711) | Arginine biosynthesis (FDR = 0.00083089) | |
| 4 | Amoebiasis (FDR = 0.00492) | Base excision repair (FDR = 0.0311627) | Alanine, aspartate, and glutamate metabolism (FDR = 0.0204) | Taurine and Hypotaurine Metabolism (FDR = 0.03605) | Sphingolipid metabolism (FDR = 0.0072172) | |
| 5 | AGE-RAGE signaling pathway in diabetes complications (FDR = 0.00492) | Complement and coagulation cascades (FDR = 0.0311627) | Sphingolipid metabolism (FDR = 0.027) | Cysteine and methionine metabolism (FDR = 0.000016837) | ||
| 6 | Focal adhesion (FDR = 0.00492) | Glycine, serine, and threonine metabolism (FDR = 0.0288) | Arginine biosynthesis (FDR = 0.000831) | |||
| 7 | Complement and coagulation cascades (FDR = 0.0401) | Cysteine and methionine metabolism (FDR = 0.0288) | ||||
| 8 | Valine, leucine, and isoleucine biosynthesis (FDR = 0.0354) | |||||
| 9 | Taurine and hypotaurine metabolism (FDR = 0.0354) | |||||
| Pathways | Enrichment Analysis | Pathway Analysis Based on KEGG | Joint Pathway Analysis | Final Output | ||||
|---|---|---|---|---|---|---|---|---|
| KEGG | SMPDB | Relative Betweenness Centrality (R-b C) | Out-Degree Centrality (O-d C) | Degree | Betweenness | Closeness | ||
| Aminoacyl–tRNA Biosynthesis | + | -- | -- | + | -- | -- | -- | 2 |
| Glyoxylate and dicarboxylate metabolism | + | -- | -- | -- | -- | -- | -- | 1 |
| Arginine biosynthesis | + | -- | -- | -- | + | -- | + | 3 |
| Alanine, aspartate, and glutamate metabolism | + | -- | + | + | + | + | -- | 5 |
| Sphingolipid metabolism | + | -- | -- | -- | + | -- | + | 3 |
| Glycine, serine, and threonine metabolism | + | -- | + | -- | + | + | -- | 4 |
| Cysteine and methionine metabolism | + | -- | -- | -- | -- | + | -- | 2 |
| Valine, leucine, and isoleucine biosynthesis | + | -- | -- | -- | -- | -- | -- | 1 |
| Taurine and hypotaurine metabolism | + | -- | + | + | -- | -- | -- | 3 |
| Urea cycle | -- | + | -- | -- | -- | -- | -- | 1 |
| Glycine and serine metabolism | + | + | + | -- | + | + | -- | 5 |
| Weak | Moderate | Good | Excellent | |
|---|---|---|---|---|
| Normalized Weight 1 | −1 to 0.040625 | 0.040625 to 0.178125 | 0.178125 to 0.346875 | 0.346875 to 1 |
| Normalized Weight 2 | −1 to 0.005215122 | 0.005215122 to 0.035244476 | 0.035244476 to 0.114987363 | 0.114987363 to 1 |
| Normalized Weight 3 | −1 to 0.036363636 | 0.036363636 to 0.090909091 | 0.090909091 to 0.145454545 | 0.145454545 to 1 |
| Normalized Weight 4 | −1 to 0.005028148 | 0.005028148 to 0.046520343 | 0.046520343 To 0.093742947 | 0.093742947 to 1 |
| No. | Metabolites | Normalized Weight 1 | Normalized Weight 2 | Normalized Weight 3 | Normalized Weight 4 | Calculated Merits |
|---|---|---|---|---|---|---|
| 1 | Maltotriose | 0.059375 | 0.012879969 | −1 | −1 | 12.33015695 |
| 2 | L-Glutamic acid | 0.95625 | 0.892826812 | 0.090909091 | 0.043457267 | 42.4297409 |
| 3 | Pyruvic acid | 0.778125 | 0.558492981 | 0.272727273 | 0.088754688 | 88.06311429 |
| 4 | L-Tryptophan | 0.296875 | 0.107651537 | 0.109090909 | 0.109603133 | 62.5 |
| 5 | Citric acid | 0.45 | 0.170635833 | 0.181818182 | 0.137017563 | 90.33333333 |
| 6 | L-Alanine | 0.39375 | 0.112928955 | 0.218181818 | 0.093742947 | 87.32827717 |
