
The Antimicrobial, Antibiofilm and Anti-Inflammatory Activities of P13#1, a Cathelicidin-like Achiral Peptoid

 ,
,  ,
,  ,
,  ,
,  , , , and
, , , and
Abstract
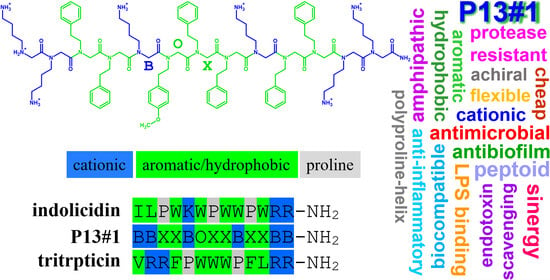
:
1. Introduction
2. Results and Discussion
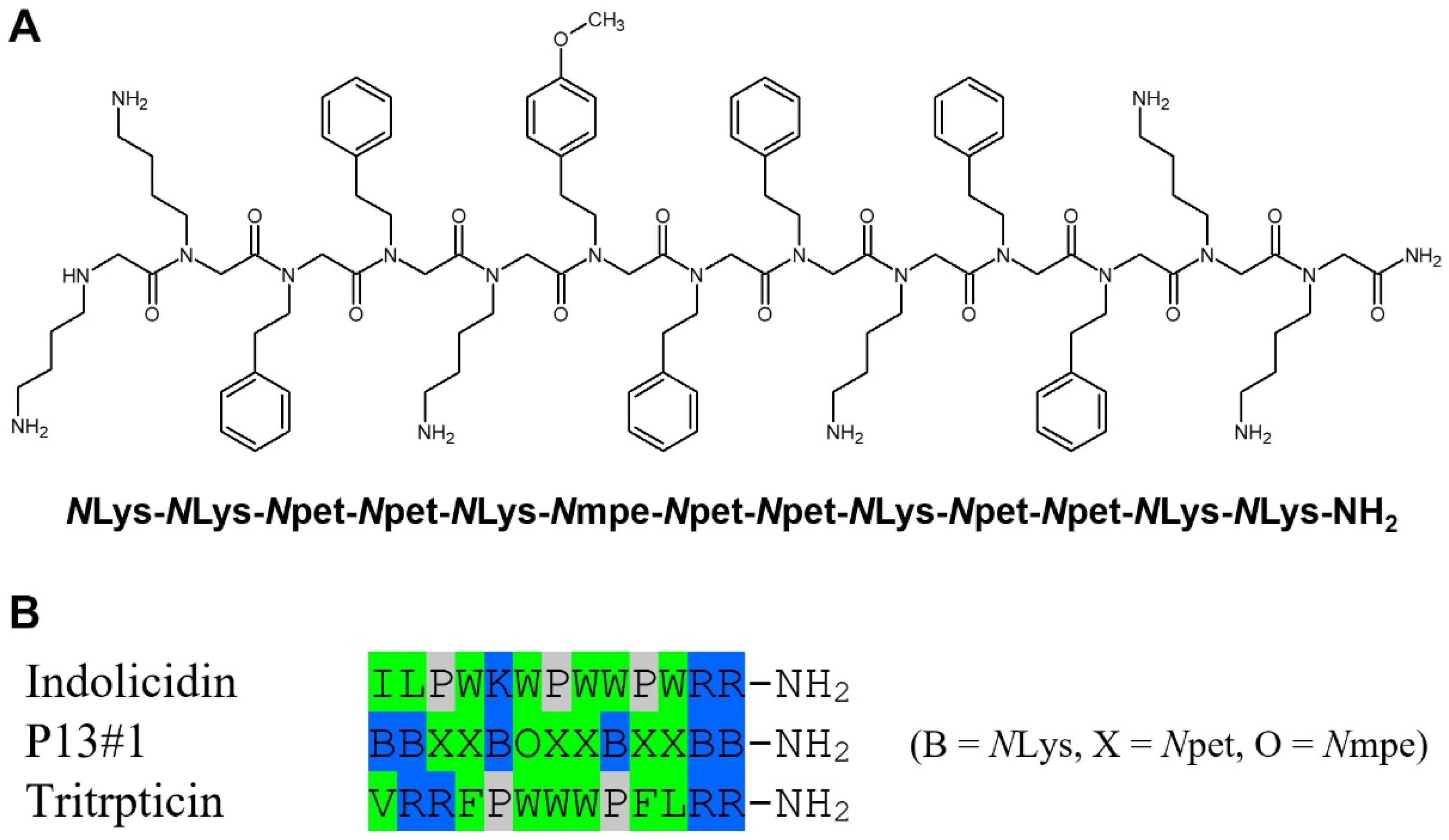
