
Effects of Escherichia coli LPS Structure on Antibacterial and Anti-Endotoxin Activities of Host Defense Peptides

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
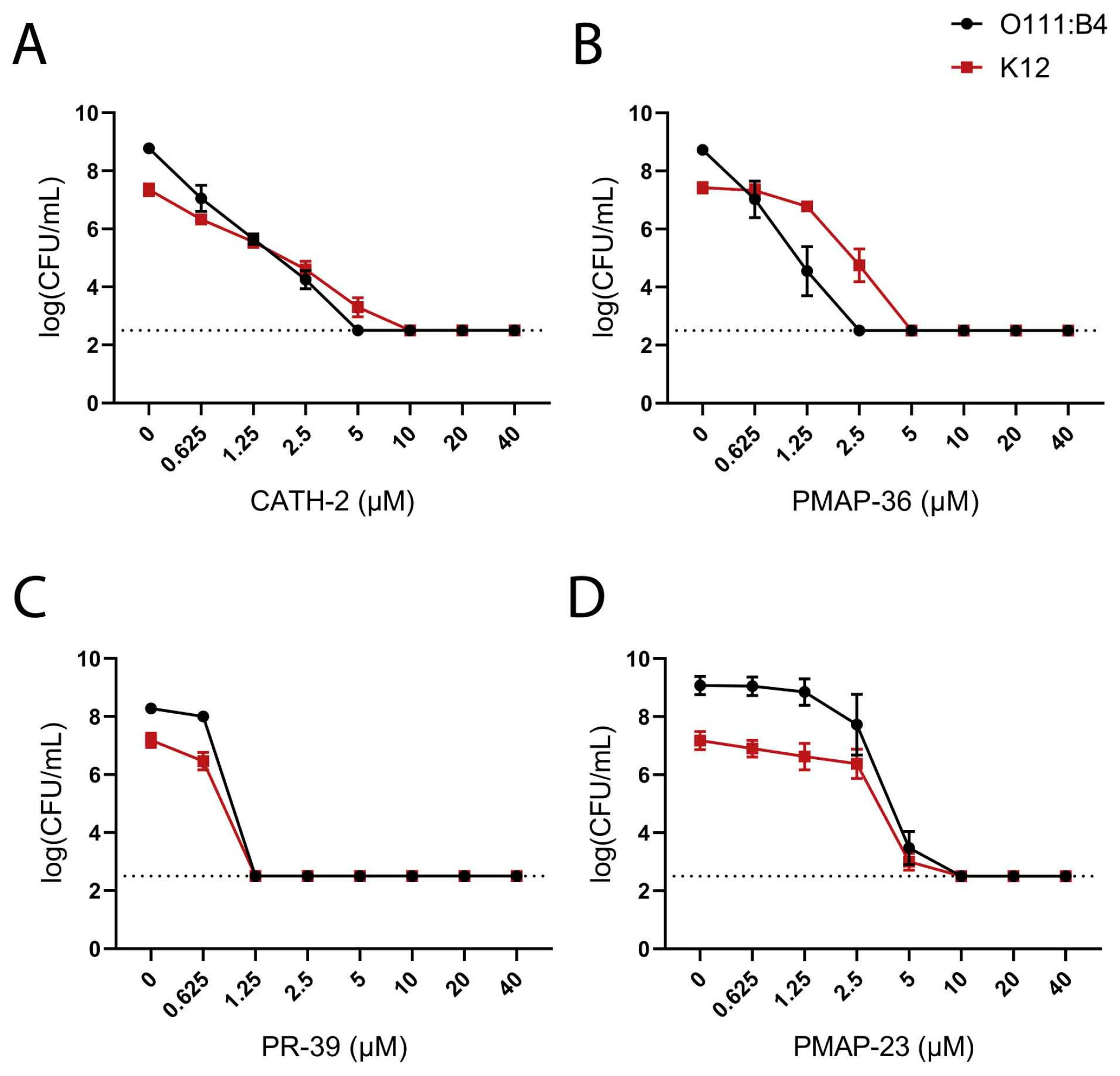
2.1. Influence of O-Antigen in Resistance to Host Defense Peptides
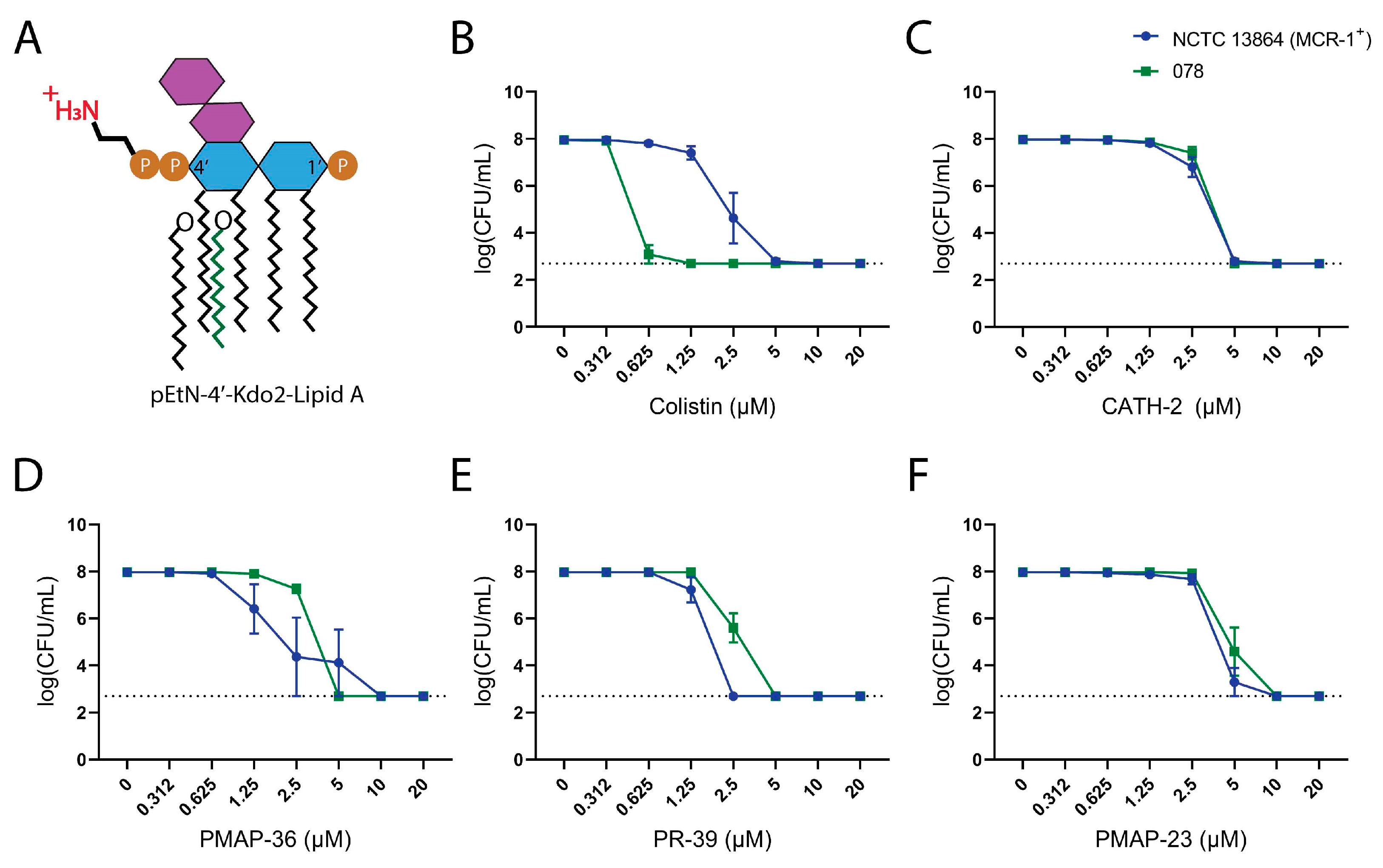
2.2. Influence of Lipid A Modification in Resistance to Host Defense Peptides
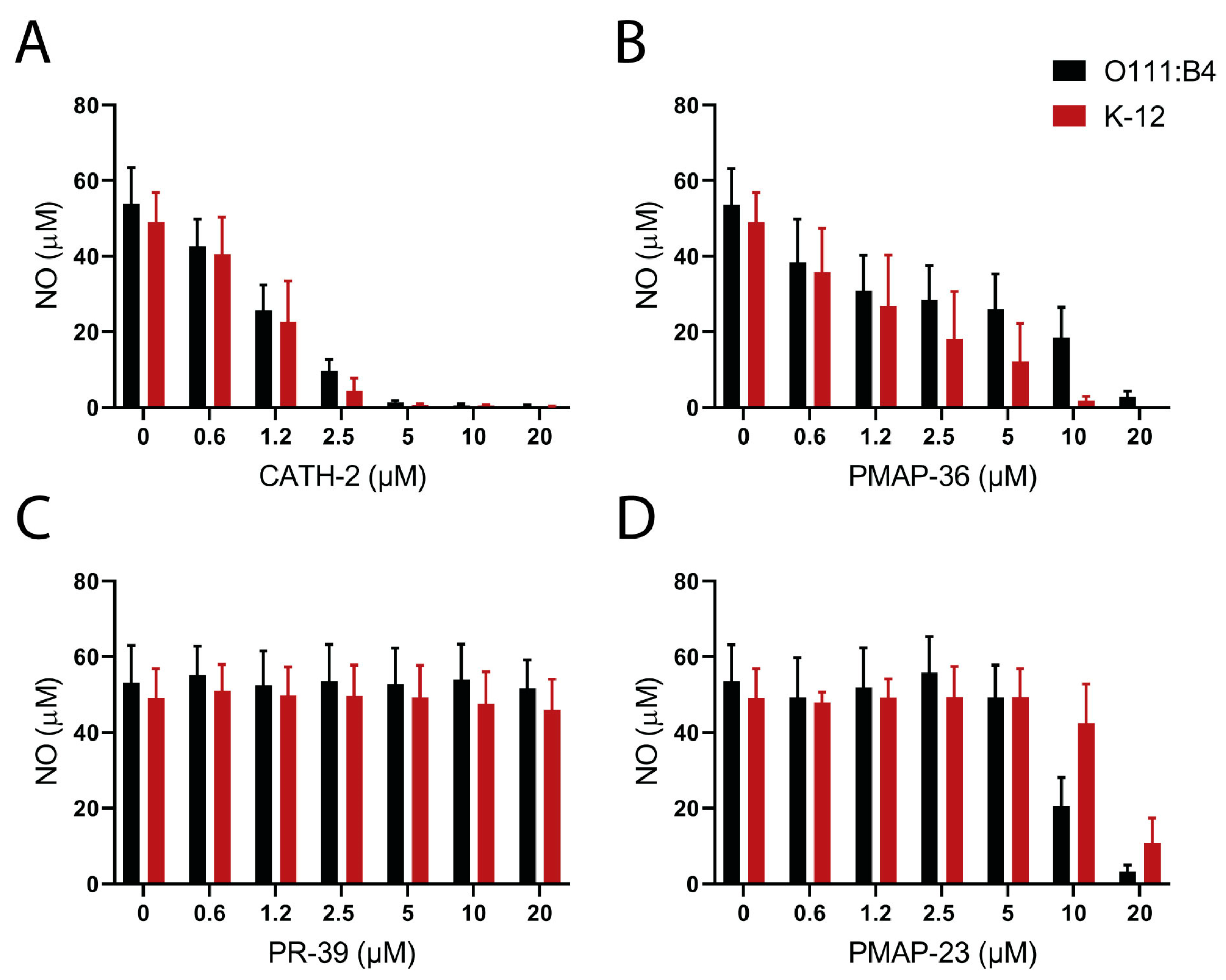
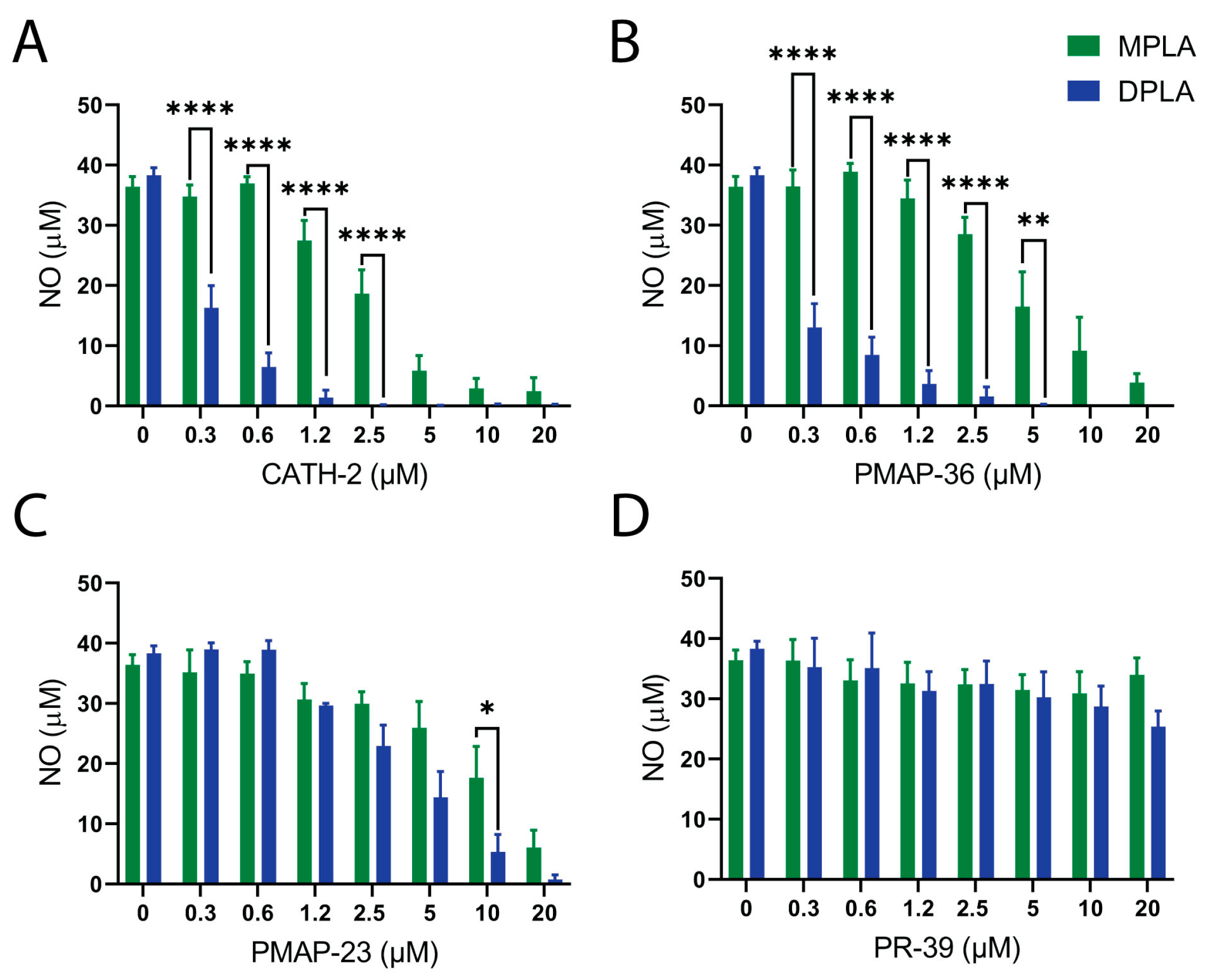
2.3. Neutralization of LPS or Lipid A Induced Macrophage Activation by Host Defense Peptides
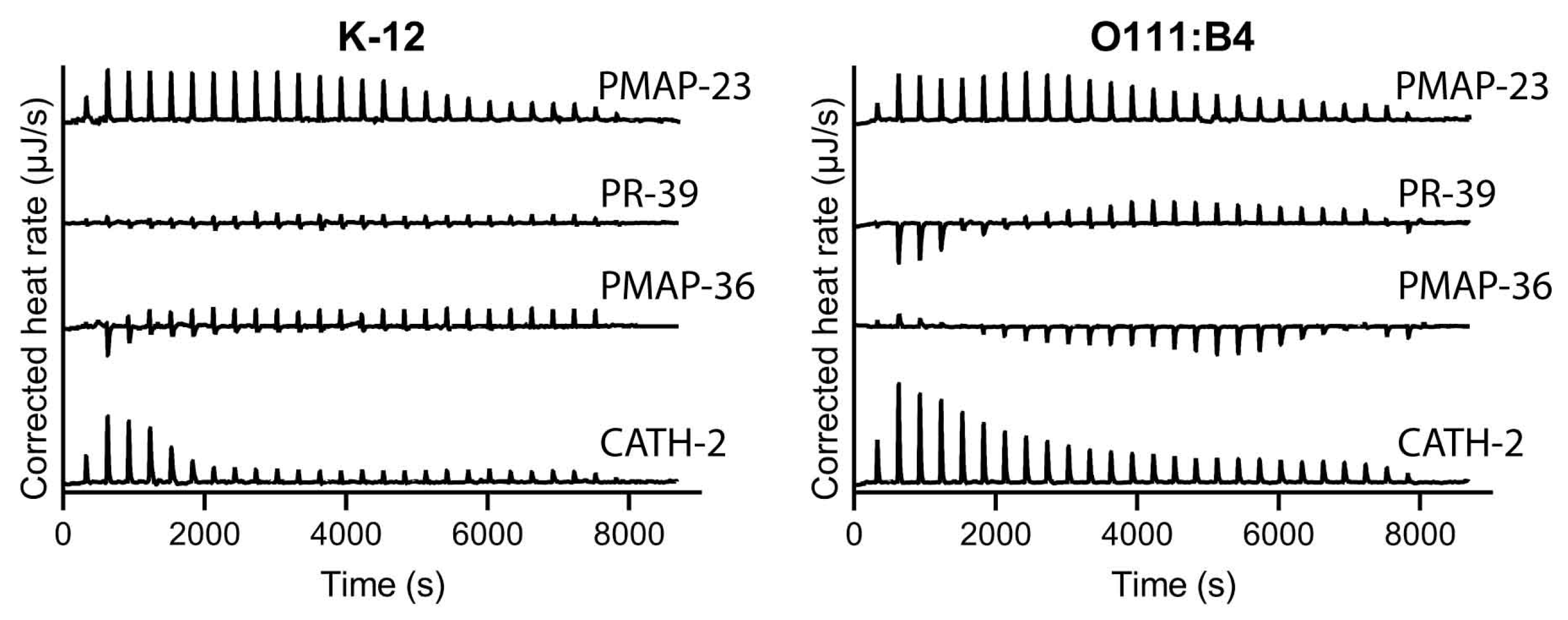
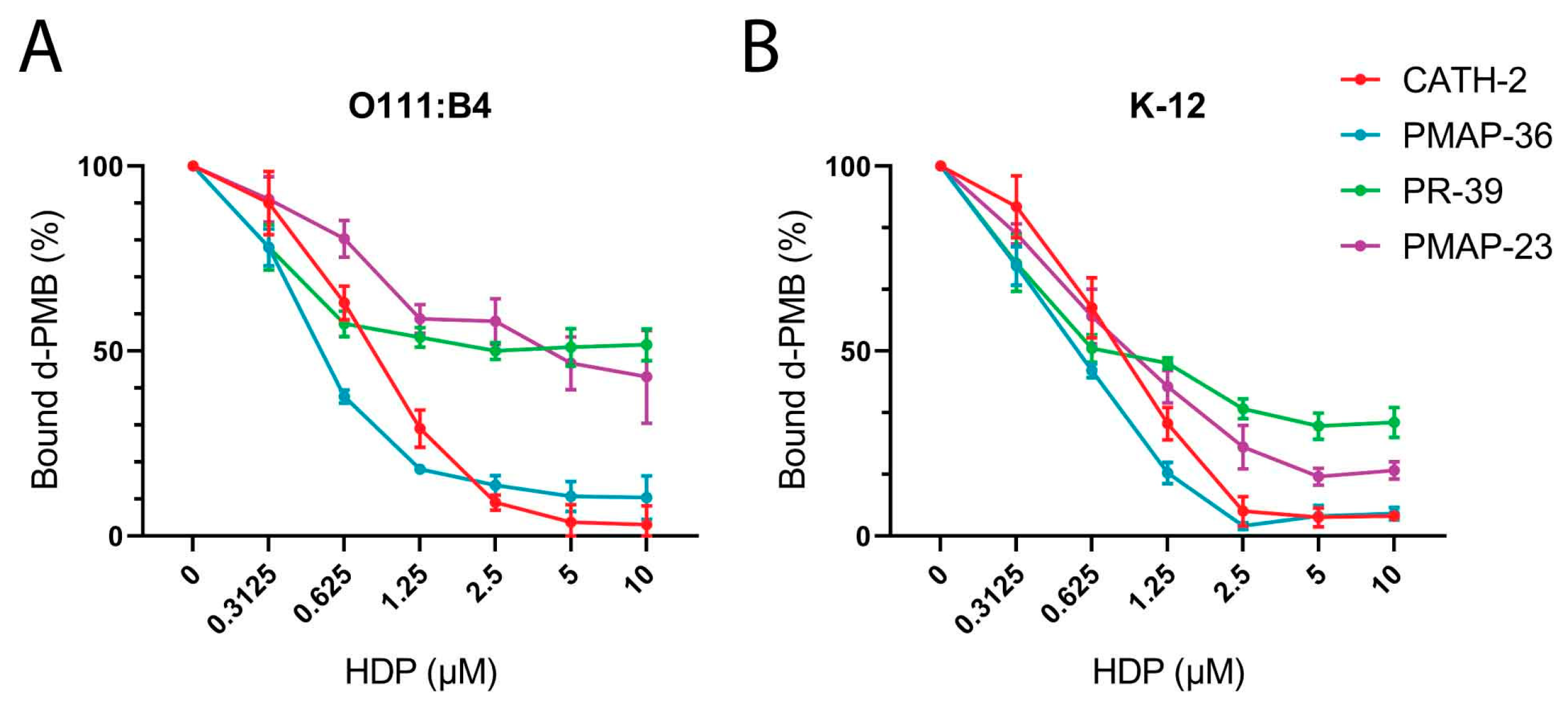
2.4. Influence of LPS Structure on Binding Affinity of Host Defense Peptides
2.5. Polymyxin B Competition Assay
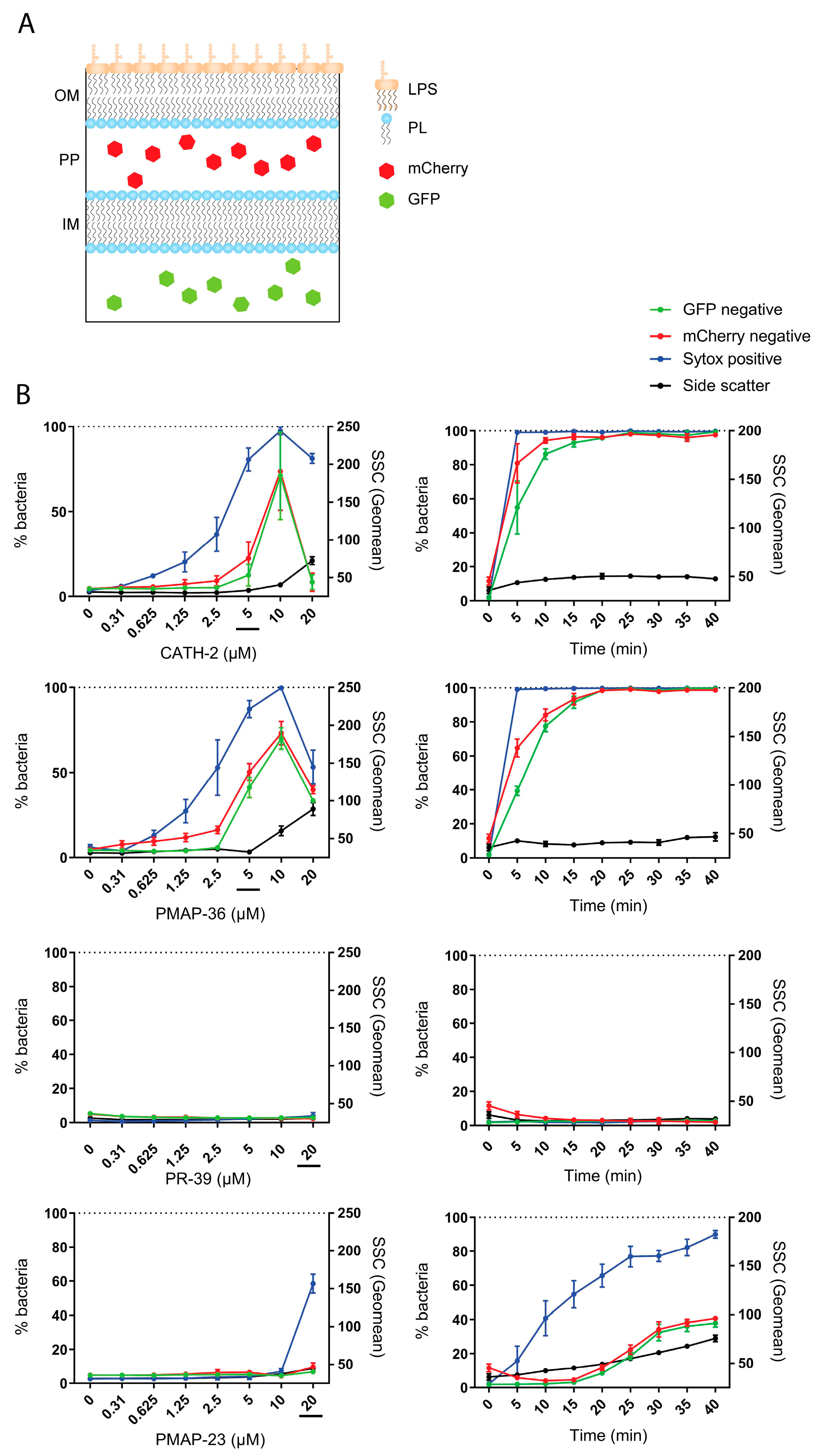
2.6. Bacterial Membrane Permeabilization by Host Defense Peptides
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Bacterial Strains
4.3. Cell Culturing
4.4. Track Dilution Assay
4.5. LPS/Lipid A Neutralization Assays
4.6. Isothermal Titration Calorimetry (ITC)
4.7. Dansyl-Polymyxin B Competition Assay
4.8. Flow Cytometry
4.9. Statistical Analysis
5. Conclusions
6. Future Trends
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Pirri, G.; Bozzi, A.; Di Giulio, A.; Aschi, M.; Rinaldi, A.C. Visions & Reflections (Minireview) Antimicrobial Peptides: Natural Templates for Synthetic Membrane-Active Compounds. Cell. Mol. Life Sci. 2008, 65, 2450–2460. [Google Scholar]
- Matsuzaki, K. Control of Cell Selectivity of Antimicrobial Peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef]
- Coorens, M.; Schneider, V.A.F.; de Groot, A.M.; van Dijk, A.; Meijerink, M.; Wells, J.M.; Scheenstra, M.R.; Veldhuizen, E.J.A.; Haagsman, H.P. Cathelicidins Inhibit Escherichia Coli-Induced TLR2 and TLR4 Activation in a Viability-Dependent Manner. J. Immunol. 2017, 199, 1418–1428. [Google Scholar] [CrossRef]
