
Preparation of Nanosized Pharmaceutical Formulations by Dual Centrifugation

 , ,
, ,
Abstract
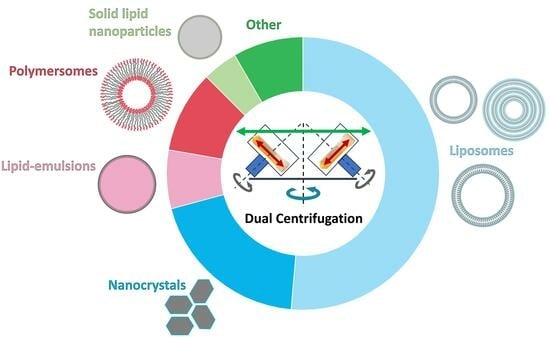
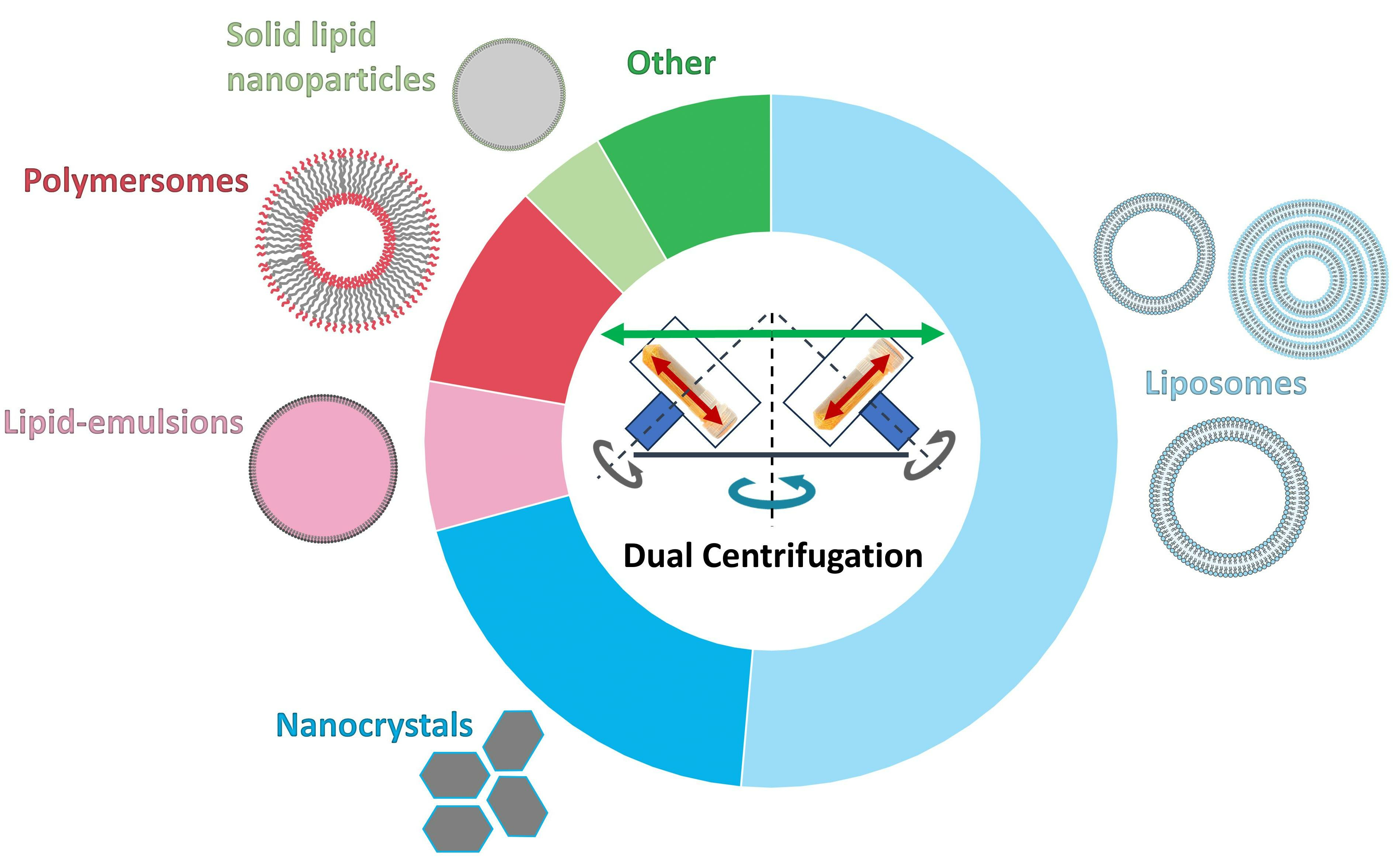
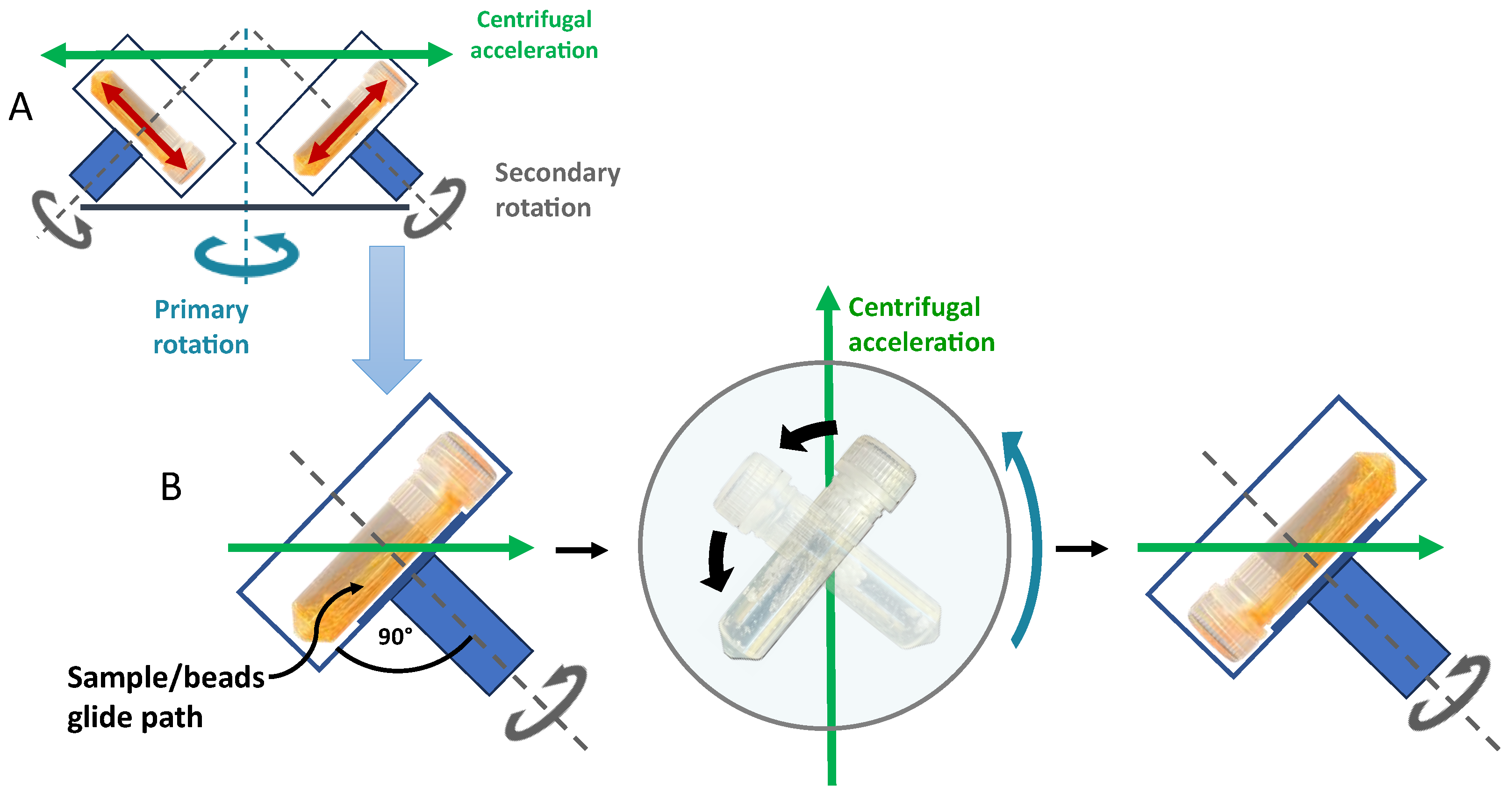
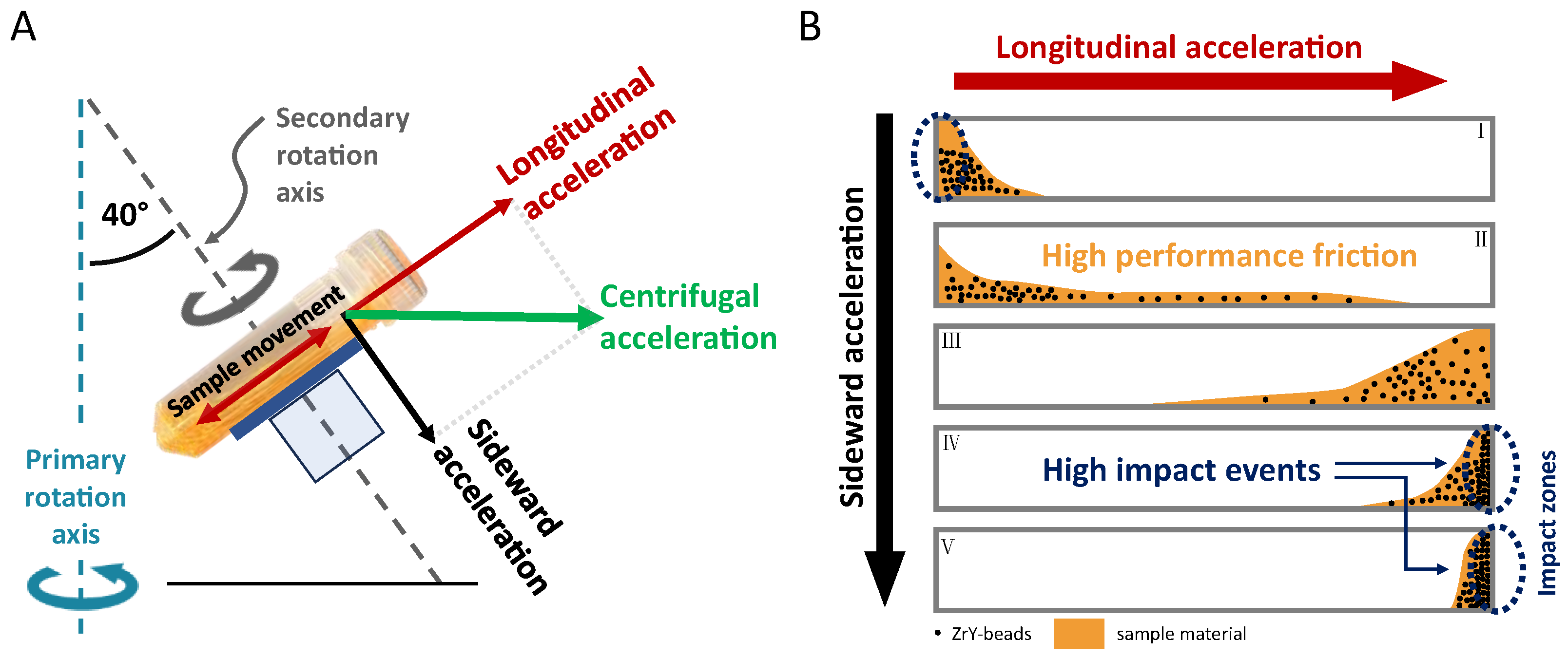
1. Basic Principles of Dual Centrifugation (DC) and Focus of the Review
2. Equipment for Dual Centrifugation
2.1. Dual Centrifuges
2.2. Beads in Use for Dual Centrifugation
2.3. Vials in Use for Dual Centrifugation
3. Nanosized Pharmaceutical Formulations Made by DC
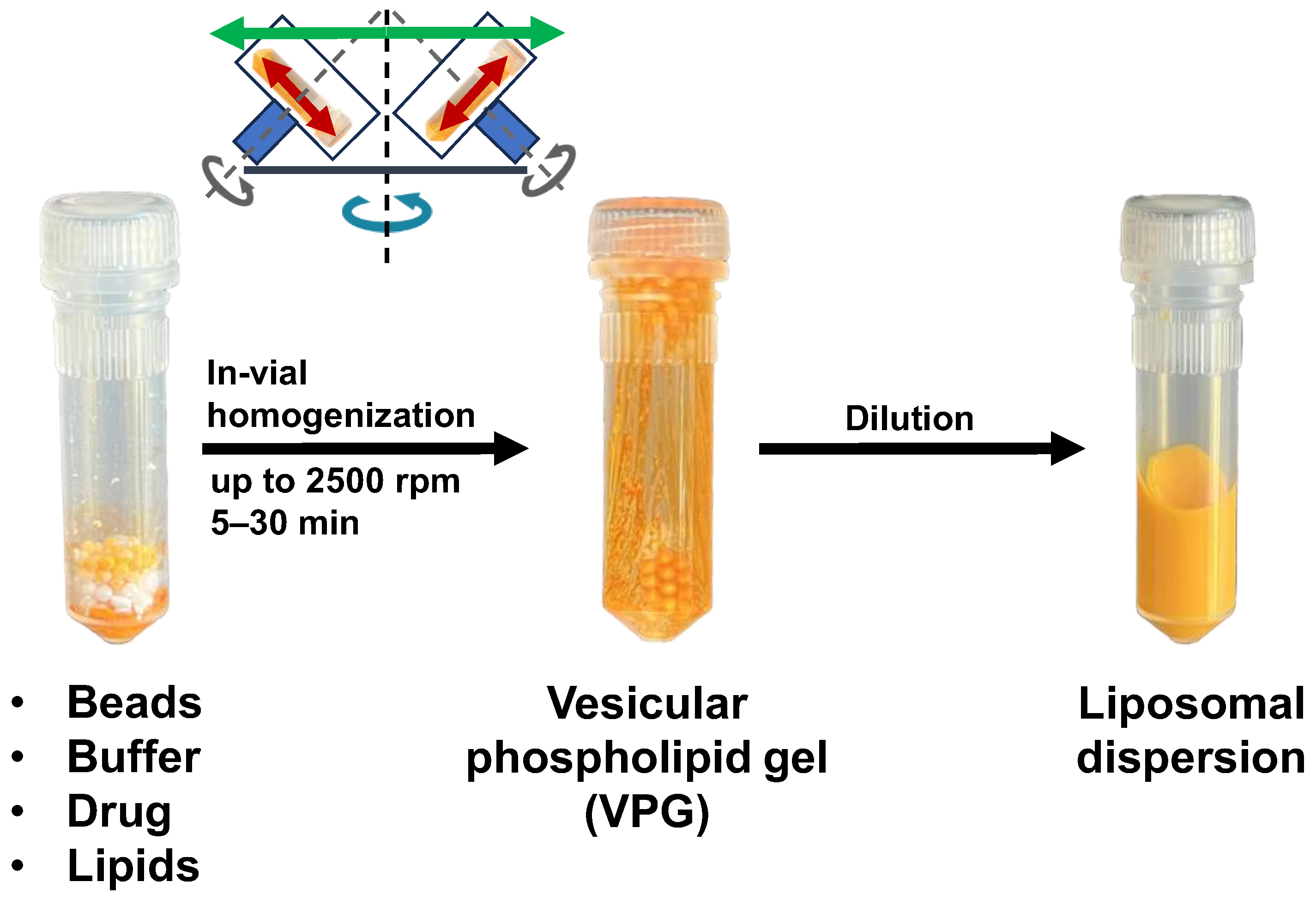
3.1. Liposomes and Vesicular Phospholipid Gels (VPGs)
3.1.1. Morphology of DC-Made Liposomes
3.1.2. Current DC-Applications in Liposome Research

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Drug | Lipid Composition | DC Device | Smallest Measured Size | Highlight | Reference |
|---|---|---|---|---|---|---|
| Analgesic | Mifepristone, FKBP51 ligand SAFit2 | E80 | DAC 150 | not applicable | SAFit2 encapsulated in a DC-made vesicular phospholipid gel (VPG) can be used to target the stress regulator FKBP51. | [40] |
| Antibiotics | Chloramphenicol | S100 | DAC 150 | approx. 130 nm | Liposome-in-hydrogels were made by DC using soy phosphatidylcholine and water-soluble β-1,3/1,6-glucan as gel component for the delivery of chloramphenicol. | [41] |
| Benzoyl peroxide and chloramphenicol | S100 | DAC 150 | 132 nm | DC-based co-encapsulation of benzoyl peroxide and chloramphenicol into liposomes without detectable influence of the two drugs on liposomal characteristics or release profiles. | [42] | |
| Chloramphenicol | E80/Phospholipon® 90H/S100/Soluthin® S90 | ZM 380R | 120 nm | DC-preparation of a liposome-in-hydrogel using various lipids and chitosan. High entrapment of chloramphenicol. | [28] | |
| Vancomycin | Lecithin/Chol/GCTE | DAC 150 | 95 nm | DC-preparation of a promising drug delivery system for oral application of peptide drugs. Potency of the drug was proven using a rat model. Long-term stable formulation was achieved. | [29] | |
| Vancomycin | EPC/Chol/head-group-modified PL | DAC 150 | 87 nm | Promising approach to overcome the mucosal barrier. Using DC-prepared liposomal nanocarriers carrying cell-penetrating peptides for oral delivery of vancomycin. | [30] | |
| Antiviral drugs | Myrcludex B | GCTE | DAC 150 | approx. 130 nm | GCTE-liposomes for efficient oral administration of Myrcludex B. Long-term storage possible due to freeze-drying. | [43] |
| Chelating agent | Copper-chelating Trientine | EPC/Chol/DSPE-mPEG2000/DSPE-PEG2000 Maleimide | DAC 150 | 139 nm | Triethylenetetramine (TETA) in DC-prepared vectorized liposomes showed an up to 16-fold higher brain uptake in in vivo experiments in rats compared to free TETA or TETA in non-vectorized liposomes. | [44] |
| Contrast agents | Superparamagnetic iron oxide particles | DPPC/POPC/DPPE m-PEG2000/Chol/DSPE-PEG2000 Maleimide | ZM 380R and DAC 150 | 127 nm | Immuno-magnetoliposomes (ML) with a high amount of entrapped superparamagnetic iron oxide particles (SPIOs) were prepared by DC. Immuno-ML can target activated platelets and are thus potentially suitable as MRI contrast agent for the detection of ruptured plaques. | [19] |
| Cytostatics | Docetaxel | S100/Chol/DC-Chol/DMPE/DMPG/DOPE/DMPC/DPPG/DOPC/DOTAP/POPC/POPE/DSPE-PEG2000/DSPE-PEG750 | DAC 150 | 58 nm | Various lipid compositions were tested to identify liposomal lipid compositions for effective docetaxel entrapment. | [35] |
| Mitotane | DOPC | ZM 380R | 117 nm | Final MT concentration 0.67 ± 0.01 mg/mL; stable for 6 months at 4–8 °C. | [17] | |
| Cytarabine | E80 | DAC 150 | 163 nm | Viscosities of VPGs were enhanced by coating the liposomes with cationic or anionic polyelectrolytes. Very slow in vitro release of cytarabine from the VPGs (up to 18 days). | [36] | |
|
Mitotane, Everolimus | DOPC/POPC/DSPC/Chol | ZM 380R | 130 nm | Simultaneous analysis of hydrophobic drugs and lipids in DC-made liposomes by HPLC-DAD-CAD. | [45] | |
| Liposomal studies | Calcein | EPC/Chol/DSPE-PEG2000 | Rotanta 400 DC prototype | 110 nm | Cholesterol–polymer amphiphiles were used for steric stabilization of DC-made liposomes. Click chemistry was used for conjugation of small molecules to the liposomal surface. | [46] |
| EPC/Chol/DSPE-PEG2000 | Rotanta 400 DC prototype | 130 nm | Functionalization of DC-made liposomes by click chemistry. | [47] | ||
| Indocyanine green | EPC/DPPG/Chol/DSPE-PEG36 | DAC 150 | 216 nm | Matrix-assisted laser desorption/ionization mass spectrometry is used for ex vivo imaging of liposomal carriers in mouse tissue. Indocyanine green serves as cargo and DPPG/PEG36-DSPE as lipid marker. | [48] | |
| EPC3/DPPC/DSPC/20:0 PC/Chol | ZM 380R | 162 nm | Migration of DC-made liposomes modified with fluorescence-labeled conjugates of different lengths into biomembranes. | [49] | ||
| HEPC/Chol/DSPE-PEG2000/DPPC | ZM 380R | approx. 100 nm | Small multilamellar vesicles (SMV) can be prepared by DC in a highly reproducible way. | [18] | ||