2.1. The Design of Peptoid P13#1
2.2. Synthesis of P13#1
2.3. The Antimicrobial Activity of P13#1
2.4. Peptoid–Antibiotic Interaction Study
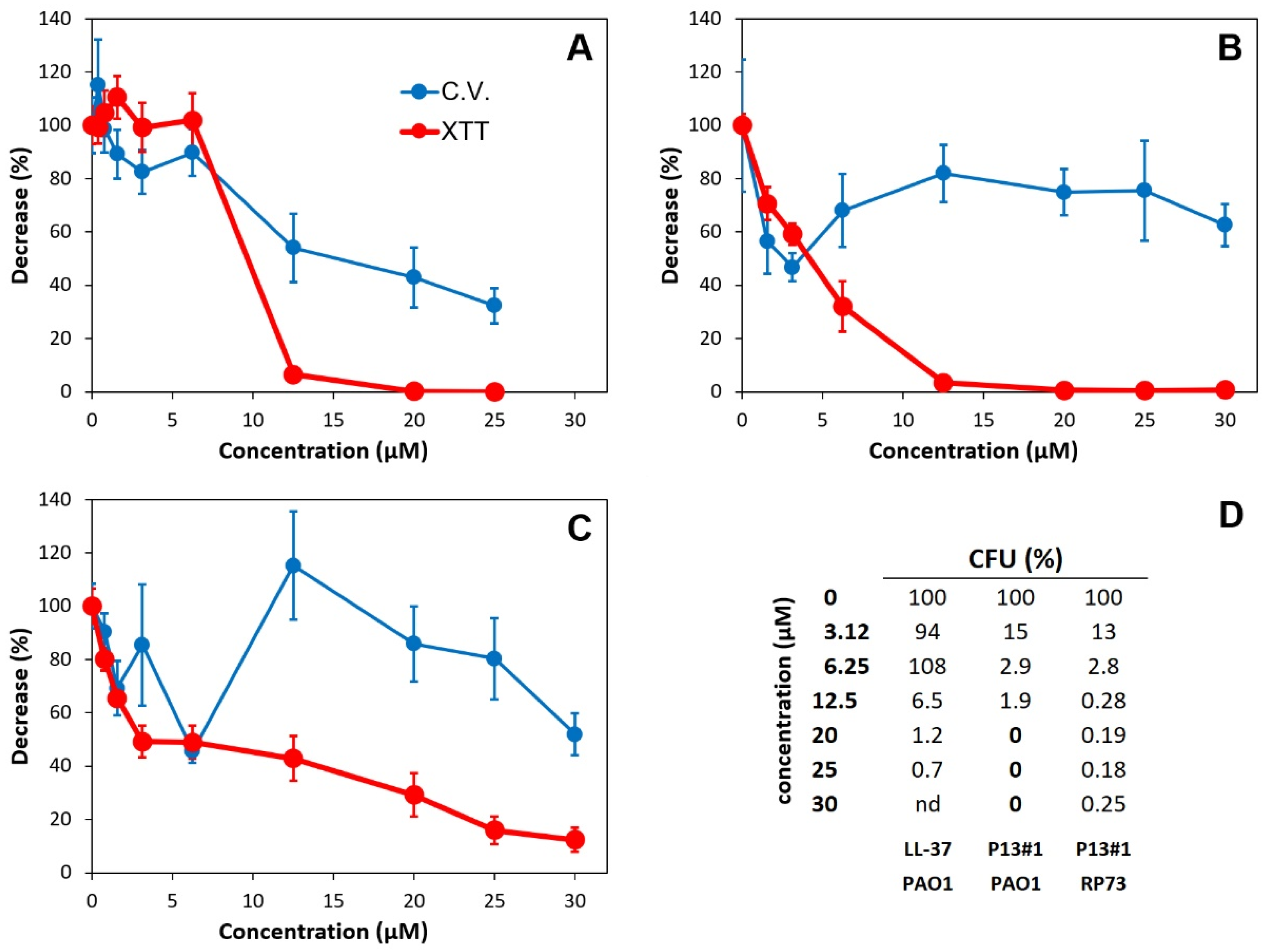
2.5. The Anti-Biofilm Activity of P13#1
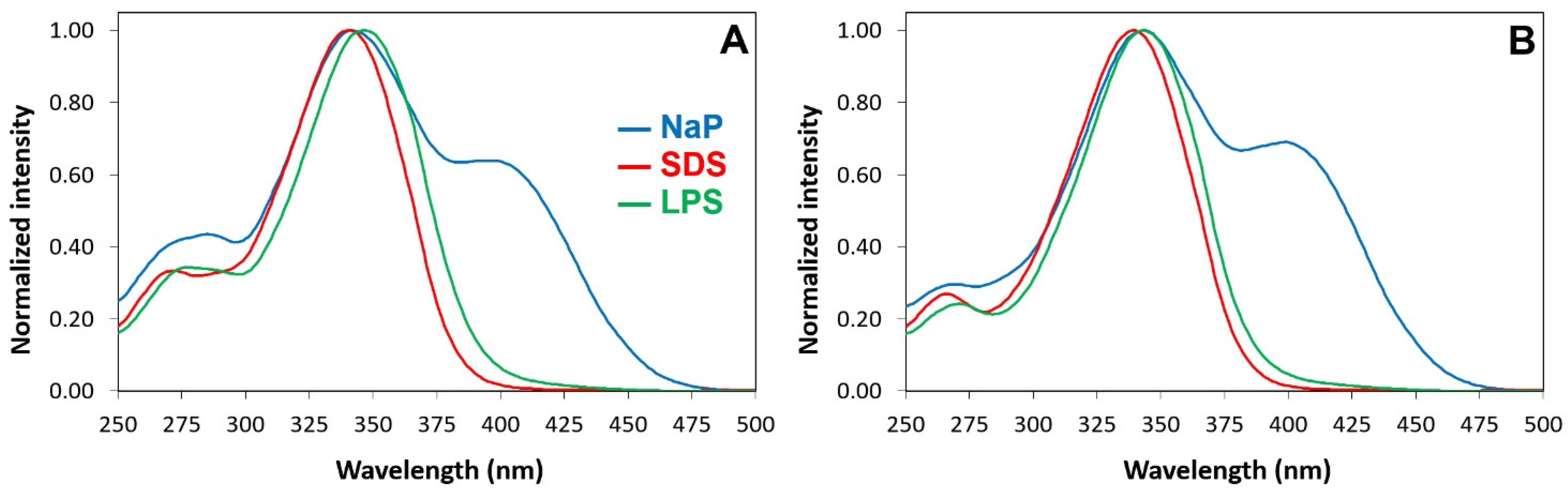