- van Harten, R.M.; van Woudenbergh, E.; van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory Antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef]
- Coorens, M.; Banaschewski, B.J.H.; Baer, B.J.; Yamashita, C.; van Dijk, A.; Haagsman, H.P.; Veldhuizen, R.A.W.; Veldhuizen, E.J.A. Killing of Pseudomonas Aeruginosa by Chicken Cathelicidin-2 Is Immunogenically Silent, Preventing Lung Inflammation In Vivo. Infect. Immun. 2017, 85, e00546-17. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F.; et al. Bacterial Endotoxin: Molecular Relationships of Structure to Activity and Function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef]
- Caroff, M.; Novikov, A. Lipopolysaccharides: Structure, Function and Bacterial Identifications. OCL 2020, 27, 31. [Google Scholar] [CrossRef]
- Simpson, B.W.; Trent, M.S. Pushing the Envelope: LPS Modifications and Their Consequences. Nat. Rev. Microbiol. 2019, 17, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Holst, M.O.; Ulmer, A.J.; Brade, H.; Flad, H.-D.; Rietschel, E.T. Biochemistry and Cell Biology of Bacterial Endotoxins. FEMS Immunol. Med. Microbiol. 1996, 16, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Ren, G.; Li, Y.; Wang, X. Influence of Core Oligosaccharide of Lipopolysaccharide to Outer Membrane Behavior of Escherichia coli. Mar. Drugs 2015, 13, 3325–3339. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-Host Defense Antibacterial Peptides Interactions: Role in Bacterial Resistance and Prevention of Sepsis. Biochim. Biophys. Acta 2006, 1758, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Holst, O.; Müller-Loennies, S.; Lindner, B.; Brade, H. Chemical Structure of the Lipid a of Escherichia coli J-5. Eur. J. Biochem. 1993, 214, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Stenutz, R.; Weintraub, A.; Widmalm, G. The Structures of Escherichia coli O-Polysaccharide Antigens. FEMS Microbiol. Rev. 2006, 30, 382–403. [Google Scholar] [CrossRef] [PubMed]