| 7 | L-Serine | 0.36875 | 0.084920841 | 0.127272727 | 0.045147312 | 62.5 |
| 8 | Betaine | 0.15625 | 0.027372191 | 0.090909091 | 0.048277571 | 42.63825352 |
| 9 | Dimethyl sulfone | 0.021875 | 0.000196837 | −1 | −1 | 11.6245581 |
| 10 | L-Arginine | 0.371875 | 0.131651201 | 0.290909091 | 0.14684817 | 89.35621737 |
| 11 | Sphinganine | 0.06875 | 0.015722582 | −1 | −1 | 11.6568779 |
| 12 | L-Glutamine | 0.371875 | 0.075654603 | 0.109090909 | 0.047897573 | 62.5 |
| 13 | L-Tyrosine | 0.25 | 0.068912558 | 0.090909091 | 0.037760448 | 42.4297409 |
| 14 | Cholesterol sulfate | 0.003125 | 0 | 0.418181818 | 0.475029838 | 90.25525526 |
| 15 | Sucrose | 0.178125 | 0.06483723 | 0.018181818 | 0 | 12.90686029 |
| 16 | L-Phenylalanine | 0.28125 | 0.049343156 | 0.109090909 | 0.091631965 | 62.5 |
| 17 | L-Cysteine | 0.4125 | 0.143670141 | 0.090909091 | 0.080663464 | 62.5 |
| 18 | L-Aspartate-semialdehyde | 0.040625 | 5.06488 × 10−5 | −1 | −1 | 13.82051282 |
| 19 | L-Methionine | 0.334375 | 0.075768988 | 0.181818182 | 0.20829445 | 86.56442358 |
| 20 | Creatine | 0.1 | 0.016946398 | 0.090909091 | 0.070195678 | 42.4297409 |
| 21 | L-Cystine | 0.00625 | 0 | 0.127272727 | 0.041291699 | 62.5 |
| 22 | L-Lysine | 0.403125 | 0.136364416 | 0.272727273 | 0.147600818 | 90.33333333 |
| 23 | L-Isoleucine | 0.2 | 0.009885279 | 0.127272727 | 0.046520343 | 44.40997593 |
| 24 | Phytosphingosine | 0.034375 | 0.005215122 | −1 | −1 | 13.61538462 |
| 25 | Adenosine | 0.346875 | 0.119206393 | 0.054545455 | 0.0280831 | 37.5 |
| 26 | L-Valine | 0.2375 | 0.020285683 | 0.145454545 | 0.068937066 | 62.5 |
| 27 | Glycine | 0.4875 | 0.217186109 | 0.363636364 | 0.259194216 | 90.33333333 |
| 28 | L-Leucine | 0.28125 | 0.039149797 | 0.145454545 | 0.080064075 | 62.5 |
| 29 | cis-Aconitic acid | 0.121875 | 0.018639401 | 0.054545455 | 0.01249794 | 37.5 |
| 30 | Hypotaurine | 0.03125 | 0.000358991 | −1 | −1 | 13.08960132 |
| 31 | S-Adenosylhomocysteine | 0.571875 | 0.575112844 | 0.036363636 | 0.005028148 | 37.5 |
| 32 | L-Aspartic acid | 0.359375 | 0.114987363 | 0.054545455 | 0.013255836 | 37.5 |
| 33 | Glutathione | 0.296875 | 0.105460029 | 0.072727273 | 0.085964538 | 62.5 |
| 34 | Arachidonic acid | 0.228125 | 0.167812851 | 0.036363636 | 0.027691555 | 37.5 |
| 35 | Adenine | 0.3 | 0.12548383 | −1 | −1 | 11.49545672 |
| 36 | Urea | 0.153125 | 0.059641216 | 0.090909091 | 0.011850264 | 42.4297409 |
| 37 | Serotonin | 0.3375 | 0.226082231 | 0.072727273 | 0.062262961 | 54.87490171 |
| 38 | Taurine | 0.153125 | 0.047879708 | 0.090909091 | 0.077994031 | 62.5 |
| 39 | L-Lactic acid | 0.1 | 0.013350728 | 1 | 1 | 89.44287908 |
| 40 | p-Hydroxyphenylacetic acid | 0.028125 | 0.000479396 | 0.054545455 | 0.035107811 | 37.5 |
| 41 | Inosine | 0.153125 | 0.021684479 | 0.054545455 | 0.029713312 | 37.5 |
| 42 | SM(d18:1/18:0) | 0.040625 | 0.014353102 | −1 | −1 | 13.82051282 |
| 43 | Hypoxanthine | 0.175 | 0.029327862 | 0.090909091 | 0.108400157 | 62.5 |
| 44 | Dopaquinone | 0.01875 | 0.000234529 | −1 | −1 | 11.19413764 |
| 45 | L-Proline | 0.2125 | 0.042078003 | 0.2 | 0.10544835 | 87.16355188 |
| 46 | L-Histidine | 0.23125 | 0.035244476 | 0.127272727 | 0.048363647 | 53.99094217 |