| Calcein | HEPC/Chol | DAC 150 | approx. 50 nm | It was shown for the first time that DC can successfully be used for the preparation of VPGs and liposomes. | [1] | |
| Vancomycin, Insulin | Lecithin/Chol | DAC 150 | 124 nm | It has been demonstrated that potential cross-reactions between macromolecular drugs and activated lipids during DC-preparation of liposomes cannot be neglected. | [50] | |
| Membrane dye DiI | EPC/Chol/DSPE-mPEG | Rotanta 400 centrifuge with a prototype DC-rotor | approx. 100 nm | Investigation of the protein corona of DC-prepared liposomes (unfunctionalized vs. PEGylated vs. hyperbranched polyglycerol functionalized) and their influence on uptake by macrophages. | [51] | |
|
Calcein-AM, Tamoxifen | Essential phospholipids/polyenylphosphatidylcholine (PPC)/phosphatidylinositol (PI) | ZM 380R | not specified | The effect of essential phospholipids (EPL) on hepatocyte function in vitro was investigated and valuable insights into the mechanism of action of EPL were gained. | [52] | |
| EPC/DSPE-mPEG2000/DSPE-PEG2000 Maleimide/DPPE-RH/Chol | DAC 150 | 118 nm | DC-made liposomes are conjugated with cationized bovine serum albumin as a transport vector to penetrate the blood–brain barrier (BBB). | [53] | ||
| Lyso-PC | Saturated and mono-unsaturated Lyso-PC | DSPC/Chol | DAC 150 | not specified |
Liposomes made by DC using saturated phospholipids caused an increase in saturated lyso-phosphatidylcholine (Lyso-PC) in plasma of tumor bearing mice, which caused a decrease in metastases. | [54] |
| Model drug | Carboxyfluorescein | E80/Chol | ZM 380R | 134 nm | An ex vivo rat intestine model is used to investigate the effects of DC-made matrix-liposomes on intestinal tissue. | [55] |
| Calcein | HEPC/EPC/Chol/DSPE-PEG2000 | ZM 380R | approx. 100 | Liposomes can be prepared by DC without lipid film preparation. Encapsulation efficiency can be determined without separation of the free calcein. | [16] | |
| Peptide/Protein-based | Fluorescein isothiocyanate-dextran (FITC), 5(6)-carboxyfluorescein (CF) | E80/SPC | DAC 150 | approx. 200 nm | DC-made matrix-liposomes for controlled peroral delivery of peptides. | [56] |
| Human growth hormone (hGH), Omeprazol | EPC/Chol/RH-DPPE | DAC 150 | 203 nm | Oligolamellar vesicles for oral delivery of human growth factor were prepared by DC. | [57] | |
| Erythropoietin (EPO) | E 80/DOTAP/DPPA | DAC 150 | approx. 160 nm | VPGs are presented as promising alternative depot systems for protein drugs. | [15] | |
| EPO | E80 | DAC 150 | not applicable | Needle-free injections of DC-made VPGs with various phospholipid contents were tested. Pig skin as an in vitro model as well as gelatine blocks were used for the release studies. | [13] | |
| Granulocyte colony-stimulating factor (G-CSF) | E80 | DAC 150 | not applicable | DC-made VPGs were used for controlled release of G-CSF. A continuous release over > 4 weeks could be observed in vitro. | [58] | |
| Exenatide | POPC/POPG | DAC 150 | not applicable | Investigation of DC-prepared VPGs made from POPC and/or POPG carrying exenatide. The release of exenatide, as well as the release of phospholipids were investigated in vitro. | [37] | |
| Vancomycin, Exenatide | Lecithin/Cholesterol/Glycerylcaldityl tetraetherlipid/cell-penetrating peptide-phospholipid conjugate | DAC 150 | 122 nm | DC-prepared liposomes containing a cell-penetrating peptide achieved high oral bioavailability for the peptides vancomycin and exenatide. | [59] | |
| RNA | siRNA | HSPC/POPC/DDAB/DSPE-PEG2000 | DAC 150 | 185 nm | DC-prepared liposomes containing siRNA were modified by sterol-based post-insertion technique (SPIT) to couple anti-GD2 antibodies for targeting neuroblastoma cells. | [22] |
| Calcein or siRNA | EPC-3/S80/DMPG/DPPG/Chol/DSPE-mPEG2000 | DAC 150 | 79 nm | Investigation of siRNA integrity by FRET during liposomal preparation by DC. | [21] | |
| siRNA | EPC-3/Chol/DSPE-mPEG2000 | DAC 150 | approx. 100 nm | FRET-based visualization of fluorescence-labeled siRNAs in cells after microinjection, lipoplex-mediated transfection, and liposome-mediated transfection. | [38] | |
| Luciferase-coding mRNA | DOTAP | DAC 150 | 147 nm | DC-prepared hybrid nanoparticles from lipidic and polymeric components were used for the mRNA-transfection. Different ratios of the cationic lipid DOTAP and the cationic biopolymer protamine were compared. | [39] |
3.1.3. Discussion—DC-Preparation of Liposomes/VPGs and Their Applications
3.2. Polymersomes
3.3. (Lipid) Emulsions
3.4. Solid Lipid Nanoparticles
3.5. Nanocrystals
4. Other DC-Applications in Pharmaceutical Nanotechnology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massing, U.; Cicko, S.; Ziroli, V. Dual Asymmetric Centrifugation (DAC)—A New Technique for Liposome Preparation. J. Control. Release Off. J. Control. Release Soc. 2008, 125, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Massing, U.; Ingebrigtsen, S.G.; Škalko-Basnet, N.; Holsæter, A.M. Dual Centrifugation—A Novel “in-Vial” Liposome Processing Technique. In Liposomes; Catala, A., Ed.; InTech: London, UK, 2017; ISBN 978-953-51-3579-1. [Google Scholar]
