
Benzimidazoles Containing Piperazine Skeleton at C-2 Position as Promising Tubulin Modulators with Anthelmintic and Antineoplastic Activity

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
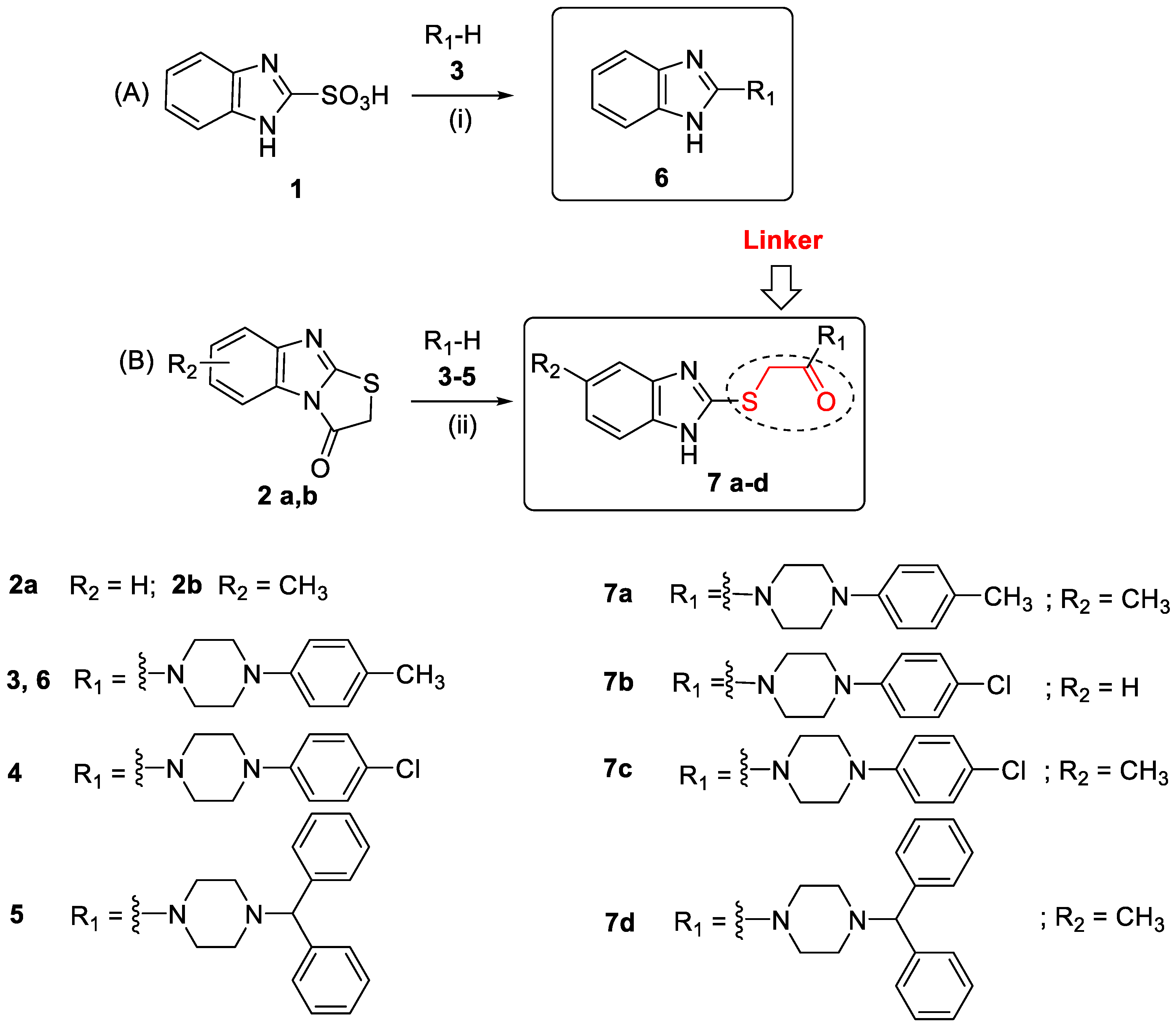
2.1. Synthesis
2.2. Antihelmintic Activity
2.3. Cytotoxic Activity
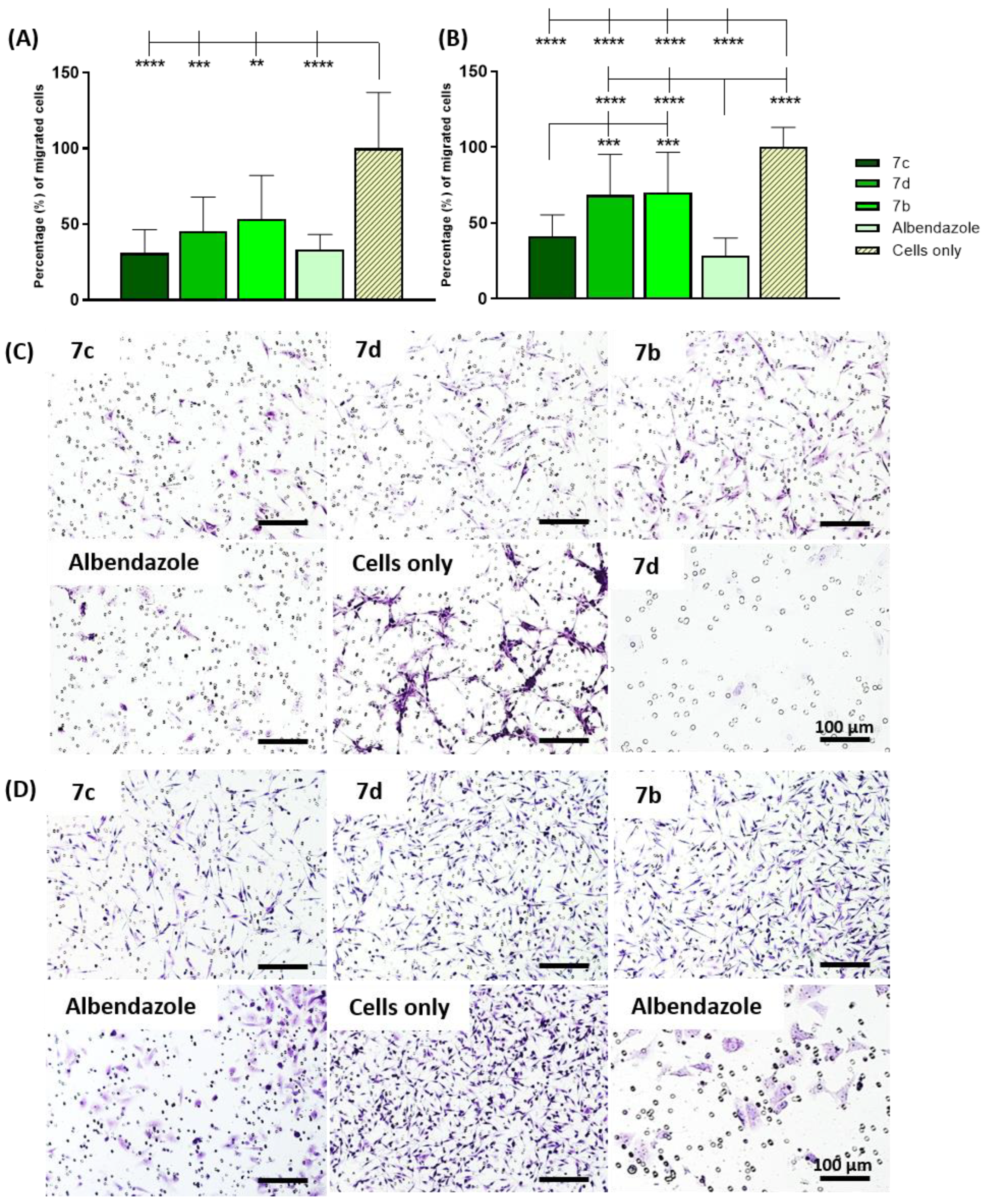
2.4. Lactate Dehydrogenase (LDH) Release, Cell Migration, and Morphological Changes
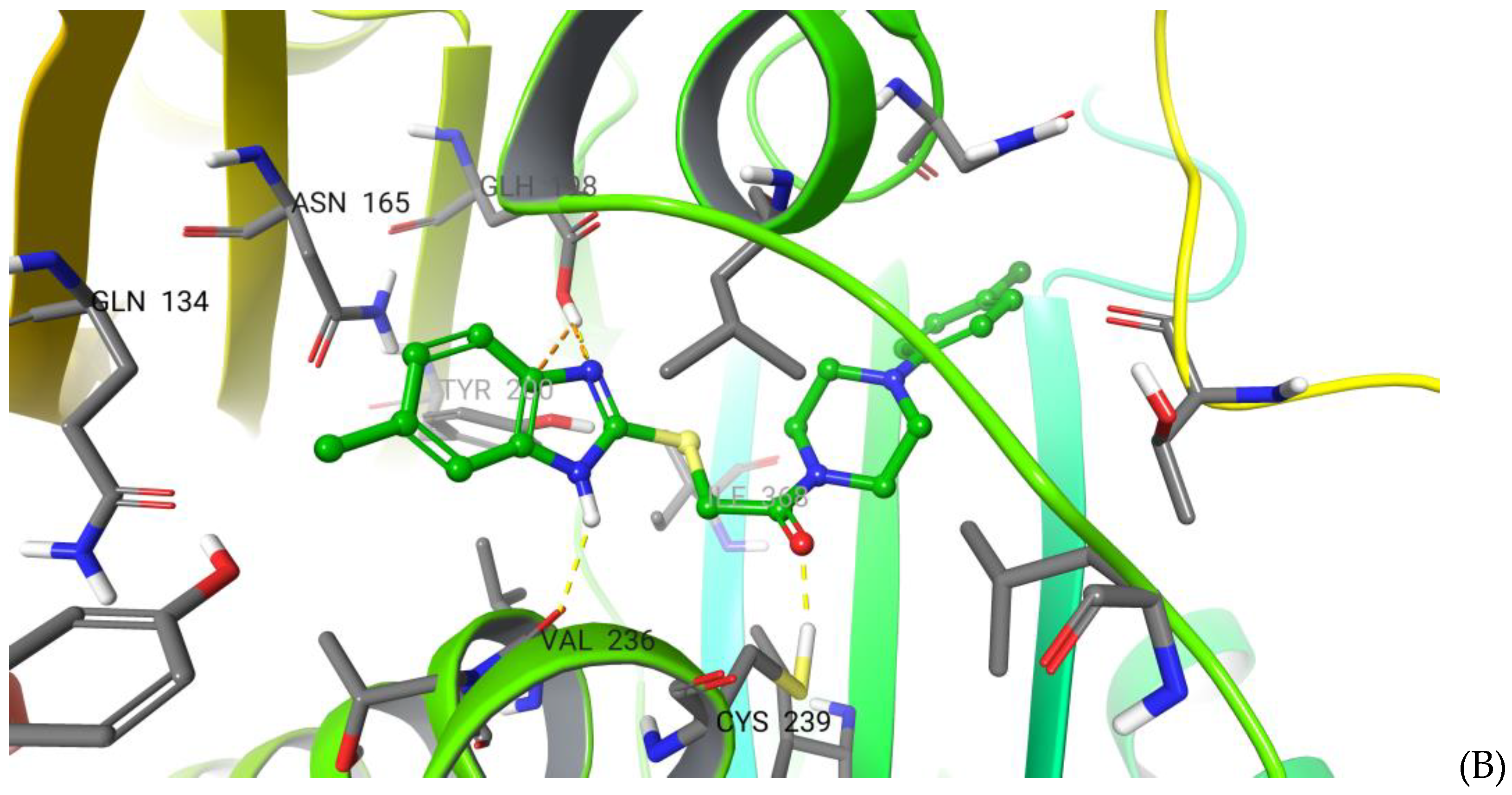
2.5. Molecular Modelling and Absolute Free Energy of Perturbations Studies
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Compounds 6
3.2.2. General Procedure for the Synthesis of Compounds 7a–d [30]
3.2.3. Compound Data
3.3. Anthelmintic Study
3.4. In Vitro Antineoplastic Study
3.4.1. Cell Culture
3.4.2. MTT Cell Viability Assay and IC50 Calculation
3.4.3. LDH Release
3.4.4. Cell Migration Evaluation
3.4.5. Cell Morphology Evaluation
3.4.6. Statistical Analysis
3.5. Molecular Modelling and Absolute Free Energy of Perturbation Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Talev, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2019, 15, 397–401. [Google Scholar] [CrossRef]
- Jourdan, J.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Gualtieri, G.; Raimondi, M.V.; Eugenio Gaudio, E.; Bortolozzi, R.; Manfreda, L.; Bai, R.; et al. Identification of pyr-rolo[3′,4′:3,4]cyclohepta[1,2-d][1,2]oxazoles as promising new candidates for the treatment of lymphomas. Eur. J. Med. Chem. 2023, 254, 115372. [Google Scholar] [CrossRef] [PubMed]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef]