| 47 | Guanine | 0.165625 | 0.023106832 | 0.018181818 | 0 | 11.96511859 |
| 48 | Cytidine | 0.134375 | 0.02976407 | 0.018181818 | 0 | 11.41056611 |
| 49 | Glycerol | 1 | 1 | 0.109090909 | 0.169397158 | 89.55878284 |
| 50 | Calcium | −1 | −1 | 0.327272727 | 0.318269183 | 90.33333333 |
| 51 | Acetoacetic acid | −1 | −1 | 0.127272727 | 0.064575489 | 62.5 |
| 52 | Formic acid | −1 | −1 | 0.018181818 | 0 | 11.41056611 |
| 53 | Zinc (II) ion | −1 | −1 | 0.072727273 | 0.025154387 | 37.5 |
| 54 | Acetic acid | −1 | −1 | 0.054545455 | 0.012651199 | 37.5 |
| 55 | Cortisol | −1 | −1 | 0.418181818 | 0.65988514 | 90.33333333 |
| D | C | B | A | |
|---|---|---|---|---|
| Degree Impact | 0 to 0.044118 | 0.044118 to 0.11111 | 0.11111 to 0.213095 | 0.213095 to 0.4918 |
| Betweenness Impact | 0 to 0 | 0 to 0 | 0 to 0.02748 | 0.02748 to 0.17737 |
| Closeness Impact | 0.062116 to 0.0994065 | 0.0994065 to 0.13354 | 0.13354 to 0.21584 | 0.21584 to 0.67273 |
| A | B | C | D | |
| FDR | 4.3423 × 10−27 to 2.41705 × 10−5 | 2.41705 × 10−5 to 0.0092785 | 0.0092785 to 0.0282315 | 0.0282315 to 0.046353 |
| No. | Metabolic Pathway | Degree Impact | Betweenness Impact | Closeness Impact | FDR | Calculated Merits |
|---|---|---|---|---|---|---|
| 1 | ABC transporters | 0 | 0 | 0.2 | 4.3423 × 10−27 | 90.04878049 |
| 2 | Protein digestion and absorption | 0 | 0 | 0.26027 | 5.1706 × 10−26 | 90.04878049 |
| 3 | Central carbon metabolism in cancer | 0 | 0 | 0.15789 | 5.2636 × 10−20 | 88.17843178 |
| 4 | Aminoacyl–tRNA biosynthesis | 0.18557 | 0 | 0.1887 | 3.0666 × 10−16 | 89.04243328 |
| 5 | Mineral absorption | 0.058824 | 0 | 0.16361 | 7.8109 × 10−13 | 88.29666352 |
| 6 | Glyoxylate and dicarboxylate metabolism | 0.19318 | 0.005094 | 0.14279 | 2.6159 × 10−7 | 65 |
| 7 | Taurine and hypotaurine metabolism | 0.27586 | 0 | 0.26261 | 4.1941 × 10−7 | 90.04862 |
| 8 | Cysteine and methionine metabolism | 0.23301 | 0.17737 | 0.17003 | 0.000016837 | 88.72511315 |
| 9 | Glycine, serine, and threonine metabolism | 0.37647 | 0.14892 | 0.23869 | 0.000031504 | 90.03666656 |
| 10 | Alanine, aspartate, and glutamate metabolism | 0.4918 | 0.12764 | 0.1311 | 0.0005232 | 88.6812472 |
| 11 | Ferroptosis | 0.094595 | 0 | 0.16278 | 0.00080319 | 50 |
| 12 | Sulfur metabolism | 0 | 0 | 0.10345 | 0.00080319 | 36.6802727 |
| 13 | Arginine biosynthesis | 0.37143 | 0.067227 | 0.42256 | 0.00083089 | 89.70463799 |
| 14 | Amoebiasis | 0.016667 | 0 | 0.13142 | 0.0012241 | 36.7479835 |
| 15 | Valine, leucine, and isoleucine biosynthesis | 0.15385 | 0 | 0.15685 | 0.0016043 | 88 |
| 16 | Sphingolipid metabolism | 0.34426 | 0.11199 | 0.67273 | 0.0072172 | 89.00877813 |
| 17 | Phenylalanine metabolism | 0.11111 | 0.011281 | 0.076586 | 0.0092785 | 43.91079969 |
| 18 | Purine metabolism | 0.16832 | 0.014208 | 0.11081 | 0.010746 | 66.68460826 |
| 19 | Thiamine metabolism | 0.026316 | 0 | 0.11902 | 0.010746 | 36.7661384 |
| 20 | Pantothenate and CoA biosynthesis | 0.095238 | 0 | 0.1303 | 0.010746 | 36.78520447 |
| 21 | Arginine and proline metabolism | 0.1453 | 0.039309 | 0.10489 | 0.01168 | 65 |
| 22 | Staphylococcus aureus infection | 0.093023 | 0.0011074 | 0.095363 | 0.021438 | 36.83966667 |
| 23 | Gap junction | 0.076923 | 0 | 0.24965 | 0.021454 | 50 |