- Hagedorn, M.; Liebich, L.; Bögershausen, A.; Massing, U.; Hoffmann, S.; Mende, S.; Rischer, M. Rapid Development of API Nano-Formulations from Screening to Production Combining Dual Centrifugation and Wet Agitator Bead Milling. Int. J. Pharm. 2019, 565, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Horne, R.W. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Hill, M.W.; Miller, N.G.A. Preparation and Use of Liposomes as Models of Biological Membranes. In Methods in Membrane Biology; Korn, E.D., Ed.; Springer: Boston, MA, USA, 1974; Volume 1, pp. 1–68. ISBN 978-1-4615-7422-4. [Google Scholar]
- Torchilin, V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Maeda, H. Enhanced Permeability and Retention (EPR) Efect: Basis for Drug Targeting to Tumor. In Biomedical Aspects of Drug Targeting; Muzykantov, V., Torchilin, V., Eds.; Springer: Boston, MA, USA, 2002; pp. 211–228. ISBN 978-1-4757-4627-3. [Google Scholar]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs1. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Mayhew, E.; Lazo, R.; Vail, W.J.; King, J.; Green, A.M. Characterization of Liposomes Prepared Using a Microemulsifier. Biochim. Biophys. Acta (BBA) Biomembr. 1984, 775, 169–174. [Google Scholar] [CrossRef]
- Brandl, M.; Bachmann, D.; Drechsler, M.; Bauer, K.H. Liposome Preparation by a New High Pressure Homogenizer Gaulin Micron Lab 40. Drug Dev. Ind. Pharm. 1990, 16, 2167–2191. [Google Scholar] [CrossRef]
- Purmann, T.; Mentrup, E.; Kreuter, J. Preparation of SUV-Liposomes by High-Pressure Homogenization. Eur. J. Pharm. Biopharm 1993, 39, 45–52. [Google Scholar]
- Barenholzt, Y.; Amselem, S.; Lichtenberg, D. A New Method for Preparation of Phospholipid Vesicles (Liposomes)—French Press. FEBS Lett. 1979, 99, 210–214. [Google Scholar] [CrossRef]
- Breitsamer, M.; Winter, G. Needle-Free Injection of Vesicular Phospholipid Gels—A Novel Approach to Overcome an Administration Hurdle for Semisolid Depot Systems. J. Pharm. Sci. 2017, 106, 968–972. [Google Scholar] [CrossRef]
- Breitsamer, M.; Winter, G. Vesicular Phospholipid Gels as Drug Delivery Systems for Small Molecular Weight Drugs, Peptides and Proteins: State of the Art Review. Int. J. Pharm. 2019, 557, 1–8. [Google Scholar] [CrossRef]
- Tian, W.; Schulze, S.; Brandl, M.; Winter, G. Vesicular Phospholipid Gel-Based Depot Formulations for Pharmaceutical Proteins: Development and in Vitro Evaluation. J. Control. Release 2010, 142, 319–325. [Google Scholar] [CrossRef]
- Koehler, J.K.; Schnur, J.; Heerklotz, H.; Massing, U. Screening for Optimal Liposome Preparation Conditions by Using Dual Centrifugation and Time-Resolved Fluorescence Measurements. Pharmaceutics 2021, 13, 2046. [Google Scholar] [CrossRef]
- Langer, C.; Köll-Weber, M.; Holzer, M.; Hantel, C.; Süss, R. Mitotane Nanocarriers for the Treatment of Adrenocortical Carcinoma: Evaluation of Albumin-Stabilized Nanoparticles and Liposomes in a Preclinical In Vitro Study with 3D Spheroids. Pharmaceutics 2022, 14, 1891. [Google Scholar] [CrossRef]
- Koehler, J.K.; Gedda, L.; Wurster, L.; Schnur, J.; Edwards, K.; Heerklotz, H.; Massing, U. Tailoring the Lamellarity of Liposomes Prepared by Dual Centrifugation. Pharmaceutics 2023, 15, 706. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Pütz, G.; Massing, U.; Hagemeyer, C.E.; von Elverfeldt, D.; Meißner, M.; Ardipradja, K.; Barnert, S.; Peter, K.; Bode, C.; et al. Immuno-Magnetoliposomes Targeting Activated Platelets as a Potentially Human-Compatible MRI Contrast Agent for Targeting Atherothrombosis. Biomaterials 2015, 53, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Moog, R.; Burger, A.; Brandl, M.; Schüler, J.; Schubert, R.; Unger, C.; Fiebig, H.; Massing, U. Change in Pharmacokinetic and Pharmacodynamic Behavior of Gemcitabine in Human Tumor Xenografts upon Entrapment in Vesicular Phospholipid Gels. Cancer Chemother. Pharmacol. 2002, 49, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.; Ziroli, V.; Helm, M.; Massing, U. Preparation of Small Amounts of Sterile siRNA-Liposomes with High Entrapping Efficiency by Dual Asymmetric Centrifugation (DAC). J. Control. Release 2009, 135, 80–88. [Google Scholar] [CrossRef]
- Adrian, J.E.; Wolf, A.; Steinbach, A.; Rössler, J.; Süss, R. Targeted Delivery to Neuroblastoma of Novel siRNA-Anti-GD2-Liposomes Prepared by Dual Asymmetric Centrifugation and Sterol-Based Post-Insertion Method. Pharm. Res. 2011, 28, 2261–2272. [Google Scholar] [CrossRef]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of Large Unilamellar Vesicles by a Rapid Extrusion Procedure. Characterization of Size Distribution, Trapped Volume and Ability to Maintain a Membrane Potential. Biochim. Biophys. Acta (BBA) Biomembr. 1985, 812, 55–65. [Google Scholar] [CrossRef]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of Variable Sizes Produced by a Rapid Extrusion Procedure. Biochim. Biophys. Acta (BBA) Biomembr. 1986, 858, 161–168. [Google Scholar] [CrossRef]