- Kenne, L.; Lindberg, B.; Söderholm, E.; Bundle, D.R.; Griffith, D.W. Structural Studies of the O-Antigens from Salmonella greenside and Salmonella adelaide. Carbohydr. Res. 1983, 111, 289–296. [Google Scholar] [CrossRef]
- Edstrom, R.D.; Heath, E.C. The Biosynthesis of Cell Wall Lipopolysaccharide in Escherichia coli: VII. Studies on the Structure of the O-Antigenic Polysaccharide. J. Biol. Chem. 1967, 242, 4125–4133. [Google Scholar] [CrossRef]
- Müller-Loennies, S.; Lindner, B.; Brade, H. Structural Analysis of Oligosaccharides from Lipopolysaccharide (LPS) of Escherichia coli K12 Strain W3100 Reveals a Link between Inner and Outer Core LPS Biosynthesis. J. Biol. Chem. 2003, 278, 34090–34101. [Google Scholar] [CrossRef]
- Amor, K.; Heinrichs, D.E.; Frirdich, E.; Ziebell, K.; Johnson, R.P.; Whitfield, C. Distribution of Core Oligosaccharide Types in Lipopolysaccharides from Escherichia coli. Infect. Immun. 2000, 68, 1116–1124. [Google Scholar] [CrossRef]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef]
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2015, 4, 18–41. [Google Scholar] [CrossRef]
- Balhuizen, M.D.; van Dijk, A.; Jansen, J.W.A.; van de Lest, C.H.A.; Veldhuizen, E.J.A.; Haagsman, H.P. Outer Membrane Vesicles Protect Gram-Negative Bacteria against Host Defense Peptides. mSphere 2021, 6, e0052321. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.O.; Dawson, R.A.; Alsharaf, L.M.; Winter, J.A. Protective Effects of Helicobacter Pylori Membrane Vesicles against Stress and Antimicrobial Agents. Microbiology 2020, 166, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.A.F.; Coorens, M.; Ordonez, S.R.; Tjeerdsma-Van Bokhoven, J.L.M.; Posthuma, G.; Van Dijk, A.; Haagsman, H.P.; Veldhuizen, E.J.A. Imaging the Antimicrobial Mechanism(s) of Cathelicidin-2. Sci. Rep. 2016, 6, 32948. [Google Scholar] [CrossRef] [PubMed]
