
Thiadiazolidinone (TDZD) Analogs Inhibit Aggregation-Mediated Pathology in Diverse Neurodegeneration Models, and Extend C. elegans Life- and Healthspan

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
2.1. PNR886 and PNR962 Reduce Aggregation in C. elegans Models of Neurodegeneration
2.2. TDZD Analog Treatments Extend Lifespan and Healthspan of Wild-Type Nematodes
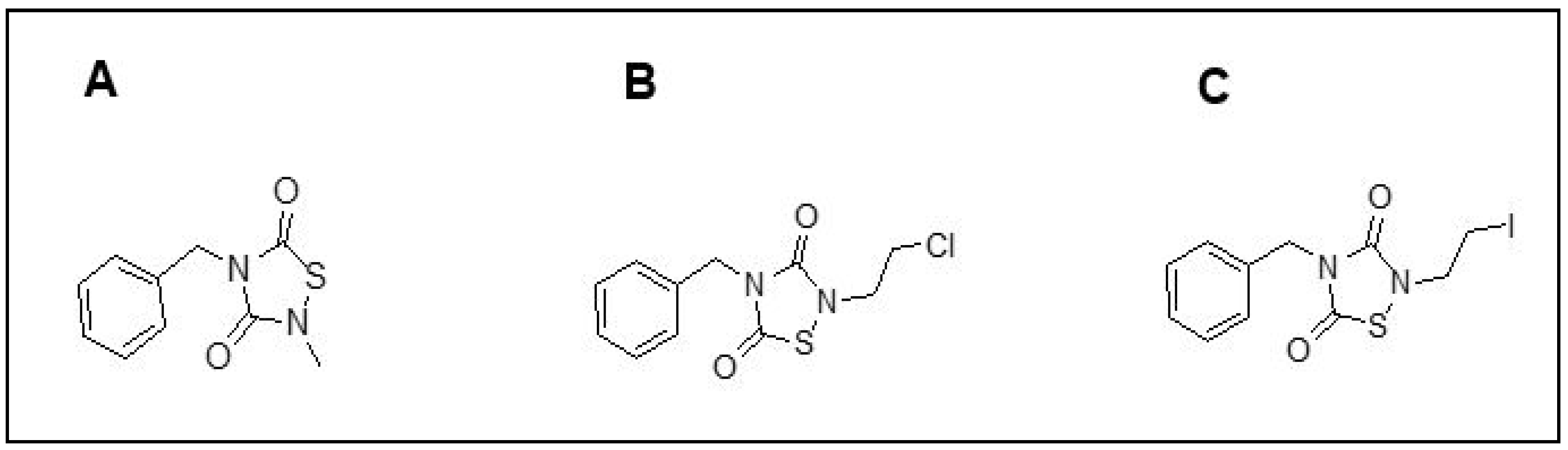
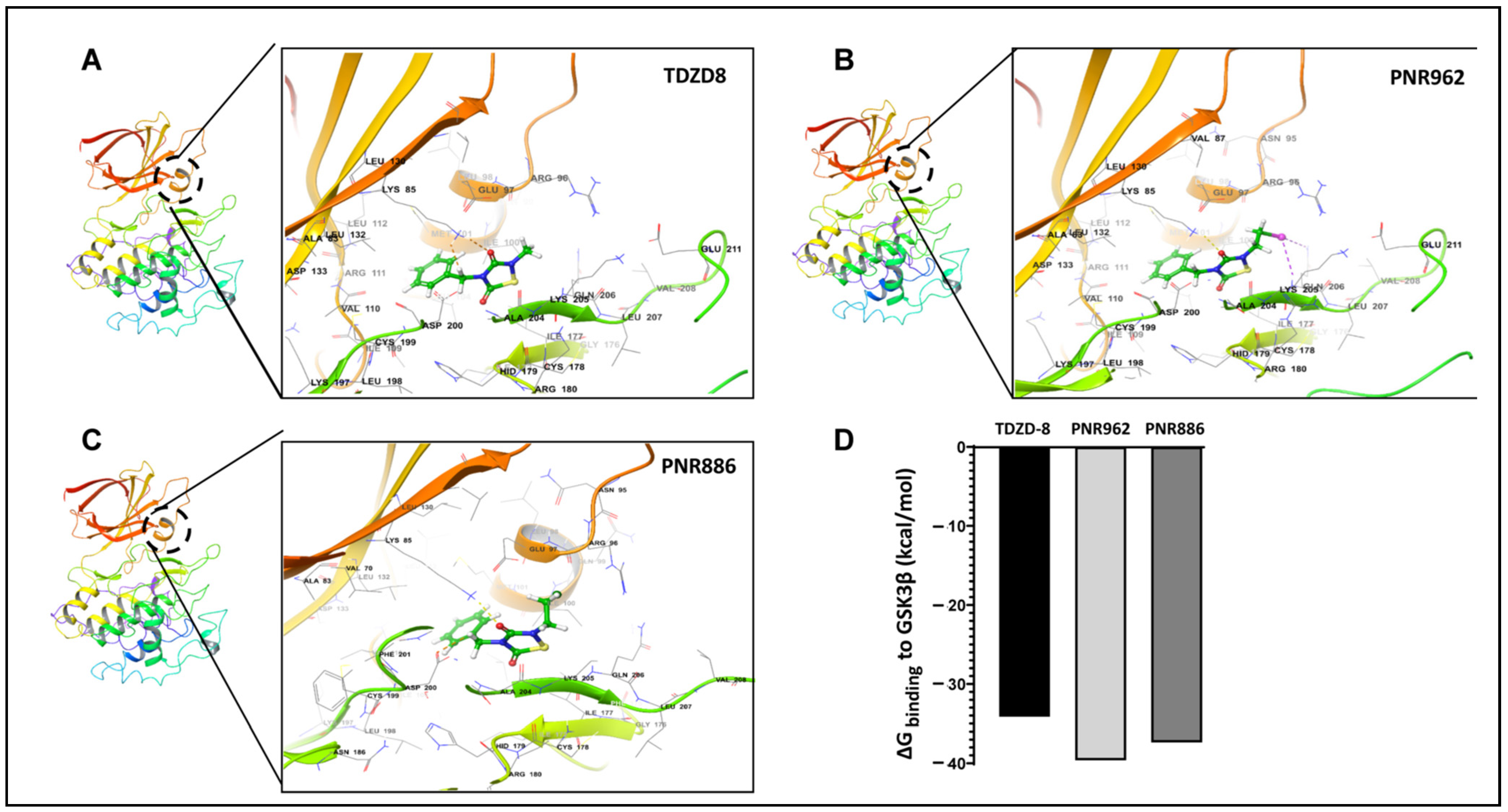
2.3. PNR886 and PNR962 Bind the Inactive-GSK3β Allosteric Pocket More Avidly than TDZD-8
2.4. Ligand Free-Energy Perturbation Predicts Improved GSK3β Binding by New Analogs
3. Discussion
4. Materials and Methods
4.1. Effects of PNR886 and PNR962 on Protein Aggregation in Human Cells
4.2. Thioflavin T Staining of Cultured Human Cells to Quantify Relative Aggregation Levels
4.3. C. elegans Strains
4.4. Effects of TDZD Analogs PNR886 and PNR962 on Aggregation in C. elegans Strain AM141
4.5. Paralysis Assay in the Aβ-Transgenic Nematode Strain CL4176
4.6. Lifespan Studies on the Nematode C. elegans Wild-Type Strain, Bristol-N2/DRM
4.7. Structural Modeling of TDZD Analogs and GSK3β
4.8. Docking of TDZD Analogs to the Inactive Conformation of GSK3β
4.9. Atomistic Molecular-Dynamic Simulations
4.10. Binding Energy (ΔGbinding) Computation in MM-GBSA
4.11. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of Metabolism in Neurodegenerative Disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.; Antonelli, A.C.; Afridi, A.; Vatsia, S.; Joshi, G.; Romanov, V.; Murray, I.V.J.; Khan, S.A. Protein Misfolding and Aggregation in Neurodegenerative Diseases: A Review of Pathogeneses, Novel Detection Strategies, and Potential Therapeutics. Rev. Neurosci. 2019, 30, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Jorm, A.F.; Jolley, D. The Incidence of Dementia: A Meta-Analysis. Neurology 1998, 51, 728–733. [Google Scholar] [CrossRef]
- Bertram, L. The Genetic Epidemiology of Neurodegenerative Disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Buchner, J. Supervising the Fold: Functional Principles of Molecular Chaperones. FASEB J. 1996, 10, 10–19. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10 (Suppl. S7), S10–S17. [Google Scholar] [CrossRef]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread Protein Aggregation as an Inherent Part of Aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Protein Aggregates in Huntington’s Disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein Homeostasis, Aging and Alzheimer’s Disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, G.; Greig, N.; Khan, T.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.; Zaidi, S.; Akram, M.; Jabir, N.; et al. Protein Misfolding and Aggregation in Alzheimer’s Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 Diabetes as a Protein Misfolding Disease. Trends Mol. Med. 2015, 21, 439–449. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-Talk between Amyloidogenic Proteins in Type-2 Diabetes and Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Kakraba, S.; Alla, R.; Mehta, J.L.; Shmookler Reis, R.J. Aspirin-Mediated Acetylation Protects Against Multiple Neurodegenerative Pathologies by Impeding Protein Aggregation. Antioxid. Redox Signal 2017, 27, 1383–1396. [Google Scholar] [CrossRef]