- Bivacqua, R.; Barreca, M.; Spanò, V.; Raimondi, M.V.; Romeo, I.; Alcaro, S.; Andrei, G.; Barraja, P.; Montalbano, A. Insight into non-nucleoside triazole-based systems as viral polymerases inhibitors. Eur. J. Med. Chem. 2023, 249, 115136. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Patziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)-mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443. [Google Scholar] [CrossRef]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers 2019, 11, 1284. [Google Scholar] [CrossRef]
- Son, D.-S.; Lee, E.-S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, e29. [Google Scholar] [CrossRef]
- Song, B.; Park, E.Y.; Kim, K.J.; Ki, S.H. Repurposing of Benzimidazole Anthelmintic Drugs as Cancer Therapeutics. Cancers 2022, 14, 4601. [Google Scholar] [CrossRef]
- Narasimhan, P.B.; Akabas, L.; Tariq, S.; Huda, N.; Bennuru, S.; Sabzevari, H.; Hofmeister, R.; Thomas, B.; Nutman, T.B.; Semnani, R.T. Similarities and differences between helminth parasites and cancer cell lines in shaping human monocytes: Insights into parallel mechanisms of immune evasion. PLoS Negl. Trop. Dis. 2018, 12, e0006404. [Google Scholar] [CrossRef] [PubMed]
- Surkau, G.; Böhm, K.J.; Müller, K.; Prinz, H. Synthesis, antiproliferative activity and inhibition of tubulin polymerization by anthracene-based oxime derivatives. Eur. J. Med. Chem. 2010, 45, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Pourgholami, M.H.; Szwajcer, M.; Chin, M.; Liauw, W.; Seef, J.; Galettis, P.; Morris, D.L.; Links, M. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in patients with advanced cancer. Cancer Chemother. Pharmacol. 2010, 65, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, S.; Fryknäs, M.; Alvfors, C.; Loskog, A.; Larsson, R.; Nygren, P. A phase 2a clinical study on the safety and efficacy of individualized dosed mebendazole in patients with advanced gastrointestinal cancer. Sci. Rep. 2021, 11, 8981. [Google Scholar] [CrossRef]