- Scheenstra, M.R.; van den Belt, M.; Tjeerdsma-van Bokhoven, J.L.M.; Schneider, V.A.F.; Ordonez, S.R.; van Dijk, A.; Veldhuizen, E.J.A.; Haagsman, H.P. Cathelicidins PMAP-36, LL-37 and CATH-2 Are Similar Peptides with Different Modes of Action. Sci. Rep. 2019, 9, 4780. [Google Scholar] [CrossRef]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a Putative Human Peptide Antibiotic, Is Cysteine-Free and Expressed in Bone Marrow and Testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A.; Lee, Y.; Boman, A.; Sun, C.; Andersson, M.; Jornvall, H.; Mutt, V.; Boman, H.G.; et al. Mechanisms of Action on Escherichia coli of Cecropin P1 and PR-39, Two Antibacterial Peptides from Pig Intestine. Infect. Immun. 1993, 61, 2978. [Google Scholar] [CrossRef]
- Veldhuizen, E.J.A.; Schneider, V.A.F.; Agustiandari, H.; Van Dijk, A.; Tjeerdsma-van Bokhoven, J.L.M.; Bikker, F.J.; Haagsman, H.P. Antimicrobial and Immunomodulatory Activities of PR-39 Derived Peptides. PLoS ONE 2014, 9, e95939. [Google Scholar]
- Veldhuizen, E.J.A.; Scheenstra, M.R.; Tjeerdsma-van Bokhoven, J.L.M.; Coorens, M.; Schneider, V.A.F.; Bikker, F.J.; van Dijk, A.; Haagsman, H.P. Antimicrobial and Immunomodulatory Activity of PMAP-23 Derived Peptides. Protein Pept. Lett. 2017, 24, 609–616. [Google Scholar] [CrossRef]
- Orioni, B.; Bocchinfuso, G.; Kim, J.Y.; Palleschi, A.; Grande, G.; Bobone, S.; Park, Y.; Kim, J.I.L.; Hahm, K.S.; Stella, L. Membrane Perturbation by the Antimicrobial Peptide PMAP-23: A Fluorescence and Molecular Dynamics Study. Biochim. Biophys. Acta 2009, 1788, 1523–1533. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.G. PMAP-23 Triggers Cell Death by Nitric Oxide-Induced Redox Imbalance in Escherichia coli. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 1187–1195. [Google Scholar] [CrossRef]