- Press, M.; Jung, T.; König, J.; Grune, T.; Höhn, A. Protein Aggregates and Proteostasis in Aging: Amylin and β-Cell Function. Mech. Ageing Dev. 2019, 177, 46–54. [Google Scholar] [CrossRef]
- Thériault, P.; ElAli, A.; Rivest, S. High Fat Diet Exacerbates Alzheimer’s Disease-Related Pathology in APPswe/PS1 Mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef]
- Zheng, L.; Duan, J.; Duan, X.; Zhou, W.; Chen, C.; Li, Y.; Chen, J.; Zhou, W.; Wang, Y.-J.; Li, T.; et al. Association of Apolipoprotein E (ApoE) Polymorphism with Alzheimer’s Disease in a Chinese Population. Curr. Alzheimer Res. 2016, 13, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, J.-P.; Mononen, N.; Hutri-Kähönen, N.; Lehtimäki, M.; Juonala, M.; Ala-Korpela, M.; Kähönen, M.; Raitakari, O.; Lehtimäki, T. The Effect of Apolipoprotein E Polymorphism on Serum Metabolome—A Population-Based 10-Year Follow-up Study. Sci. Rep. 2019, 9, 458. [Google Scholar] [CrossRef]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of Sex and High-Fat Diet in Metabolic and Hypothalamic Disturbances in the 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Neuroinflamm. 2020, 17, 285. [Google Scholar] [CrossRef] [PubMed]
- Okusaga, O. Accelerated Aging in Schizophrenia Patients: The Potential Role of Oxidative Stress. Aging Dis. 2014, 5, 256–262. [Google Scholar] [CrossRef] [PubMed]
- King, E.; O’Brien, J.T.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.-P.; Thomas, A.J. Peripheral Inflammation in Prodromal Alzheimer’s and Lewy Body Dementias. J. Neurol. Neurosurg. Psychiatry 2018, 89, 339–345. [Google Scholar] [CrossRef]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative Frequencies of Alzheimer Disease, Lewy Body, Vascular and Frontotemporal Dementia, and Hippocampal Sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease:: Getting On and Staying On. Curr. Ther. Res. 2003, 64, 216–235. [Google Scholar] [CrossRef]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and Safety of Donepezil, Galantamine, and Rivastigmine for the Treatment of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar]
- Li, D.-D.; Zhang, Y.-H.; Zhang, W.; Zhao, P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 472. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-Inflammation and Anti-Inflammatory Treatment Options for Alzheimer’s Disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef]