- Batzri, S.; Korn, E.D. Single Bilayer Liposomes Prepared without Sonication. Biochim. Biophys. Acta (BBA) Biomembr. 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Khadke, S.; Roces, C.B.; Donaghey, R.; Giacobbo, V.; Su, Y.; Perrie, Y. Scalable Solvent-Free Production of Liposomes. J. Pharm. Pharmacol. 2020, 72, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Zhang, Y.; Tang, X.; Li, A. Cationic/Anionic Polyelectrolyte (PLL/PGA) Coated Vesicular Phospholipid Gels (VPGs) Loaded with Cytarabine for Sustained Release and Anti-Glioma Effects. Drug Des. Dev. Ther. 2020, 14, 1825–1836. [Google Scholar] [CrossRef]
- Ingebrigtsen, S.G.; Didriksen, A.; Johannessen, M.; Škalko-Basnet, N.; Holsæter, A.M. Old Drug, New Wrapping—A Possible Comeback for Chloramphenicol? Int. J. Pharm. 2017, 526, 538–546. [Google Scholar] [CrossRef]
- Uhl, P.; Pantze, S.; Storck, P.; Parmentier, J.; Witzigmann, D.; Hofhaus, G.; Huwyler, J.; Mier, W.; Fricker, G. Oral Delivery of Vancomycin by Tetraether Lipid Liposomes. Eur. J. Pharm. Sci. 2017, 108, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Sauter, M.; Hertlein, T.; Witzigmann, D.; Laffleur, F.; Hofhaus, G.; Fidelj, V.; Tursch, A.; Özbek, S.; Hopke, E.; et al. Overcoming the Mucosal Barrier: Tetraether Lipid-Stabilized Liposomal Nanocarriers Decorated with Cell-Penetrating Peptides Enable Oral Delivery of Vancomycin. Adv. Ther. 2021, 4, 2000247. [Google Scholar] [CrossRef]
- Pereira, S.; Egbu, R.; Jannati, G.; Al-Jamal, W.T. Docetaxel-Loaded Liposomes: The Effect of Lipid Composition and Purification on Drug Encapsulation and in Vitro Toxicity. Int. J. Pharm. 2016, 514, 150–159. [Google Scholar] [CrossRef]
- Storm, G.; van Bloois, L.; Brouwer, M.; Crommelin, D.J.A. The Interaction of Cytostatic Drugs with Adsorbents in Aqueous Media. The Potential Implications for Liposome Preparation. Biochim. Biophys. Acta (BBA) Biomembr. 1985, 818, 343–351. [Google Scholar] [CrossRef]
- van der Meel, R.; Oliveira, S.; Altintas, I.; Haselberg, R.; van der Veeken, J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.P.; Storm, G.; Hennink, W.E.; Schiffelers, R.M.; et al. Tumor-Targeted Nanobullets: Anti-EGFR Nanobody-Liposomes Loaded with Anti-IGF-1R Kinase Inhibitor for Cancer Treatment. J. Control. Release 2012, 159, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Dimov, N.; Kastner, E.; Hussain, M.; Perrie, Y.; Szita, N. Formation and Purification of Tailored Liposomes for Drug Delivery Using a Module-Based Micro Continuous-Flow System. Sci. Rep. 2017, 7, 12045. [Google Scholar] [CrossRef]
- Holsæter, A.M.; Wizgird, K.; Karlsen, I.; Hemmingsen, J.F.; Brandl, M.; Škalko-Basnet, N. How Docetaxel Entrapment, Vesicle Size, Zeta Potential and Stability Change with Liposome Composition–A Formulation Screening Study. Eur. J. Pharm. Sci. 2022, 177, 106267. [Google Scholar] [CrossRef]
- Qi, N.; Cai, C.; Zhang, W.; Niu, Y.; Yang, J.; Wang, L.; Tian, B.; Liu, X.; Lin, X.; Zhang, Y.; et al. Sustained Delivery of Cytarabine-Loaded Vesicular Phospholipid Gels for Treatment of Xenografted Glioma. Int. J. Pharm. 2014, 472, 48–55. [Google Scholar] [CrossRef]
- Breitsamer, M.; Stulz, A.; Heerklotz, H.H.; Winter, G. Do Interactions between Protein and Phospholipids Influence the Release Behavior from Lipid-Based Exenatide Depot Systems? Eur. J. Pharm. Biopharm. 2019, 142, 61–69. [Google Scholar] [CrossRef]
- Järve, A.; Müller, J.; Kim, I.-H.; Rohr, K.; MacLean, C.; Fricker, G.; Massing, U.; Eberle, F.; Dalpke, A.; Fischer, R.; et al. Surveillance of siRNA Integrity by FRET Imaging. Nucleic Acids Res. 2007, 35, e124. [Google Scholar] [CrossRef][Green Version]
- Siewert, C.D.; Haas, H.; Cornet, V.; Nogueira, S.S.; Nawroth, T.; Uebbing, L.; Ziller, A.; Al-Gousous, J.; Radulescu, A.; Schroer, M.A.; et al. Hybrid Biopolymer and Lipid Nanoparticles with Improved Transfection Efficacy for mRNA. Cells 2020, 9, 2034. [Google Scholar] [CrossRef]
- Maiarù, M.; Morgan, O.B.; Mao, T.; Breitsamer, M.; Bamber, H.; Pöhlmann, M.; Schmidt, M.V.; Winter, G.; Hausch, F.; Géranton, S.M. The Stress Regulator FKBP51: A Novel and Promising Druggable Target for the Treatment of Persistent Pain States across Sexes. Pain 2018, 159, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Ingebrigtsen, S.G.; Škalko-Basnet, N.; Holsæter, A.M. Development and Optimization of a New Processing Approach for Manufacturing Topical Liposomes-in-Hydrogel Drug Formulations by Dual Asymmetric Centrifugation. Drug Dev. Ind. Pharm. 2016, 42, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Ingebrigtsen, S.G.; Škalko-Basnet, N.; Jacobsen, C.d.A.C.; Holsæter, A.M. Successful Co-Encapsulation of Benzoyl Peroxide and Chloramphenicol in Liposomes by a Novel Manufacturing Method-Dual Asymmetric Centrifugation. Eur. J. Pharm. Sci. 2017, 97, 192–199. [Google Scholar] [CrossRef]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A Liposomal Formulation for the Oral Application of the Investigational Hepatitis B Drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, R.; Uhl, P.; Helm, F.; Wupperfeld, D.; Sauter, M.; Mier, W.; Stremmel, W.; Hofhaus, G.; Fricker, G. Delivery of Copper-Chelating Trientine (TETA) to the Central Nervous System by Surface Modified Liposomes. Int. J. Pharm. 2016, 512, 87–95. [Google Scholar] [CrossRef]