- Heesterbeek, D.A.; Bardoel, B.W.; Parsons, E.S.; Bennett, I.; Ruyken, M.; Doorduijn, D.J.; Gorham, R.D.; Berends, E.T.; Pyne, A.L.; Hoogenboom, B.W.; et al. Bacterial Killing by Complement Requires Membrane Attack Complex Formation via Surface-Bound C5 Convertases. EMBO J. 2019, 38, e99852. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L.T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Curello, J.; MacDougall, C. Beyond Susceptible and Resistant, Part II: Treatment of Infections Due to Gram-Negative Organisms Producing Extended-Spectrum β-Lactamases. J. Pediatr. Pharmacol. Ther. 2014, 19, 156–164. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of Plasmid-Mediated Colistin Resistance Mechanism MCR-1 in Animals and Human Beings in China: A Microbiological and Molecular Biological Study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.C.; Yu, P.L. Factors Affecting the Antimicrobial Activity of Ovine-Derived Cathelicidins against E. coli 0157:H7. Int. J. Antimicrob. Agents 2005, 25, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ön, A.; Vejzovic, D.; Jennings, J.; Parigger, L.; Cordfunke, R.A.; Drijfhout, J.W.; Lohner, K.; Malanovic, N. Bactericidal Activity to Escherichia coli: Different Modes of Action of Two 24-Mer Peptides SAAP-148 and OP-145, Both Derived from Human Cathelicidine LL-37. Antibiotics 2023, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Khadka, N.K.; Aryal, C.M.; Pan, J. Lipopolysaccharide-Dependent Membrane Permeation and Lipid Clustering Caused by Cyclic Lipopeptide Colistin. ACS Omega 2018, 3, 17828–17834. [Google Scholar] [CrossRef]
- Dobias, J.; Poirel, L.; Nordmann, P. Cross-Resistance to Human Cationic Antimicrobial Peptides and to Polymyxins Mediated by the Plasmid-Encoded MCR-1? Clin. Microbiol. Infect. 2017, 23, e1–e676. [Google Scholar] [CrossRef]
- Kao, C.; Lin, X.; Yi, G.; Zhang, Y.; Rowe-Magnus, D.A.; Bush, K. Cathelicidin Antimicrobial Peptides with Reduced Activation of Toll-like Receptor Signaling Have Potent Bactericidal Activity against Colistin-Resistant Bacteria. mBio 2016, 7, e01418-16. [Google Scholar] [CrossRef]