- He, L.; Shi, L.; Du, Z.; Huang, H.; Gong, R.; Ma, L.; Chen, L.; Gao, S.; Lyu, J.; Gu, H. Mebendazole exhibits potent anti-leukemia activity on acute myeloid leukemia. Exp. Cell Res. 2018, 369, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Xinggang, M.; Xiaoyan, X.; Xiang, Z.; Luo’an, F. Application of Albendazole in Preparation of Medicines for Treating Glioblastoma. Patent CN 108078987(A), 29 May 2018. [Google Scholar]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef]
- Petersen, J.S.S.M.; Baird, S.K. Treatment of breast and colon cancer cell lines with anti-helmintic benzimidazoles mebendazole or albendazole results in selective apoptotic cell death. J. Cancer Res. Clin. Oncol. 2021, 147, 2945–2953. [Google Scholar] [CrossRef]
- Kim, U.; Changsoo, S.; Kim, C.-Y.; Bokyeong, R.; Kim, J.; Bang, J.; Park, J.-H. Albendazole exerts antiproliferative effects on prostate cancer cells by inducing reactive oxygen species generation. Oncol. Lett. 2021, 21, 395. [Google Scholar] [CrossRef]
- Anichina, K.; Argirova, M.; Tzoneva, R.; Uzunova, V.; Mavrova, A.; Vuchev, D.; Popova-Daskalova, G.V.; Fratev, F.; Guncheva, M.; Yancheva, D. 1H-benzimidazole-2-yl hydrazones as tubulin-targeting agents: Synthesis, structural characterization, anthelmintic activity and antiproliferative activity against MCF-7 breast carcinoma cells and molecular docking studies. Chem. Biol. Interact. 2021, 345, 109540. [Google Scholar] [CrossRef]
- Fisher, M.H. Chemistry of Antinematodal Agents. In Chemotherapy of Parasitic Diseases, 1st ed.; Campbell, W.C., Rew, R.C., Eds.; Springer: Boston, MA, USA, 1986; pp. 253–255. [Google Scholar]
- Akkoç, M.K.; Yüksel, M.Y.; Durmaz, İ.; Atalay, R.Ç. Design, synthesis, and biological evaluation of indole-based 1,4-disubstituted piperazines as cytotoxic agents. Turk. J. Chem. 2012, 36, 515–525. [Google Scholar] [CrossRef]