- Langer, C.; Süss, R. HPLC-DAD-CAD-Based Approach for the Simultaneous Analysis of Hydrophobic Drugs and Lipid Compounds in Liposomes and for Cyclodextrin/Drug Inclusion Complexes. J. Pharm. Biomed. Anal. 2021, 201, 114120. [Google Scholar] [CrossRef] [PubMed]
- Fritz, T.; Hirsch, M.; Richter, F.C.; Müller, S.S.; Hofmann, A.M.; Rusitzka, K.A.K.; Markl, J.; Massing, U.; Frey, H.; Helm, M. Click Modification of Multifunctional Liposomes Bearing Hyperbranched Polyether Chains. Biomacromolecules 2014, 15, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Fritz, T.; Voigt, M.; Worm, M.; Negwer, I.; Müller, S.S.; Kettenbach, K.; Ross, T.L.; Roesch, F.; Koynov, K.; Frey, H.; et al. Orthogonal Click Conjugation to the Liposomal Surface Reveals the Stability of the Lipid Anchorage as Crucial for Targeting. Chem. Eur. J. 2016, 22, 11578–11582. [Google Scholar] [CrossRef]
- Fülöp, A.; Sammour, D.A.; Erich, K.; von Gerichten, J.; van Hoogevest, P.; Sandhoff, R.; Hopf, C. Molecular Imaging of Brain Localization of Liposomes in Mice Using MALDI Mass Spectrometry. Sci. Rep. 2016, 6, 33791. [Google Scholar] [CrossRef]
- Gleue, L.; Schupp, J.; Zimmer, N.; Becker, E.; Frey, H.; Tuettenberg, A.; Helm, M. Stability of Alkyl Chain-Mediated Lipid Anchoring in Liposomal Membranes. Cells 2020, 9, 2213. [Google Scholar] [CrossRef]
- Sauter, M.; Burhenne, J.; Haefeli, W.E.; Uhl, P. An Underestimated Factor: The Extent of Cross-Reactions Modifying APIs in Surface-Modified Liposomal Preparations Caused by Comprised Activated Lipids. Molecules 2020, 25, 4436. [Google Scholar] [CrossRef]
- Weber, C.; Voigt, M.; Simon, J.; Danner, A.-K.; Frey, H.; Mailänder, V.; Helm, M.; Morsbach, S.; Landfester, K. Functionalization of Liposomes with Hydrophilic Polymers Results in Macrophage Uptake Independent of the Protein Corona. Biomacromolecules 2019, 20, 2989–2999. [Google Scholar] [CrossRef]
- Wupperfeld, D.; Fricker, G.; Bois De Fer, B.; Frank, L.; Wehrle, A.; Popovic, B. Essential Phospholipids Decrease Apoptosis and Increase Membrane Transport in Human Hepatocyte Cell Lines. Lipids Health Dis. 2022, 21, 91. [Google Scholar] [CrossRef]
- Helm, F.; Fricker, G. Liposomal Conjugates for Drug Delivery to the Central Nervous System. Pharmaceutics 2015, 7, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Raynor, A.; Jantscheff, P.; Ross, T.; Schlesinger, M.; Wilde, M.; Haasis, S.; Dreckmann, T.; Bendas, G.; Massing, U. Saturated and Mono-Unsaturated Lysophosphatidylcholine Metabolism in Tumour Cells: A Potential Therapeutic Target for Preventing Metastases. Lipids Health Dis. 2015, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Binnefeld, M.; Fritz, S.; Balzer, V.; Skalická, V.; Witzigmann, D.; Kauczor, H.-U.; Fricker, G.; Salomon, J.J. Physicochemical and Biopharmaceutical Characterization of Novel Matrix-Liposomes. Eur. J. Pharm. Biopharm. 2020, 153, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Pantze, S.F.; Parmentier, J.; Hofhaus, G.; Fricker, G. Matrix Liposomes: A Solid Liposomal Formulation for Oral Administration. Eur. J. Lipid Sci. Technol. 2014, 116, 1145–1154. [Google Scholar] [CrossRef]
- Parmentier, J.; Hofhaus, G.; Thomas, S.; Cuesta, L.C.; Gropp, F.; Schröder, R.; Hartmann, K.; Fricker, G. Improved Oral Bioavailability of Human Growth Hormone by a Combination of Liposomes Containing Bio-Enhancers and Tetraether Lipids and Omeprazole. J. Pharm. Sci. 2014, 103, 3985–3993. [Google Scholar] [CrossRef]
- Buchmann, S.; Sandmann, G.H.; Walz, L.; Reichel, T.; Beitzel, K.; Wexel, G.; Tian, W.; Battmann, A.; Vogt, S.; Winter, G.; et al. Growth Factor Release by Vesicular Phospholipid Gels: In-Vitro Results and Application for Rotator Cuff Repair in a Rat Model. BMC Musculoskelet. Disord. 2015, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Bajraktari-Sylejmani, G.; Witzigmann, D.; Bay, C.; Zimmermann, S.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Sauter, M. A Nanocarrier Approach for Oral Peptide Delivery: Evaluation of Cell-Penetrating-Peptide-Modified Liposomal Formulations in Dogs. Adv. Ther. 2023, 2300021. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol Injection Method for Hydrophilic and Lipophilic Drug-Loaded Liposome Preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef]
- Scherer, M.; Kappel, C.; Mohr, N.; Fischer, K.; Heller, P.; Forst, R.; Depoix, F.; Bros, M.; Zentel, R. Functionalization of Active Ester-Based Polymersomes for Enhanced Cell Uptake and Stimuli-Responsive Cargo Release. Biomacromolecules 2016, 17, 3305–3317. [Google Scholar] [CrossRef]
- Köthe, T.; Martin, S.; Reich, G.; Fricker, G. Dual Asymmetric Centrifugation as a Novel Method to Prepare Highly Concentrated Dispersions of PEG-b-PCL Polymersomes as Drug Carriers. Int. J. Pharm. 2020, 579, 119087. [Google Scholar] [CrossRef]
- Scherer, M.; Fischer, K.; Depoix, F.; Fritz, T.; Thiermann, R.; Mohr, K.; Zentel, R. Pentafluorophenyl Ester-Based Polymersomes as Nanosized Drug-Delivery Vehicles. Macromol. Rapid Commun. 2016, 37, 60–66. [Google Scholar] [CrossRef]