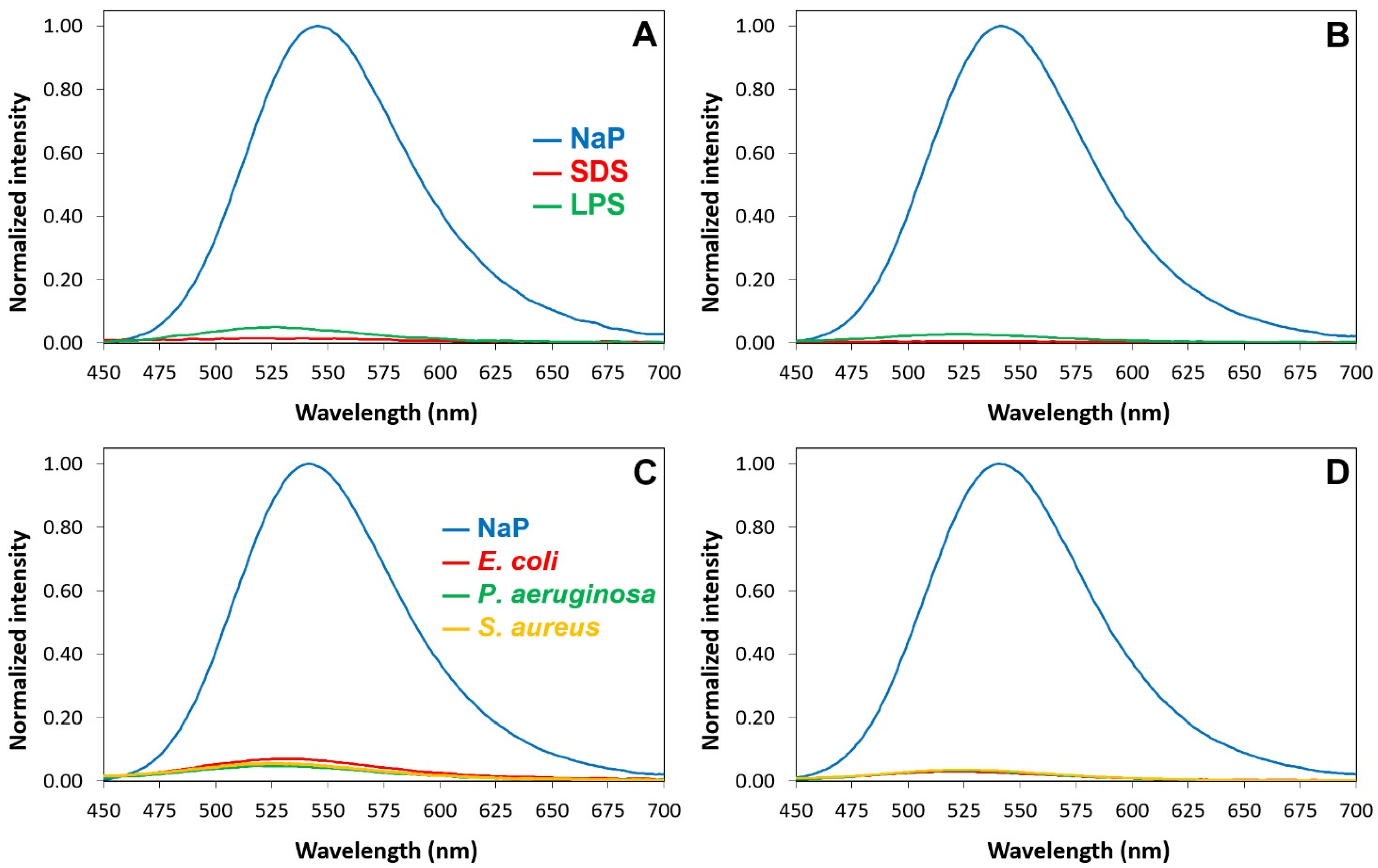
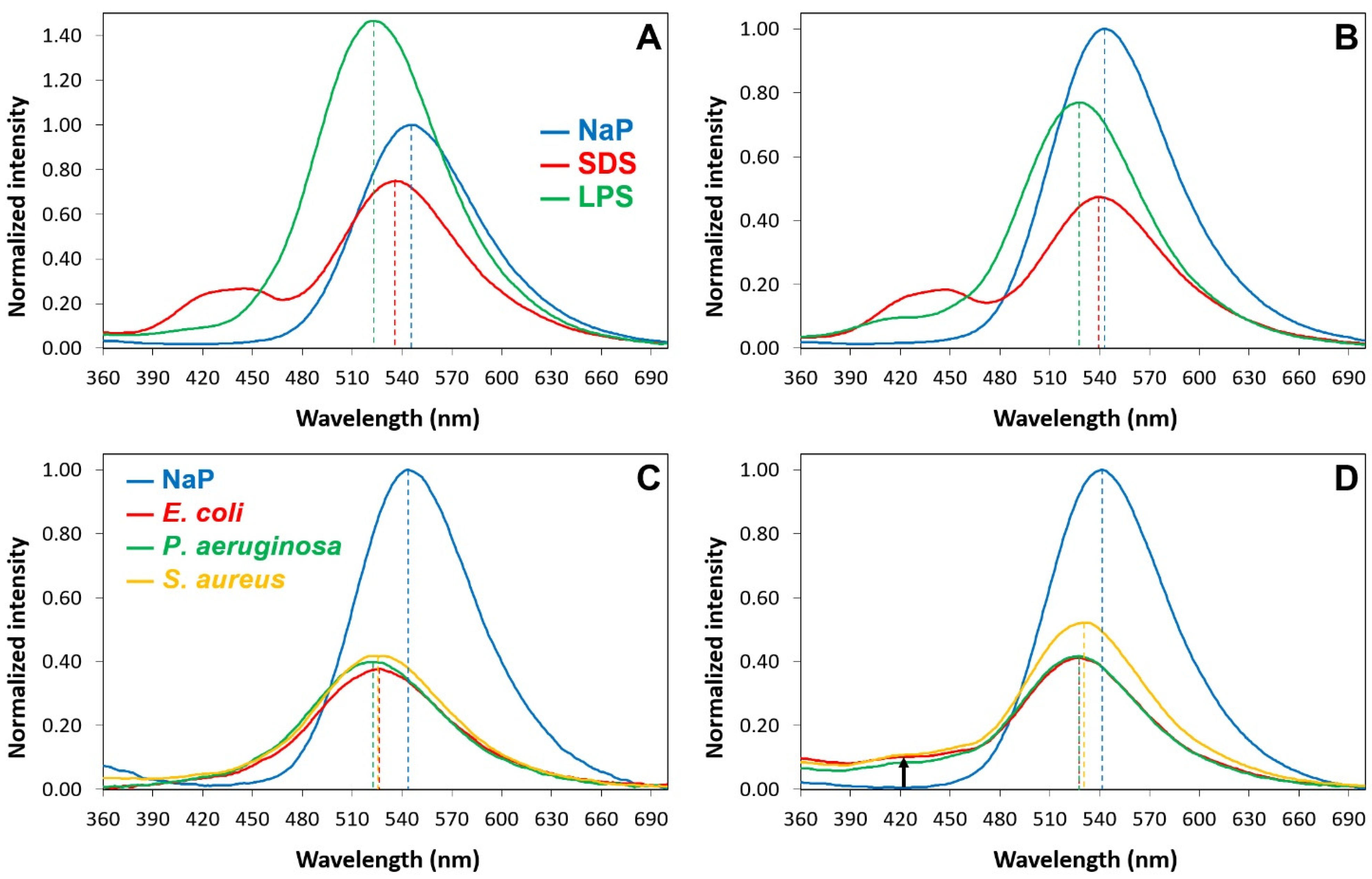
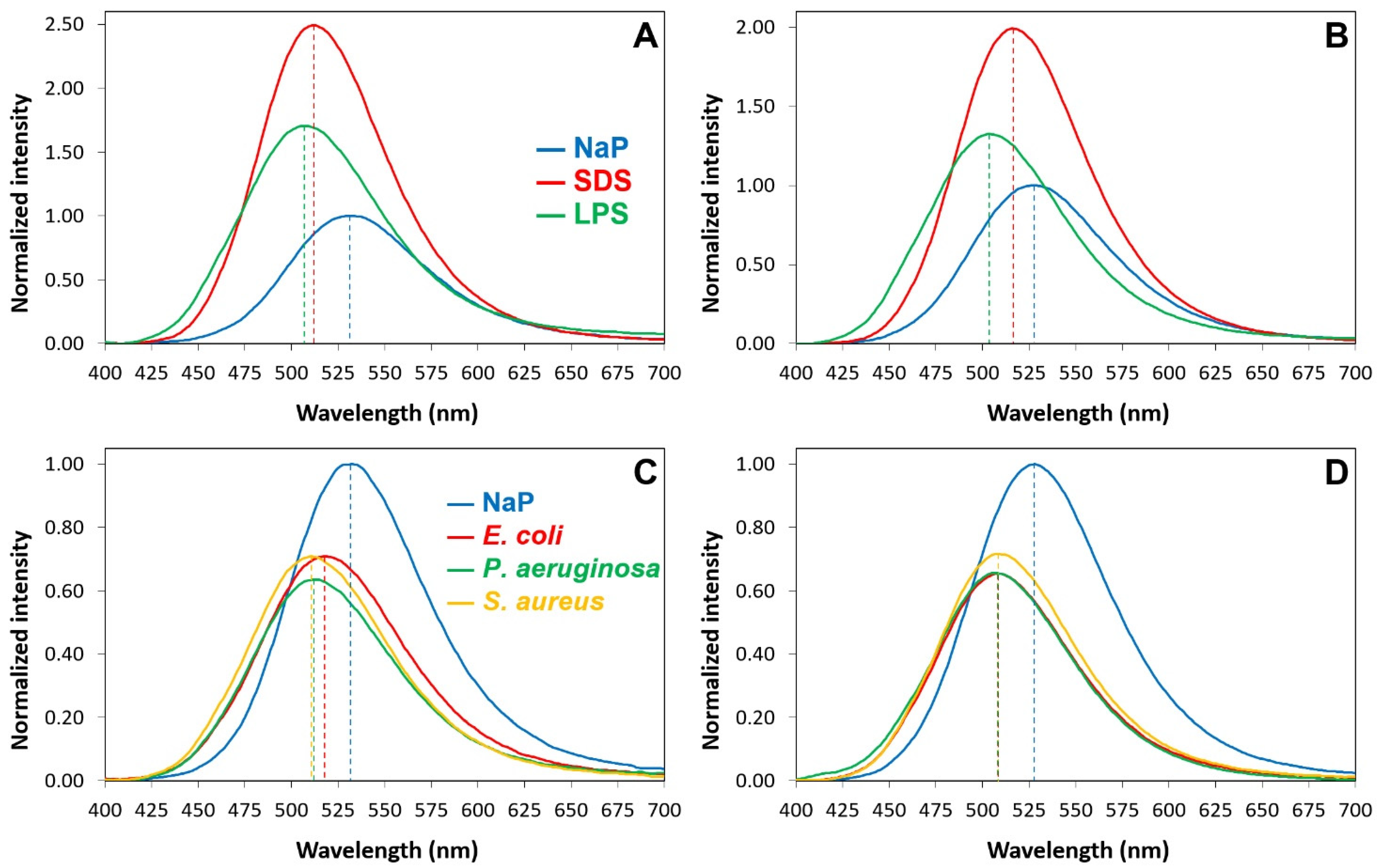