- Waszkielewicz, A.M.; Gunia, A.; Szkaradek, N.; Pytka, K.; Siwek, A.; Satała, G.; Bojarski, A.J.; Szneler, E.; Marona, H. Synthesis and evaluation of pharmacological properties of some new xanthone derivatives with piperazine moiety. Bioorganic Med. Chem. Lett. 2013, 23, 4419–4423. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Jadala, C.; Sathish, M.; Pratibha, A.; Tokala, R.; Lakshmi, U.J.; Reddy, V.G.; Shankaraiah, N.; Godugu, C. Synthesis of CA4 carboxamides mimicking with sulfonyl piperazines by a molecular hybridization approach: In vitro cytotoxicity evaluation and tubulin polymerization inhibition. Chem. Med. Chem. 2019, 14, 2052–2060. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, D.W.; Kwon, H.S.; Lee, S.H.; Park, S.K.; Chung, S.G.; Cho, E.H.; Paik, S.Y.; Lee, J.H. Microtubule inhibitory effects of various SJ compounds on tissue culture cells. Arch. Pharm. Res. 2004, 27, 436–441. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Ana, G.; Kelly, P.M.; Nathwani, S.M.; Noorani, S.; Fayne, D.; Bright, S.A.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Synthesis and Evaluation of Antiproliferative Microtubule-Destabilising Combretastatin A-4 Piperazine Conjugates. Org. Biomol. Chem. 2019, 17, 6184–6200. [Google Scholar] [CrossRef]
- Özdemir, A.; Turanli, S.; Çalişkan, B.; Arka, M.; Banoglu, E. Evaluation of Cytotoxic Activity of New Benzimidazole-Piperazine Hybrids Against Human MCF-7 and A549 Cancer Cells. Pharm. Chem. J. 2020, 53, 1036–1045. [Google Scholar] [CrossRef]
- Argirova, M.A.; Georgieva, M.K.; Hristova-Avakumova, N.G.; Vuchev, D.I.; Popova-Daskalova, G.V.; Anichina, K.K.; Yancheva, D.Y. New 1H-benzimidazole-2-yl hydrazones with combined antiparasitic and antioxidant activity. RSC Adv. 2021, 11, 39848–39868. [Google Scholar] [CrossRef]
- Winn, M.; Kyncl, J. N-(2-benzimidazolyl)-piperazines. Patent US 4093726 (A), 6 June 1978. [Google Scholar]