- Weber, B.; Kappel, C.; Scherer, M.; Helm, M.; Bros, M.; Grabbe, S.; Barz, M. PeptoSomes for Vaccination: Combining Antigen and Adjuvant in Polypept(o)Ide-Based Polymersomes. Macromol. Biosci. 2017, 17, 1700061. [Google Scholar] [CrossRef] [PubMed]
- Fenaroli, F.; Repnik, U.; Xu, Y.; Johann, K.; Van Herck, S.; Dey, P.; Skjeldal, F.M.; Frei, D.M.; Bagherifam, S.; Kocere, A.; et al. Enhanced Permeability and Retention-like Extravasation of Nanoparticles from the Vasculature into Tuberculosis Granulomas in Zebrafish and Mouse Models. ACS Nano 2018, 12, 8646–8661. [Google Scholar] [CrossRef] [PubMed]
- Dal, N.-J.K.; Schäfer, G.; Thompson, A.M.; Schmitt, S.; Redinger, N.; Alonso-Rodriguez, N.; Johann, K.; Ojong, J.; Wohlmann, J.; Best, A.; et al. Π-Π Interactions Stabilize PeptoMicelle-Based Formulations of Pretomanid Derivatives Leading to Promising Therapy against Tuberculosis in Zebrafish and Mouse Models. J. Control. Release 2023, 354, 851–868. [Google Scholar] [CrossRef] [PubMed]
- Johann, K.; Bohn, T.; Shahneh, F.; Luther, N.; Birke, A.; Jaurich, H.; Helm, M.; Klein, M.; Raker, V.K.; Bopp, T.; et al. Therapeutic Melanoma Inhibition by Local Micelle-Mediated Cyclic Nucleotide Repression. Nat. Commun. 2021, 12, 5981. [Google Scholar] [CrossRef]
- Massing, U.; Krämer, W.; Deuringer, B.; Hagedorn, M.; Ziroli, V. Herstellung von Nanopartikeln Zur Wirkstoffapplikation. Biospektrum 2018, 24, 720–721. [Google Scholar] [CrossRef]
- Tenambergen, F.; Maruiama, C.H.; Mäder, K. Dual Asymmetric Centrifugation as an Alternative Preparation Method for Parenteral Fat Emulsions in Preformulation Development. Int. J. Pharm. 2013, 447, 31–37. [Google Scholar] [CrossRef]
- Chung, E.P.; Wells, A.R.; Kiamco, M.M.; Leung, K.P. Dual Asymmetric Centrifugation Efficiently Produces a Poloxamer-Based Nanoemulsion Gel for Topical Delivery of Pirfenidone. AAPS PharmSciTech 2020, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Francke, N.M.; Schneider, F.; Baumann, K.; Bunjes, H. Formulation of Cannabidiol in Colloidal Lipid Carriers. Molecules 2021, 26, 1469. [Google Scholar] [CrossRef]
- Krämer, W.; Schubert, R.; Massing, U. Small-Scale Preparation of Perfluorocarbon-Nanoemulsions Utilizing Dual Centrifugation. Int. J. Pharm. 2019, 572, 118753. [Google Scholar] [CrossRef]
- Steiner, D.; Bunjes, H. Influence of Process and Formulation Parameters on the Preparation of Solid Lipid Nanoparticles by Dual Centrifugation. Int. J. Pharm. X 2021, 3, 100085. [Google Scholar] [CrossRef]
- Steiner, D.; Massing, U.; Bunjes, H. Feste Lipidnanopartikel zur Applikation unlöslicher Wirkstoffe. Biospektrum 2020, 26, 520–521. [Google Scholar] [CrossRef]
- Steiner, D.; Emmendörffer, J.F.; Bunjes, H. Orodispersible Films: A Delivery Platform for Solid Lipid Nanoparticles? Pharmaceutics 2021, 13, 2162. [Google Scholar] [CrossRef]
- Ostwald, W. Über die vermeintliche Isomerie des roten und gelben Quecksilberoxyds und die Oberflächenspannung fester Körper. Z. Phys. Chem. 1900, 34U, 495–503. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, S.; Gokhale, R.; Burgess, D.J. Physical Stability of Nanosuspensions: Investigation of the Role of Stabilizers on Ostwald Ripening. Int. J. Pharm. 2011, 406, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Juhnke, M.; Berghausen, J.; Timpe, C. Accelerated Formulation Development for Nanomilled Active Pharmaceutical Ingredients Using a Screening Approach. Chem. Eng. Technol. 2010, 33, 1412–1418. [Google Scholar] [CrossRef]
- Bitterlich, A.; Laabs, C.; Krautstrunk, I.; Dengler, M.; Juhnke, M.; Grandeury, A.; Bunjes, H.; Kwade, A. Process Parameter Dependent Growth Phenomena of Naproxen Nanosuspension Manufactured by Wet Media Milling. Eur. J. Pharm. Biopharm. 2015, 92, 171–179. [Google Scholar] [CrossRef]
- Steiner, D.; Finke, J.H.; Breitung-Faes, S.; Kwade, A. Breakage, Temperature Dependency and Contamination of Lactose during Ball Milling in Ethanol. Adv. Powder Technol. 2016, 27, 1700–1709. [Google Scholar] [CrossRef]
- Hagedorn, M.; Bögershausen, A.; Rischer, M.; Schubert, R.; Massing, U. Dual Centrifugation—A New Technique for Nanomilling of Poorly Soluble Drugs and Formulation Screening by an DoE-Approach. Int. J. Pharm. 2017, 530, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Willmann, A.-C.; Berkenfeld, K.; Faber, T.; Wachtel, H.; Boeck, G.; Wagner, K.G. Itraconazole Nanosuspensions via Dual Centrifugation Media Milling: Impact of Formulation and Process Parameters on Particle Size and Solid-State Conversion as Well as Storage Stability. Pharmaceutics 2022, 14, 1528. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.A.; Park, J.S.; Kim, M.S.; Jeong, M.Y.; Park, H.J.; Choi, J.H.; Seo, J.H.; Choi, Y.S.; Kang, M.J. High-Payload Nanosuspension of Centella Asiatica Extract for Improved Skin Delivery with No Irritation. Int. J. Nanomed. 2021, 16, 7417–7432. [Google Scholar] [CrossRef]
- Kim, M.S.; Ho, M.J.; Joung, M.Y.; Choi, Y.S.; Kang, M.J. Effect of Dispersion Medium on Pharmacokinetic Profile of Rotigotine Crystalline Suspension Following Subcutaneous Injection. Pharmaceutics 2022, 14, 2630. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, M.S.; Joung, M.Y.; Park, H.J.; Ho, M.-J.; Choi, J.H.; Seo, J.H.; Song, W.H.; Choi, Y.W.; Lee, S.; et al. Design of Montelukast Nanocrystalline Suspension for Parenteral Prolonged Delivery. Int. J. Nanomed. 2022, 17, 3673–3690. [Google Scholar] [CrossRef]