- Moss, D.E. Improving Anti-Neurodegenerative Benefits of Acetylcholinesterase Inhibitors in Alzheimer’s Disease: Are Irreversible Inhibitors the Future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Fitzpatrick, A.; Ives, D.G.; Saxton, J.; Williamson, J.; Lopez, O.L.; Burke, G.; Fried, L.; Kuller, L.H.; Robbins, J.; et al. The Ginkgo Evaluation of Memory (GEM) Study: Design and Baseline Data of a Randomized Trial of Ginkgo Biloba Extract in Prevention of Dementia. Contemp. Clin. Trials 2006, 27, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A. Current Therapeutic Options for Alzheimers Disease. Curr. Genom. 2007, 8, 550–558. [Google Scholar] [CrossRef]
- Thomas, J.; Thomas, C.J.; Radcliffe, J.; Itsiopoulos, C. Omega-3 Fatty Acids in Early Prevention of Inflammatory Neurodegenerative Disease: A Focus on Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef] [PubMed]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug Treatments in Alzheimer’s Disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Trépanier, M.-O.; Giuliano, V.; Bazinet, R.P. Brain Omega-3 Polyunsaturated Fatty Acids Modulate Microglia Cell Number and Morphology in Response to Intracerebroventricular Amyloid-β 1-40 in Mice. J. Neuroinflamm. 2016, 13, 257. [Google Scholar] [CrossRef]
- Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Discovery of 1,2,4-Thiadiazolidine-3,5-Dione Analogs That Exhibit Unusual and Selective Rapid Cell Death Kinetics against Acute Myelogenous Leukemia Cells in Culture. Bioorg Med. Chem. Lett. 2011, 21, 4879–4883. [Google Scholar] [CrossRef][Green Version]
- Castro, A.; Encinas, A.; Gil, C.; Bräse, S.; Porcal, W.; Pérez, C.; Moreno, F.J.; Martínez, A. Non-ATP Competitive Glycogen Synthase Kinase 3β (GSK-3β) Inhibitors: Study of Structural Requirements for Thiadiazolidinone Derivatives. Bioorg Med. Chem. 2008, 16, 495–510. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Mainali, N.; Bowroju, S.K.; Atluri, P.; Penthala, N.R.; Ayyadevera, S.; Crooks, P.A.; Shmookler Reis, R.J. Structural Modeling of GSK3β Implicates the Inactive (DFG-out) Conformation as the Target Bound by TDZD Analogs. Sci. Rep. 2020, 10, 18326. [Google Scholar] [CrossRef]
- Domínguez, J.M.; Fuertes, A.; Orozco, L.; del Monte-Millán, M.; Delgado, E.; Medina, M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef]
- Noori, M.S.; Bhatt, P.M.; Courreges, M.C.; Ghazanfari, D.; Cuckler, C.; Orac, C.M.; McMills, M.C.; Schwartz, F.L.; Deosarkar, S.P.; Bergmeier, S.C.; et al. Identification of a Novel Selective and Potent Inhibitor of Glycogen Synthase Kinase-3. Am. J. Physiol. Cell Physiol. 2019, 317, C1289–C1303. [Google Scholar] [CrossRef] [PubMed]
- Kakraba, S.; Ayyadevara, S.; Penthala, N.R.; Balasubramaniam, M.; Ganne, A.; Liu, L.; Alla, R.; Bommagani, S.B.; Barger, S.W.; Griffin, W.S.T.; et al. A Novel Microtubule-Binding Drug Attenuates and Reverses Protein Aggregation in Animal Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 12, 310. [Google Scholar] [CrossRef] [PubMed]
- Bowroju, S.K.; Mainali, N.; Ayyadevara, S.; Penthala, N.R.; Krishnamachari, S.; Kakraba, S.; Shmookler Reis, R.J.; Crooks, P.A. Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation. Molecules 2020, 25, 3610. [Google Scholar] [CrossRef] [PubMed]