- Mavrova, A.T.; Anichina, K.K.; Vuchev, D.I.; Tsenov, J.A.; Denkova, P.S.; Kondeva, M.S.; Micheva, M.K. The anthelminthic activity of some newly synthesized 5(6)-(un)substituted-1H-benzimidazol-2-ylthioacetylpiperazine derivatives. Eur. J. Med. Chem. 2006, 41, 1412–1420. [Google Scholar] [CrossRef]
- Anichina, K.; Georgiev, N.; Lumov, N.; Vuchev, D.; Popova-Daskalova, G.; Momekov, G.; Cherneva, E.; Mihaylova, R.; Mavrova, A.; Atanasova-Vladimirova, S.; et al. Fused Triazinobenzimidazoles Bearing Heterocyclic Moiety: Synthesis, Structure Investigations, and In Silico and In Vitro Biological Activity. Molecules 2023, 28, 5034. [Google Scholar] [CrossRef]
- Chai, J.-Y.; Jung, B.-K.; Hong, S.-J. Albendazole and Mebendazole as Anti-Parasitic and Anti-Cancer Agents: An Update. Korean J. Parasitol. 2021, 59, 189–225. [Google Scholar] [CrossRef]
- Mavrova, A.T.; Anichina, K.K.; Vuchev, D.I.; Tsenov, J.A.; Kondeva, M.S.; Micheva, M.K. Synthesis and an-titrichinellosis activity of some 2-substituted-[1,3]thiazolo[3,2-a]benzimidazol-3(2H)-ones. Bioorg. Med. Chem. 2005, 13, 5550–5559. [Google Scholar] [CrossRef]
- Schrödinger Suite, 2023–3; Schrödinger, LLC: New York, NY, USA. 2023. Available online: https://www.schrodinger.com/ (accessed on 26 September 2023).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein-Ligand Binding. J. Chem. Theory. Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef]
- Chen, W.; Cui, D.; Jerome, S.V.; Michino, M.; Lenselink, E.B.; Huggins, D.J.; Beautrait, A.; Vendome, J.; Abel, R.; Friesner, R.A.; et al. Enhancing Hit Discovery in Virtual Screening through Absolute Protein-Ligand Binding Free-Energy Calculations. J. Chem. Inf. Model. 2023, 63, 3171–3185. [Google Scholar] [CrossRef] [PubMed]