- Lynnerup, J.T.; Eriksen, J.B.; Bauer-Brandl, A.; Holsæter, A.M.; Brandl, M. Insight into the Mechanism behind Oral Bioavailability-Enhancement by Nanosuspensions through Combined Dissolution/Permeation Studies. Eur. J. Pharm. Sci. 2023, 184, 106417. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Staufenbiel, S.; Bodmeier, R. Combination of Co-Crystal and Nanocrystal Techniques to Improve the Solubility and Dissolution Rate of Poorly Soluble Drugs. Pharm. Res. 2022, 39, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z. Development and Evaluation of Nano-Formulations for Immediate Release Oral Dosage Forms of Poorly Soluble Drugs. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2022. [Google Scholar]
- Zhang, C.; Bodmeier, R. A Comparative Study of PLGA Microparticle Properties Loaded with Micronized, Nanosized or Dissolved Drug. Int. J. Pharm. 2022, 628, 122313. [Google Scholar] [CrossRef] [PubMed]
- Zulbeari, N.; Holm, R. Wet Bead Milling by Dual Centrifugation—An Approach to Obtain Reproducible and Differentiable Suspensions. Int. J. Pharm. 2023, 646, 123455. [Google Scholar] [CrossRef]
- Huang, Z.; Staufenbiel, S.; Bodmeier, R. Incorporation of Itraconazole Nano-Co-Crystals into Multiparticulate Oral Dosage Forms. Eur. J. Pharm. Biopharm. 2022, 176, 75–86. [Google Scholar] [CrossRef]
- Huang, Z.; Staufenbiel, S.; Bodmeier, R. Kinetic Solubility Improvement and Influence of Polymers on Controlled Supersaturation of Itraconazole-Succinic Acid Nano-Co-Crystals. Int. J. Pharm. 2022, 616, 121536. [Google Scholar] [CrossRef]
- Klein, T.; Gruschwitz, F.V.; Kuchenbrod, M.T.; Nischang, I.; Hoeppener, S.; Brendel, J.C. Adjusting the Length of Supramolecular Polymer Bottlebrushes by Top-down Approaches. Beilstein J. Org. Chem. 2021, 17, 2621–2628. [Google Scholar] [CrossRef]
- Agate, S.; Tyagi, P.; Naithani, V.; Lucia, L.; Pal, L. Innovating Generation of Nanocellulose from Industrial Hemp by Dual Asymmetric Centrifugation. ACS Sustain. Chem. Eng. 2020, 8, 1850–1858. [Google Scholar] [CrossRef]
- Raghav, N.; Sharma, M.R.; Kennedy, J.F. Nanocellulose: A Mini-Review on Types and Use in Drug Delivery Systems. Carbohydr. Polym. Technol. Appl. 2021, 2, 100031. [Google Scholar] [CrossRef]
- Deuringer, B.; Härdtner, C.; Krebs, K.; Thomann, R.; Holzer, M.; Hilgendorf, I.; Süss, R. Everolimus-Loaded Reconstituted High-Density Lipoprotein Prepared by a Novel Dual Centrifugation Approach for Anti-Atherosclerotic Therapy. Int. J. Nanomed. 2022, 17, 5081–5097. [Google Scholar] [CrossRef]
- Roerig, J.; Mitrach, F.; Schmid, M.; Hause, G.; Hacker, M.C.; Wölk, C.; Schulz-Siegmund, M. Synergistic siRNA Loading of Extracellular Vesicles Enables Functional Delivery into Cells. Small Methods 2022, 6, 2201001. [Google Scholar] [CrossRef]
- Pohlit, H.; Bellinghausen, I.; Schömer, M.; Heydenreich, B.; Saloga, J.; Frey, H. Biodegradable pH-Sensitive Poly(Ethylene Glycol) Nanocarriers for Allergen Encapsulation and Controlled Release. Biomacromolecules 2015, 16, 3103–3111. [Google Scholar] [CrossRef]
- Kutza, C.; Metz, H.; Kutza, J.; Syrowatka, F.; Mäder, K. Toward a Detailed Characterization of Oil Adsorbates as “Solid Liquids”. Eur. J. Pharm. Biopharm. 2013, 84, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal mRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Gurzeler, L.-A.; Ziegelmüller, J.; Mühlemann, O.; Karousis, E.D. Production of Human Translation-Competent Lysates Using Dual Centrifugation. RNA Biol. 2022, 19, 78–88. [Google Scholar] [CrossRef] [PubMed]





| Drug | Stabilizers | DC Device | Smallest Median or Mean Particle Size 1 | Highlights | Reference |
|---|---|---|---|---|---|
| Centella asiatica extract | Carbopol 934NF, HPMC E50LV, Kollidon VA64, Kolliphor EL, Kolliphor HS15, Kolliphor RH 40, Lecithin, MC, Na-CMC, PEG 40 stearate, Poloxamer 188, Polysorbate 20, Polysorbate 80, PVP K30, Tyloxapol | ZM 380R | 145 nm PDI: 0.28 | The optimized nanosuspension formulation for improved skin absorption without occurrence of skin irritations contains centella asiatica extract (10% w/v), PVP (0.5%), and water (89.5%). HPMC was able to achieve the smallest size after preparation, but PVP exhibited higher stability. | [83] |
| Cinnarizine | HPMC, SDS | DAC 150 | 275 nm PDI: 0.29 | Particles retained their crystallinity after milling. Nanosizing shows no significant increase in thermodynamic drug solubility but in dissolution rate. A 90 min milling time is identified as the best milling duration for milling speed of 1500 rpm. | [86] |
| Poloxamer 188, Polysorbate 20, SDS | DV 1 | 587 nm | Poloxamer 188 (4%) led to the smallest particles after milling. No change in crystallinity could be observed. | [90] | |
| Curcumin | HPMC, SDS, Polysorbate 80, PVP-VA64 | ZM 380R | 113 nm | Three of four stabilizer combinations tested led to the same size distribution of nanoparticles. | [68] |