- Nasim, S.; Crooks, P.A. N-Chlorosuccinimide Is a Convenient Oxidant for the Synthesis of 2,4-Disubstituted 1,2,4-Thiadiazolidine-3,5-Diones. Tetrahedron Lett. 2009, 50, 257–259. [Google Scholar] [CrossRef]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential Transformation of Human Neuronal Cells by Human Adenoviruses and the Origin of HEK 293 Cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- David, D.C. Aging and the Aggregating Proteome. Front. Genet. 2012, 3, 247. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The Role of Protein Clearance Mechanisms in Organismal Ageing and Age-Related Diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The Proteostasis Network and Its Decline in Ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Matai, L.; Sarkar, G.C.; Chamoli, M.; Malik, Y.; Kumar, S.S.; Rautela, U.; Jana, N.R.; Chakraborty, K.; Mukhopadhyay, A. Dietary Restriction Improves Proteostasis and Increases Life Span through Endoplasmic Reticulum Hormesis. Proc. Natl. Acad. Sci. USA 2019, 116, 17383–17392. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Parcon, P.A.; Barger, S.W.; Griffin, W.S.T.; Alla, R.; Tackett, A.J.; Mackintosh, S.G.; Petricoin, E.; Zhou, W.; et al. Proteins That Mediate Protein Aggregation and Cytotoxicity Distinguish Alzheimer’s Hippocampus from Normal Controls. Aging Cell 2016, 15, 924–939. [Google Scholar] [CrossRef]
- Guimarães, C.R. MM-GB/SA Rescoring of Docking Poses. Methods Mol. Biol. 2012, 819, 255–268. [Google Scholar] [CrossRef]
- Zhang, Z.; Miteva, M.A.; Wang, L.; Alexov, E. Analyzing Effects of Naturally Occurring Missense Mutations. Comput. Math. Methods Med. 2012, 2012, 805827. [Google Scholar] [CrossRef] [PubMed]
- Netsey, E.K.; Kakraba, S.; Naandam, S.M.; Yadem, A.C. A Mathematical Graph-Theoretic Model of Single Point Mutations Associated with Sickle Cell Anemia Disease. J. Adv. Biotech. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Kakraba, S.; Knisley, D. A Graph-Theoretic Model of Single Point Mutations in the Cystic Fibrosis Transmembrane Conductance Regulator. J. Adv. Biotech. 2016, 6, 780–786. [Google Scholar] [CrossRef]
- Dobson, C.M.; Ellis, R.J. Protein Folding and Misfolding inside and Outside the Cell. EMBO J. 1998, 17, 5251–5254. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Bartlett, A.I.; Radford, S.E. An Expanding Arsenal of Experimental Methods Yields an Explosion of Insights into Protein Folding Mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 582–588. [Google Scholar] [CrossRef]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein Aggregation and Degradation Mechanisms in Neurodegenerative Diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar]
- Ayyadevara, S.; Balasubramaniam, M.; Gao, Y.; Yu, L.; Alla, R.; Shmookler Reis, R. Proteins in Aggregates Functionally Impact Multiple Neurodegenerative Disease Models by Forming Proteasome-blocking Complexes. Aging Cell 2015, 14, 35–48. [Google Scholar] [CrossRef]