- Kintses, B.; Jangir, P.K.; Fekete, G.; Számel, M.; Méhi, O.; Spohn, R.; Daruka, L.; Martins, A.; Hosseinnia, A.; Gagarinova, A.; et al. Chemical-Genetic Profiling Reveals Limited Cross-Resistance between Antimicrobial Peptides with Different Modes of Action. Nat. Commun. 2019, 10, 5731. [Google Scholar] [CrossRef]
- Jakubec, M.; Rylandsholm, F.G.; Rainsford, P.; Silk, M.; Bril’kov, M.; Kristoffersen, T.; Juskewitz, E.; Ericson, J.U.; Svendsen, J.S.M. Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli. Biomolecules 2023, 13, 1155. [Google Scholar] [CrossRef]
- Scott, A.J.; Oyler, B.L.; Goodlett, D.R.; Ernst, R.K. Lipid A Structural Modifications in Extreme Conditions and Identification of Unique Modifying Enzymes to Define the Toll-like Receptor 4 Structure-Activity Relationship. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 1439–1450. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Aidoukovitch, A.; Anders, E.; Dahl, S.; Nebel, D.; Svensson, D.; Nilsson, B.O. The Host Defense Peptide LL-37 Is Internalized by Human Periodontal Ligament Cells and Prevents LPS-Induced MCP-1 Production. J. Periodontal. Res. 2019, 54, 662–670. [Google Scholar] [CrossRef]
- Scott, A.; ad Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.K.; McAuley, D.F.; Tunney, M.M.; Irwin, C.R.; Stuart Elborn, J.; Taggart, C.C. Evaluation of the Ability of LL-37 to Neutralise LPS In Vitro and Ex Vivo. PLoS ONE 2011, 6, e26525. [Google Scholar] [CrossRef]
- Yu, H.; Lu, Y.; Qiao, X.; Wei, L.; Fu, T.; Cai, S.; Wang, C.; Liu, X.; Zhong, S.; Wang, Y. Novel Cathelicidins from Pigeon Highlights Evolutionary Convergence in Avain Cathelicidins and Functions in Modulation of Innate Immunity. Sci. Rep. 2015, 5, 11082. [Google Scholar] [CrossRef]
- Javed, A.; Slingerland, C.J.; Wood, T.M.; Martin, N.I.; Broere, F.; Weingarth, M.H.; Veldhuizen, E.J.A. Chimeric Peptidomimetic Antibiotic Efficiently Neutralizes Lipopolysaccharides (LPS) and Bacteria-Induced Activation of RAW Macrophages. ACS Infect. Dis. 2023, 9, 518–526. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, A.; Soares, T.A.; Santos, D.E.S.; Bore, S.L.; Sevink, G.J.A.; Cascella, M.; Milano, G. Aggregation of Lipid A Variants: A Hybrid Particle-Field Model. Biochim. Biophys. Acta (BBA) Gen. Subj. 2021, 1865, 129570. [Google Scholar] [CrossRef] [PubMed]