2.6. Interactions of P13#1 with SDS, LPSs and Bacterial Cells
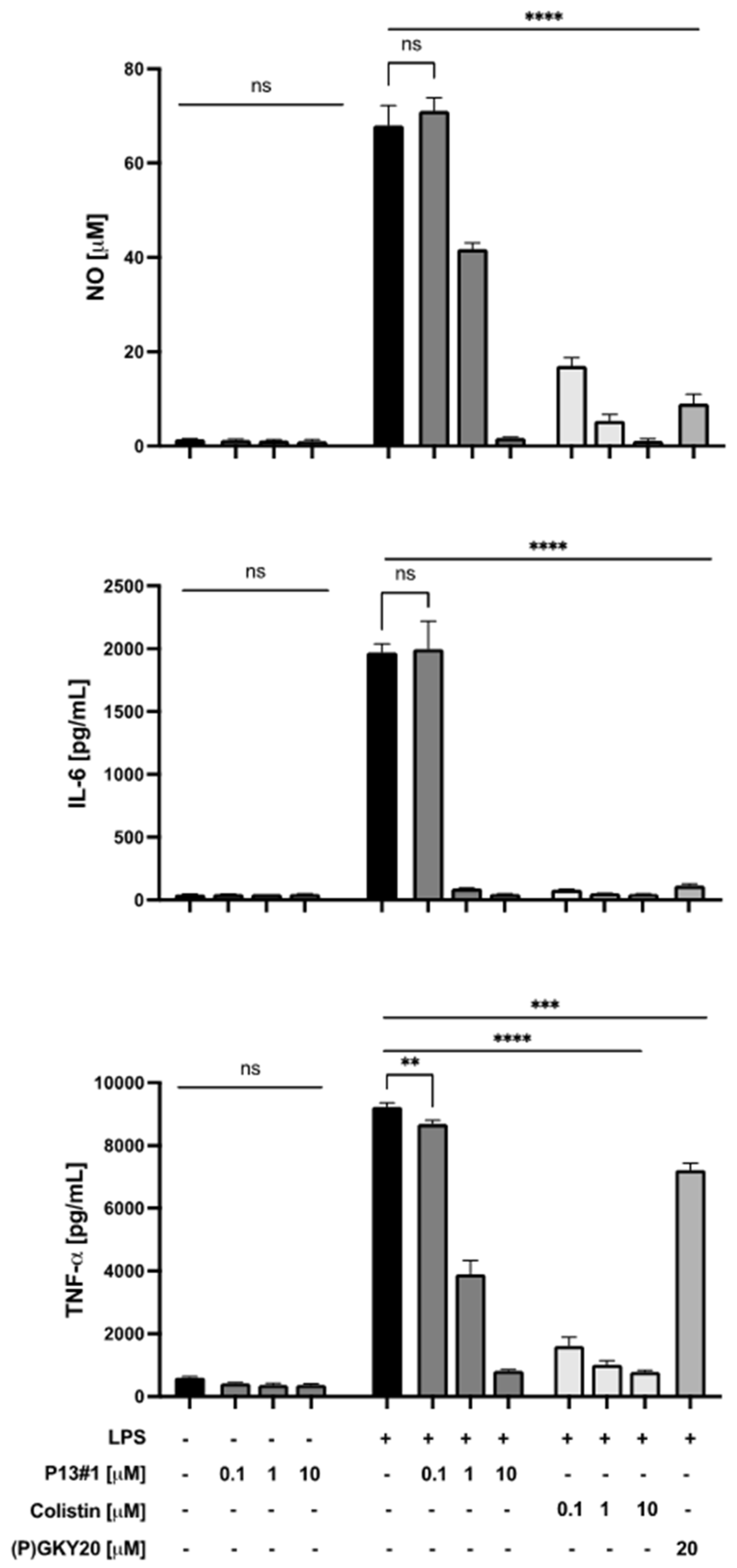
2.7. The Anti-Inflammatory Activity of