- Gejjalagere Honnappa, C.; Mazhuvancherry Kesavan, U. A Concise Review on Advances in Development of Small Molecule Anti-Inflammatory Therapeutics Emphasising AMPK: An Emerging Target. Int. J. Immunopathol. Pharmacol. 2016, 29, 562–571. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Nam, J.H.; Nam, Y.; Nam, H.Y.; Yoon, G.; Ko, E.; Kim, S.-B.; Bautista, M.R.; Capule, C.C.; Koyanagi, T.; et al. The Small Molecule CA140 Inhibits the Neuroinflammatory Response in Wild-Type Mice and a Mouse Model of AD. J. Neuroinflamm. 2018, 15, 286. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Ganne, A.; Hendrix, R.D.; Balasubramaniam, M.; Shmookler Reis, R.J.; Barger, S.W. Functional Assessments through Novel Proteomics Approaches: Application to Insulin/IGF Signaling in Neurodegenerative Disease. J. Neurosci. Methods 2019, 319, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Bharill, P.; Dandapat, A.; Hu, C.; Khaidakov, M.; Mitra, S.; Shmookler Reis, R.J.; Mehta, J.L. Aspirin Inhibits Oxidant Stress, Reduces Age-Associated Functional Declines, and Extends Lifespan of Caenorhabditis elegans. Antioxid. Redox Signal 2013, 18, 481–490. [Google Scholar] [CrossRef]
- Mócsai, A.; Kovács, L.; Gergely, P. What Is the Future of Targeted Therapy in Rheumatology: Biologics or Small Molecules? BMC Med. 2014, 12, 43. [Google Scholar] [CrossRef]
- Hanke, T.; Merk, D.; Steinhilber, D.; Geisslinger, G.; Schubert-Zsilavecz, M. Small Molecules with Anti-Inflammatory Properties in Clinical Development. Pharmacol. Ther. 2016, 157, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xiao, J.; Liu, X.; Jiang, Z.; Zhan, Y.; Yin, T.; He, L.; Zhang, F.; Xing, S.; Chen, B.; et al. AICD: An Integrated Anti-Inflammatory Compounds Database for Drug Discovery. Sci. Rep. 2019, 9, 7737. [Google Scholar] [CrossRef]
- van der Zanden, S.Y.; Luimstra, J.J.; Neefjes, J.; Borst, J.; Ovaa, H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immunol. 2020, 41, 493–511. [Google Scholar] [CrossRef]
- Hirth, F. Drosophila Melanogaster in the Study of Human Neurodegeneration. CNS Neurol. Disord. Drug Targets 2010, 9, 504–523. [Google Scholar] [CrossRef]
- Van Dam, D.; De Deyn, P.P. Animal Models in the Drug Discovery Pipeline for Alzheimer’s Disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-Tau Antibodies That Block Tau Aggregate Seeding In Vitro Markedly Decrease Pathology and Improve Cognition in Vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to Discover Therapeutic Compounds for Ageing-Associated Neurodegenerative Diseases. Chem. Cent. J. 2015, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-Y.; Lane, H.-Y.; Lin, C.-H. Medications Used for Cognitive Enhancement in Patients with Schizophrenia, Bipolar Disorder, Alzheimer’s Disease, and Parkinson’s Disease. Front. Psychiatry 2018, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Das, B.; Hou, H.; He, W.; Yan, R. BACE1 Deletion in the Adult Mouse Reverses Preformed Amyloid Deposition and Improves Cognitive Functions. J. Exp. Med. 2018, 215, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Vatolin, S.; Radivoyevitch, T.; Maciejewski, J.P. New Drugs for Pharmacological Extension of Replicative Life Span in Normal and Progeroid Cells. NPJ Aging Mech. Dis. 2019, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Nolan, A.; Frias, E.S.; Boone, M.; Ureta, G.; Grue, K.; Paladini, M.