- Japelj, B.; Pristovšek, P.; Majerle, A.; Jerala, R. Structural Origin of Endotoxin Neutralization and Antimicrobial Activity of a Lactoferrin-Based Peptide. J. Biol. Chem. 2005, 280, 16955–16961. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Domadia, P.N.; Torres, J.; Hallock, K.J.; Ramamoorthy, A.; Bhattacharjya, S. NMR Structure of Pardaxin, a Pore-Forming Antimicrobial Peptide, in Lipopolysaccharide Micelles: Mechanism of outer membrane permeabilization. J. Biol. Chem. 2010, 285, 3883–3895. [Google Scholar] [CrossRef]
- Van Dijk, A.; Van Eldik, M.; Veldhuizen, E.J.A.; Tjeerdsma-Van Bokhoven, H.L.M.; De Zoete, M.R.; Bikker, F.J.; Haagsman, H.P. Immunomodulatory and Anti-Inflammatory Activities of Chicken Cathelicidin-2 Derived Peptides. PLoS ONE 2016, 11, e0147919. [Google Scholar] [CrossRef] [PubMed]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Sang, Y.; Teresa Ortega, M.; Rune, K.; Xiau, W.; Zhang, G.; Soulages, J.L.; Lushington, G.H.; Fang, J.; Williams, T.D.; Blecha, F.; et al. Canine Cathelicidin (K9CATH): Gene Cloning, Expression, and Biochemical Activity of a Novel pro-Myeloid Antimicrobial Peptide. Dev. Comp. Immunol. 2007, 31, 1278–1296. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, E.J.A.; Rijnders, M.; Claassen, E.A.; van Dijk, A.; Haagsman, H.P. Porcine Beta-Defensin 2 Displays Broad Antimicrobial Activity against Pathogenic Intestinal Bacteria. Mol. Immunol. 2008, 45, 386–394. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, M.; van Dijk, A.; van der Ent, C.K.; Arets, H.G.M.; Breukink, E.; van Os, N.; Adrichem, R.; van der Water, S.; Lino Gómez, R.; Kristensen, M.; et al. PepBiotics, Novel Cathelicidin-Inspired Antimicrobials to Fight Pulmonary Bacterial Infections. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865, 129951. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of Nitrate, Nitrite, and [15N]Nitrate in Biological Fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | No. aa | Charge | Mass (Da) |