| Dexamethasone | SDS | ZM 380R | 180 nm after freeze-drying | Encapsulation of nanosized drug in PLGA microparticles with high encapsulation efficiencies (>85%) and good release properties. | [89] |
| Fenofibrate | DOSS, HPMC, Polysorbate 80, PVP-K25, SDS | ZM 380R | 127 nm | First use of dual centrifugation for API-nanomilling. No change in the crystal structure during milling. Results of DC-nanomilling are similar to results obtained by agitator mills. | [3,81] |
| HPMC, SDS | DAC 150 | 259 nm PDI: approx. 0.2 | Particles retained their crystallinity. No significant increase in thermodynamic drug solubility could be observed but in dissolution rate. The study shows that in vitro dissolution/permeation studies can be employed to better understand oral absorption enhancement of nanocrystal formulations. | [86] | |
| Hydrocortisone | SDS | ZM 380R | 160 nm after freeze-drying | The nanosized drugs were encapsulated in PLGA microparticles showing a more continuous release than the micronized drug and a better encapsulation efficiency than with dissolved drug. | [89] |
| Ibuprofen | HPMC, SDS | ZM 380R | approx. 190 nm | DC-milling of ibuprofen led to smaller particles than milling with a planetary ball mill. | [3,81] |
| Indomethacin | Poloxamer 188, Polysorbate 20, SDS | DV 1 | 355 nm | Nanomilling with DC led to highly reproducible results regarding particle size, distribution, and stability. SDS (1%) resulted in the smallest particles sizes after milling. However, nanosuspensions stabilized with poloxamer 188 (4%) showed better short-term stability over 28 days. | [90] |
| Poloxamer 407 | ZM 380R | 163 nm PDI: 0.14 after freeze-drying | The combination of nanocrystals and co-crystals enables a better kinetic solubility and faster dissolution rates compared to the single components. | [87] | |
| Indomethacin -nicotinamide-co-crystals | Poloxamer 407 | ZM 380R | 280 nm PDI: 0.29 after freeze-drying | [87] | |
| Indomethacin-saccharin-co-crystals | Poloxamer 407 | ZM 380R | 329 nm PDI: 0.20 after freeze-drying | [87] | |
| Itraconazole | HPC-SL, SDS, Polysorbate 80 | ZM 380R | 127 nm PDI: 0.18 | A combination of three stabilizers at minimal concentrations of 0.9% HPC-SL, 0.14% SDS, and 0.07% polysorbate 80 (all w/w) was necessary for sufficient stabilization | [82] |
| HPC, HPMC E5, Poloxamer 188, Poloxamer 407, Polysorbate 80, PVP K30, SDS, TPGS | 223 nm PDI: 0.24 after freeze-drying | PVP and HPMC were not able to form stable itraconazole nanosuspensions | [87,88] | ||
| Poloxamer 407 | 211 nm PDI: 0.22 | Addition of HPMC E5 to the itraconazole nanosuspension did not increase the crystal solubility. | [91] | ||
| Itraconazole-fumaric acid-co-crystals | Poloxamer 407 | ZM 380R | 443 nm PDI: 0.35 after freeze-drying | Itraconazole nano-co-crystals with a size of about 450 nm were successfully prepared for the first time. | [87] |
| Itraconazole-succinic acid-co-crystals | Poloxamer 407 | ZM 380R | 455 nm PDI: 0.24 after freeze-drying | [87,92] | |
| Poloxamer 407 | ZM 380R | 365 nm PDI: 0.25 | Itraconazole-succinic acid-co-crystals can be effectively integrated into an oral solid dosage form using bead layering as a downstream method without negatively affecting the drug dissolution. | [91] | |
| Mitotane | Bovine serum albumin | ZM 380R | 359 nm PDI: 0.14 | A stable and storable mitotane formulation with a high drug content and good in vitro characteristics was developed. | [17] |
| Montelukast | Cremophor EL, Cremophor RH40, Na-CMC, PEG 4000, Poloxamer 188, Polysorbate 80, PVP K17, Solutol HS15, Tyloxapol | ZM 380R | 190 nm PDI: not indicated | Polysorbate 80 leads to the smallest and most homogeneous particles. | [85] |
| Naproxen | DOSS, HPMC | ZM 380R | approx. 130 nm | No difference in crystal structure after milling could be observed. | [3,81] |
| Polysorbate 80, PVP-K25 | [3] | ||||
| Rotigotine | Kolliphor EL, Kolliphor HS 15, Kolliphor RH 40, Na-CMC, PEG 4000, Poloxamer 188, Polysorbate 80, PVP K17 | ZM 380R | 375 nm PDI: 0.206 | Na-CMC turned out to be the best stabilizer leading to a small particle size with an appropriate homogeneity. | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koehler, J.K.; Schmager, S.; Bender, V.; Steiner, D.; Massing, U. Preparation of Nanosized Pharmaceutical Formulations by Dual Centrifugation. Pharmaceuticals 2023, 16, 1519. https://doi.org/10.3390/ph16111519
Koehler JK, Schmager S, Bender V, Steiner D, Massing U. Preparation of Nanosized Pharmaceutical Formulations by Dual Centrifugation. Pharmaceuticals. 2023; 16(11):1519. https://doi.org/10.3390/ph16111519
Chicago/Turabian StyleKoehler, Jonas K., Stefanie Schmager, Valentin Bender, Denise Steiner, and Ulrich Massing. 2023. "Preparation of Nanosized Pharmaceutical Formulations by Dual Centrifugation" Pharmaceuticals 16, no. 11: 1519. https://doi.org/10.3390/ph16111519
APA StyleKoehler, J. K., Schmager, S., Bender, V., Steiner, D., & Massing, U. (2023). Preparation of Nanosized Pharmaceutical Formulations by Dual Centrifugation. Pharmaceuticals, 16(11), 1519. https://doi.org/10.3390/ph16111519