-S.; Elizarraras, E.; Delgado, L.; Bernales, S.; et al. Small Molecule Cognitive Enhancer Reverses Age-Related Memory Decline in Mice. eLife 2020, 9, e62048. [Google Scholar] [CrossRef] [PubMed]
- Mazanetz, M.P.; Fischer, P.M. Untangling Tau Hyperphosphorylation in Drug Design for Neurodegenerative Diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An Unbiased Approach to Identifying Tau Kinases That Phosphorylate Tau at Sites Associated with Alzheimer Disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid Beta-Induced Glycogen Synthase Kinase 3β Phosphorylated VDAC1 in Alzheimer’s Disease: Implications for Synaptic Dysfunction and Neuronal Damage. Biochim. Biophys. Acta–Mol. Basis Dis. 2013, 1832, 1913–1921. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef] [PubMed]
- Yadikar, H.; Torres, I.; Aiello, G.; Kurup, M.; Yang, Z.; Lin, F.; Kobeissy, F.; Yost, R.; Wang, K.K. Screening of Tau Protein Kinase Inhibitors in a Tauopathy-Relevant Cell-Based Model of Tau Hyperphosphorylation and Oligomerization. PLoS ONE 2020, 15, e0224952. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J. Decreased Nuclear Beta-Catenin, Tau Hyperphosphorylation and Neurodegeneration in GSK-3beta Conditional Transgenic Mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef]
- Li, H.-L.; Wang, H.-H.; Liu, S.-J.; Deng, Y.-Q.; Zhang, Y.-J.; Tian, Q.; Wang, X.-C.; Chen, X.-Q.; Yang, Y.; Zhang, J.-Y.; et al. Phosphorylation of Tau Antagonizes Apoptosis by Stabilizing β-Catenin, a Mechanism Involved in Alzheimer’s Neurodegeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 3591–3596. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Involvement of Aberrant Glycosylation in Phosphorylation of Tau by Cdk5 and GSK-3β. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Su, Y.; Ryder, J.; Li, B.; Wu, X.; Fox, N.; Solenberg, P.; Brune, K.; Paul, S.; Zhou, Y.; Liu, F.; et al. Lithium, a Common Drug for Bipolar Disorder Treatment, Regulates Amyloid-β Precursor Protein Processing. Biochemistry 2004, 43, 6899–6908. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Zhang, J.-S.; Herreros-Villanueva, M.; Koenig, A.; Deng, Z.; de Narvajas, A.A.-M.; Gomez, T.S.; Meng, X.; Bujanda, L.; Ellenrieder, V.; Li, X.K.; et al. Differential Activity of GSK-3 Isoforms Regulates NF-ΚB and TRAIL- or TNFα Induced Apoptosis in Pancreatic Cancer Cells. Cell Death Dis. 2014, 5, e1142. [Google Scholar] [CrossRef]
- Medunjanin, S.; Schleithoff, L.; Fiegehenn, C.; Weinert, S.; Zuschratter, W.; Braun-Dullaeus, R.C. GSK-3β Controls NF-KappaB Activity via IKKγ/NEMO. Sci. Rep. 2016, 6, 38553. [Google Scholar] [CrossRef] [PubMed]