- Khalak, Y.; Tresadern, G.; Aldeghi, M.; Baumann, H.M.; Mobley, D.L.; de Groot, B.L.; Gapsys, V. Alchemical absolute protein-ligand binding free energies for drug design. Chem. Sci. 2021, 12, 13958–13971. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Steinbrecher, T.; Jónsdóttir, S.Ó. Prediction of Accurate Binding Modes Using Combination of Classical and Accelerated Molecular Dynamics and Free-Energy Perturbation Calculations: An Application to Toxicity Studies. ACS Omega 2018, 3, 4357–4371. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F. PPARγ non-covalent antagonists exhibit mutable binding modes with a similar free energy of binding: A case study. J. Biomol. Struct. Dyn. 2017, 35, 476–485. [Google Scholar] [CrossRef]
- Adhikari, S.; Daftardar, S.; Fratev, F.; Rivera, M.; Sirimulla, S.; Alexander, K.; Boddu, S.H.S. Elucidation of the orientation of selected drugs with 2-hydroxylpropyl-β-cyclodextrin using 2D-NMR spectroscopy and molecular modeling. Int. J. Pharm. 2018, 545, 357–365. [Google Scholar] [CrossRef]
- Fratev, F. PPARγ helix 12 exhibits an antagonist conformation. Phys. Chem. Chem. Phys. 2016, 18, 9272–9280. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration 50 (100) µg/mL in µM | Efficacy (%) b After 24 h | Efficacy (%) b After 48 h | ||
|---|---|---|---|---|---|
| 50 µg/mL | 100 µg/mL | 50 µg/mL | 100 µg/mL | ||
| 6 | 0.171(0.342) | 13.5 | 19.5 | 14.8 | 33.8 |
| 7a c | 0.131 (0.263) | 26.5 | 56.8 | 34.7 | 63.0 |
| 7b c | 0.129 (0.358) | 29.6 | 64.7 | 33.5 | 83.0 |
| 7c c | 0.125 (0.249) | 35.8 | 72.0 | 41.0 | 92.7 |
| 7d c | 0.110 (0.219) | 37.4 | 75.8 | 44.0 | 88.0 |
| Albendazole | 0.188 (0.377) | 10.7 | 10.9 | 14.4 | 15.6 |
| Ivermectin | 0.057 (0.114) | 45.4 | 49.2 | 62.0 | 78.3 |
| MDA-MB-231 Cell | U-87 MG Cells | |||
|---|---|---|---|---|
| Compound | IC50 (µM) | 95% CI (µM) | IC50 (µM) | 95% CI (µM) |