|---|---|---|---|---|
| CATH-2 | RFGRFLRKIRRFRPKVTITIQGSARF-NH2 | 26 | 9+ | 3208 |
| PMAP-36 | Ac-GRFRRLRKKTRKRLKKIGKVLKWIPPIVGSIPLGCG | 36 | 13+ | 4198 |
| PR-39 | RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP | 39 | 10+ | 4721 |
| PMAP-23 | RIIDLLWRVRRPQKPKFVTVWVR | 23 | 6+ | 2963 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javed, A.; Balhuizen, M.D.; Pannekoek, A.; Bikker, F.J.; Heesterbeek, D.A.C.; Haagsman, H.P.; Broere, F.; Weingarth, M.; Veldhuizen, E.J.A. Effects of Escherichia coli LPS Structure on Antibacterial and Anti-Endotoxin Activities of Host Defense Peptides. Pharmaceuticals 2023, 16, 1485. https://doi.org/10.3390/ph16101485
Javed A, Balhuizen MD, Pannekoek A, Bikker FJ, Heesterbeek DAC, Haagsman HP, Broere F, Weingarth M, Veldhuizen EJA. Effects of Escherichia coli LPS Structure on Antibacterial and Anti-Endotoxin Activities of Host Defense Peptides. Pharmaceuticals. 2023; 16(10):1485. https://doi.org/10.3390/ph16101485
Chicago/Turabian StyleJaved, Ali, Melanie D. Balhuizen, Arianne Pannekoek, Floris J. Bikker, Dani A. C. Heesterbeek, Henk P. Haagsman, Femke Broere, Markus Weingarth, and Edwin J. A. Veldhuizen. 2023. "Effects of Escherichia coli LPS Structure on Antibacterial and Anti-Endotoxin Activities of Host Defense Peptides" Pharmaceuticals 16, no. 10: 1485. https://doi.org/10.3390/ph16101485
APA StyleJaved, A., Balhuizen, M. D., Pannekoek, A., Bikker, F. J., Heesterbeek, D. A. C., Haagsman, H. P., Broere, F., Weingarth, M., & Veldhuizen, E. J. A. (2023). Effects of Escherichia coli LPS Structure on Antibacterial and Anti-Endotoxin Activities of Host Defense Peptides. Pharmaceuticals, 16(10), 1485. https://doi.org/10.3390/ph16101485