| 24 | Neuroactive ligand–receptor interaction | 0.061798 | 0 | 0.062116 | 0.023787 | 30.30921831 |
| 25 | Primary bile acid biosynthesis | 0.09375 | 0 | 0.23168 | 0.027448 | 50 |
| 26 | AGE-RAGE signaling pathway in diabetic complications | 0.029412 | 0 | 0.13354 | 0.029015 | 36.71982118 |
| 27 | Tyrosine metabolism | 0.1129 | 0.04149 | 0.094005 | 0.032855 | 65 |
| 28 | Butanoate metabolism | 0.16364 | 0.016498 | 0.089484 | 0.032855 | 88 |
| 29 | Pyruvate metabolism | 0.33962 | 0.038462 | 0.25425 | 0.032855 | 88.14549064 |
| 30 | Taste transduction | 0.14085 | 0 | 0.12007 | 0.032961 | 36.73783459 |
| 31 | Nitrogen metabolism | 0.065217 | 0 | 0.069877 | 0.036419 | 14.3852384 |
| 32 | Carbohydrate digestion and absorption | 0 | 0 | 0.080645 | 0.036419 | 13.94718423 |
| 33 | Phenylalanine, tyrosine, and tryptophan biosynthesis | 0.29268 | 0.0093496 | 0.085978 | 0.046353 | 50 |
| Maximum iteration | 300 |
| Population size | 100 |
| Number of chromosomes | 55 |
| Mutation coefficient | 0.09 |
| Proportion of crossover | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latifi-Navid, H.; Barzegar Behrooz, A.; Jamehdor, S.; Davari, M.; Latifinavid, M.; Zolfaghari, N.; Piroozmand, S.; Taghizadeh, S.; Bourbour, M.; Shemshaki, G.; et al. Construction of an Exudative Age-Related Macular Degeneration Diagnostic and Therapeutic Molecular Network Using Multi-Layer Network Analysis, a Fuzzy Logic Model, and Deep Learning Techniques: Are Retinal and Brain Neurodegenerative Disorders Related? Pharmaceuticals 2023, 16, 1555. https://doi.org/10.3390/ph16111555
Latifi-Navid H, Barzegar Behrooz A, Jamehdor S, Davari M, Latifinavid M, Zolfaghari N, Piroozmand S, Taghizadeh S, Bourbour M, Shemshaki G, et al. Construction of an Exudative Age-Related Macular Degeneration Diagnostic and Therapeutic Molecular Network Using Multi-Layer Network Analysis, a Fuzzy Logic Model, and Deep Learning Techniques: Are Retinal and Brain Neurodegenerative Disorders Related? Pharmaceuticals. 2023; 16(11):1555. https://doi.org/10.3390/ph16111555
Chicago/Turabian StyleLatifi-Navid, Hamid, Amir Barzegar Behrooz, Saleh Jamehdor, Maliheh Davari, Masoud Latifinavid, Narges Zolfaghari, Somayeh Piroozmand, Sepideh Taghizadeh, Mahsa Bourbour, Golnaz Shemshaki, and et al. 2023. "Construction of an Exudative Age-Related Macular Degeneration Diagnostic and Therapeutic Molecular Network Using Multi-Layer Network Analysis, a Fuzzy Logic Model, and Deep Learning Techniques: Are Retinal and Brain Neurodegenerative Disorders Related?" Pharmaceuticals 16, no. 11: 1555. https://doi.org/10.3390/ph16111555
APA StyleLatifi-Navid, H., Barzegar Behrooz, A., Jamehdor, S., Davari, M., Latifinavid, M., Zolfaghari, N., Piroozmand, S., Taghizadeh, S., Bourbour, M., Shemshaki, G., Latifi-Navid, S., Arab, S. S., Soheili, Z.-S., Ahmadieh, H., & Sheibani, N. (2023). Construction of an Exudative Age-Related Macular Degeneration Diagnostic and Therapeutic Molecular Network Using Multi-Layer Network Analysis, a Fuzzy Logic Model, and Deep Learning Techniques: Are Retinal and Brain Neurodegenerative Disorders Related? Pharmaceuticals, 16(11), 1555. https://doi.org/10.3390/ph16111555