- Abd-Ellah, A.; Voogdt, C.; Krappmann, D.; Möller, P.; Marienfeld, R.B. GSK3β Modulates NF-ΚB Activation and RelB Degradation through Site-Specific Phosphorylation of BCL10. Sci. Rep. 2018, 8, 1352. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich, O.; Eldar-Finkelman, H. The Role of Glycogen Synthase Kinase-3 In Insulin Resistance and Type 2 Diabetes. Expert. Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Nagao, H.; Cai, W.; Wewer Albrechtsen, N.J.; Steger, M.; Batista, T.M.; Pan, H.; Dreyfuss, J.M.; Mann, M.; Kahn, C.R. Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. Proc. Natl. Acad. Sci. USA 2021, 118, e2019474118. [Google Scholar] [CrossRef]
- Wu, C.Y.; Wang, C.; Saskin, R.; Shah, B.R.; Kapral, M.K.; Lanctôt, K.L.; Herrmann, N.; Cogo-Moreira, H.; MacIntosh, B.J.; Edwards, J.D.; et al. No association between metformin initiation and incident dementia in older adults newly diagnosed with diabetes. J. Intern. Med. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Abner, E.L.; Nelson, P.T.; Kryscio, R.J.; Schmitt, F.A.; Fardo, D.W.; Woltjer, R.L.; Cairns, N.J.; Yu, L.; Dodge, H.H.; Xiong, C.; et al. Diabetes is Associated with Cerebrovascular but not Alzheimer’s Disease Neuropathology. Alzheimers Dement. 2016, 12, 882–889. [Google Scholar] [CrossRef]
- Dos Santos Matioli, M.N.P.; Suemoto, C.K.; Rodriguez, R.D.; Farias, D.S.; da Silva, M.M.; Leite, R.E.P.; Ferretti-Rebustini, R.E.L.; Farfel, J.M.; Pasqualucci, C.A.; Jacob Filho, W.; et al. Diabetes is Not Associated with Alzheimer’s Disease Neuropathology. J Alzheimers Dis. 2017, 60, 1035–1043. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in Neurodegenerative Diseases. Int. J. Alzheimer’s Dis. 2011, 2011, 189246. [Google Scholar] [CrossRef]
- Shmookler Reis, R.J.; Atluri, R.; Balasubramaniam, M.; Johnson, J.; Ganne, A.; Ayyadevara, S. “Protein aggregates” contain RNA and DNA, entrapped by misfolded proteins but largely rescued by blocking translational elongation. Aging Cell 2021, 2021, e13326. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Van Eldik, L.J.; Griffin, W.S.T.; Barger, S.W. S100B-Induced Microglial and Neuronal IL-1 Expression Is Mediated by Cell Type-Specific Transcription Factors. J. Neurochem. 2005, 92, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Dosanjh, L.E.; Brown, M.K.; Rao, G.; Link, C.D.; Luo, Y. Behavioral Phenotyping of a Transgenic Caenorhabditis elegans Expressing Neuronal Amyloid-β. J. Alzheimer’s Dis. 2010, 19, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Dostal, V.; Link, C.D. Assaying Beta-Amyloid Toxicity Using a Transgenic C. elegans Model. J. Vis. Exp. 2010, 44, e2252. [Google Scholar] [CrossRef] [PubMed]
- Bharill, P.; Ayyadevara, S.; Alla, R.; Shmookler Reis, R.J. Extreme Depletion of PIP3 Accompanies the Increased Life Span and Stress Tolerance of PI3K-Null C. elegans Mutants. Front. Genet. 2013, 4, 34. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Johnson, J.; Alla, R.; Mackintosh, S.G.; Shmookler Reis, R.J. PIP3-Binding Proteins Promote Age-Dependent Protein Aggregation and Limit Survival in C. elegans. Oncotarget 2016, 7, 48870–48886. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Parcon, P.A.; Bose, C.; Liu, L.; Jones, R.A.; Farlow, M.R.; Mrak, R.E.; Barger, S.W.; Griffin, W.S.T. Interleukin-1β Drives NEDD8 Nuclear-to-Cytoplasmic Translocation, Fostering Parkin Activation via NEDD8 Binding to the P-Ubiquitin Activating Site. J. Neuroinflamm. 2019, 16, 275. [Google Scholar] [CrossRef]