| 6 | 102.7 | (+309.7, −50.23) | 104.7 | (+1700, −60.69) |
| 7a | 74.37 | (+192.03, −35.18) | 54.86 | (+121.34, −20.26) |
| 7b | 39.78 | (+18.11, −9.96) | 41.42 | (+60.08, −15.49) |
| 7c | 35.84 | (+11.11, −7.11) | 42.30 | (+12.47, −7.62) |
| 7d | 34.31 | (+1.81, −1.71) | 38.29 | (+4.99, −4.25) |
| Albendazole | 83.1 | (+103.7, −33.59) | 40.59 | (+106.31, −19.92) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anichina, K.; Mavrova, A.; Vuchev, D.; Popova-Daskalova, G.; Bassi, G.; Rossi, A.; Montesi, M.; Panseri, S.; Fratev, F.; Naydenova, E. Benzimidazoles Containing Piperazine Skeleton at C-2 Position as Promising Tubulin Modulators with Anthelmintic and Antineoplastic Activity. Pharmaceuticals 2023, 16, 1518. https://doi.org/10.3390/ph16111518
Anichina K, Mavrova A, Vuchev D, Popova-Daskalova G, Bassi G, Rossi A, Montesi M, Panseri S, Fratev F, Naydenova E. Benzimidazoles Containing Piperazine Skeleton at C-2 Position as Promising Tubulin Modulators with Anthelmintic and Antineoplastic Activity. Pharmaceuticals. 2023; 16(11):1518. https://doi.org/10.3390/ph16111518
Chicago/Turabian StyleAnichina, Kameliya, Anelia Mavrova, Dimitar Vuchev, Galya Popova-Daskalova, Giada Bassi, Arianna Rossi, Monica Montesi, Silvia Panseri, Filip Fratev, and Emilia Naydenova. 2023. "Benzimidazoles Containing Piperazine Skeleton at C-2 Position as Promising Tubulin Modulators with Anthelmintic and Antineoplastic Activity" Pharmaceuticals 16, no. 11: 1518. https://doi.org/10.3390/ph16111518
APA StyleAnichina, K., Mavrova, A., Vuchev, D., Popova-Daskalova, G., Bassi, G., Rossi, A., Montesi, M., Panseri, S., Fratev, F., & Naydenova, E. (2023). Benzimidazoles Containing Piperazine Skeleton at C-2 Position as Promising Tubulin Modulators with Anthelmintic and Antineoplastic Activity. Pharmaceuticals, 16(11), 1518. https://doi.org/10.3390/ph16111518