- Lakkaniga, N.R.; Balasubramaniam, M.; Zhang, S.; Frett, B.; Li, H. Structural Characterization of the Aurora Kinase B “DFG-Flip” Using Metadynamics. AAPS J. 2020, 22, 14. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TDZD Analog | GSK3β Amino Acids Interacting with TDZD Analogs |
|---|---|
| TDZD-8 | ILE100, MET101, VAL110, ARG111, LEU112, LEU132, ASP200, ALA204, LYS205, GLN206 |
| PNR886 | LYS85, GLU97, GLN99, ILE100, MET101, ASP200, PHE201, ALA204, LYS205, GLN206 |
| PNR962 | LYS85, ARG96, GLU97, ILE100, MET101, VAL110, ARG111, LEU112, LEU132, ASP200, ALA204, LYS205, GLN206 |
| TDZD-8 & PNR886 | ILE100, MET101, ASP200, ALA204, LYS205, GLN206 |
| TDZD-8 & PNR962 | ILE100, MET101, VAL110, ARG111, LEU112, LEU132, ASP200, ALA204, LYS205, GLN206 |
| All Three Analogs | ILE100, MET101, ASP200, ALA204, LYS205, GLN206 |
| TDZD Drug | ΔGbinding (kcal/mol) | Ki (µM) |
|---|---|---|
| PNR886 | −6.2 | 2.7 |
| PNR962 | −6.2 | 2.7 |
| TDZD-8 | −6.1 | 3.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakraba, S.; Ayyadevara, S.; Mainali, N.; Balasubramaniam, M.; Bowroju, S.; Penthala, N.R.; Atluri, R.; Barger, S.W.; Griffin, S.T.; Crooks, P.A.; et al. Thiadiazolidinone (TDZD) Analogs Inhibit Aggregation-Mediated Pathology in Diverse Neurodegeneration Models, and Extend C. elegans Life- and Healthspan. Pharmaceuticals 2023, 16, 1498. https://doi.org/10.3390/ph16101498
Kakraba S, Ayyadevara S, Mainali N, Balasubramaniam M, Bowroju S, Penthala NR, Atluri R, Barger SW, Griffin ST, Crooks PA, et al. Thiadiazolidinone (TDZD) Analogs Inhibit Aggregation-Mediated Pathology in Diverse Neurodegeneration Models, and Extend C. elegans Life- and Healthspan. Pharmaceuticals. 2023; 16(10):1498. https://doi.org/10.3390/ph16101498
Chicago/Turabian StyleKakraba, Samuel, Srinivas Ayyadevara, Nirjal Mainali, Meenakshisundaram Balasubramaniam, Suresh Bowroju, Narsimha Reddy Penthala, Ramani Atluri, Steven W. Barger, Sue T. Griffin, Peter A. Crooks, and et al. 2023. "Thiadiazolidinone (TDZD) Analogs Inhibit Aggregation-Mediated Pathology in Diverse Neurodegeneration Models, and Extend C. elegans Life- and Healthspan" Pharmaceuticals 16, no. 10: 1498. https://doi.org/10.3390/ph16101498
APA StyleKakraba, S., Ayyadevara, S., Mainali, N., Balasubramaniam, M., Bowroju, S., Penthala, N. R., Atluri, R., Barger, S. W., Griffin, S. T., Crooks, P. A., & Shmookler Reis, R. J. (2023). Thiadiazolidinone (TDZD) Analogs Inhibit Aggregation-Mediated Pathology in Diverse Neurodegeneration Models, and Extend C. elegans Life- and Healthspan. Pharmaceuticals, 16(10), 1498. https://doi.org/10.3390/ph16101498