P13#1 in Cell Cultures
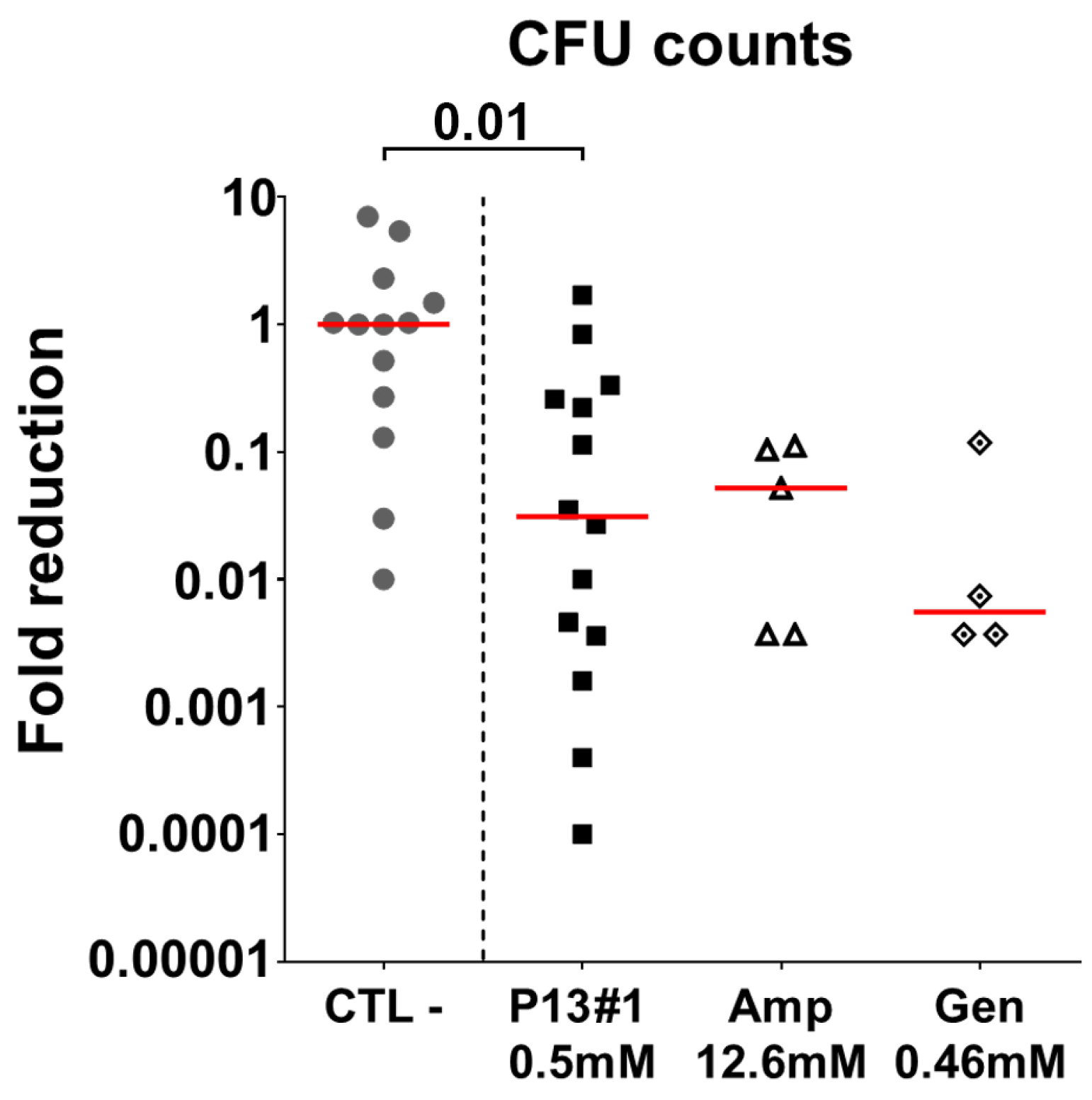
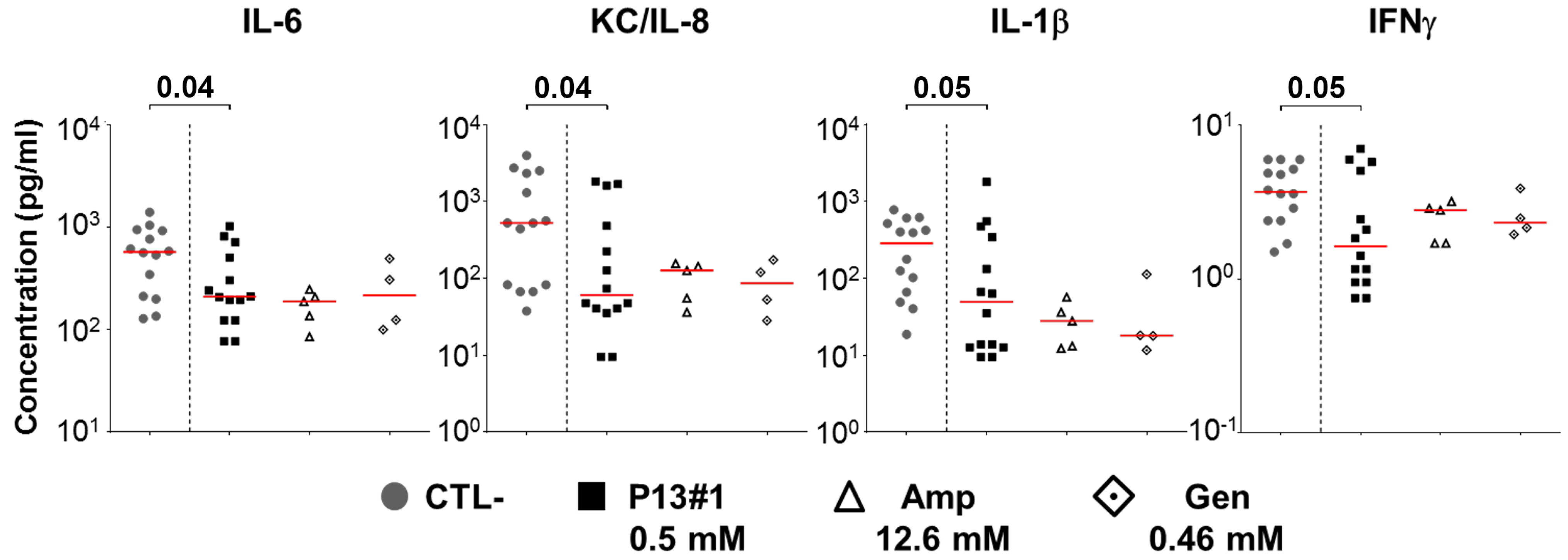
2.8. The In Vivo Efficacy of P13#1
3. Materials and Methods
3.1. Materials and General Methods
3.2. Solid-Phase Synthesis of P13#1
3.3. Solid-Phase Synthesis of CG-P13#1
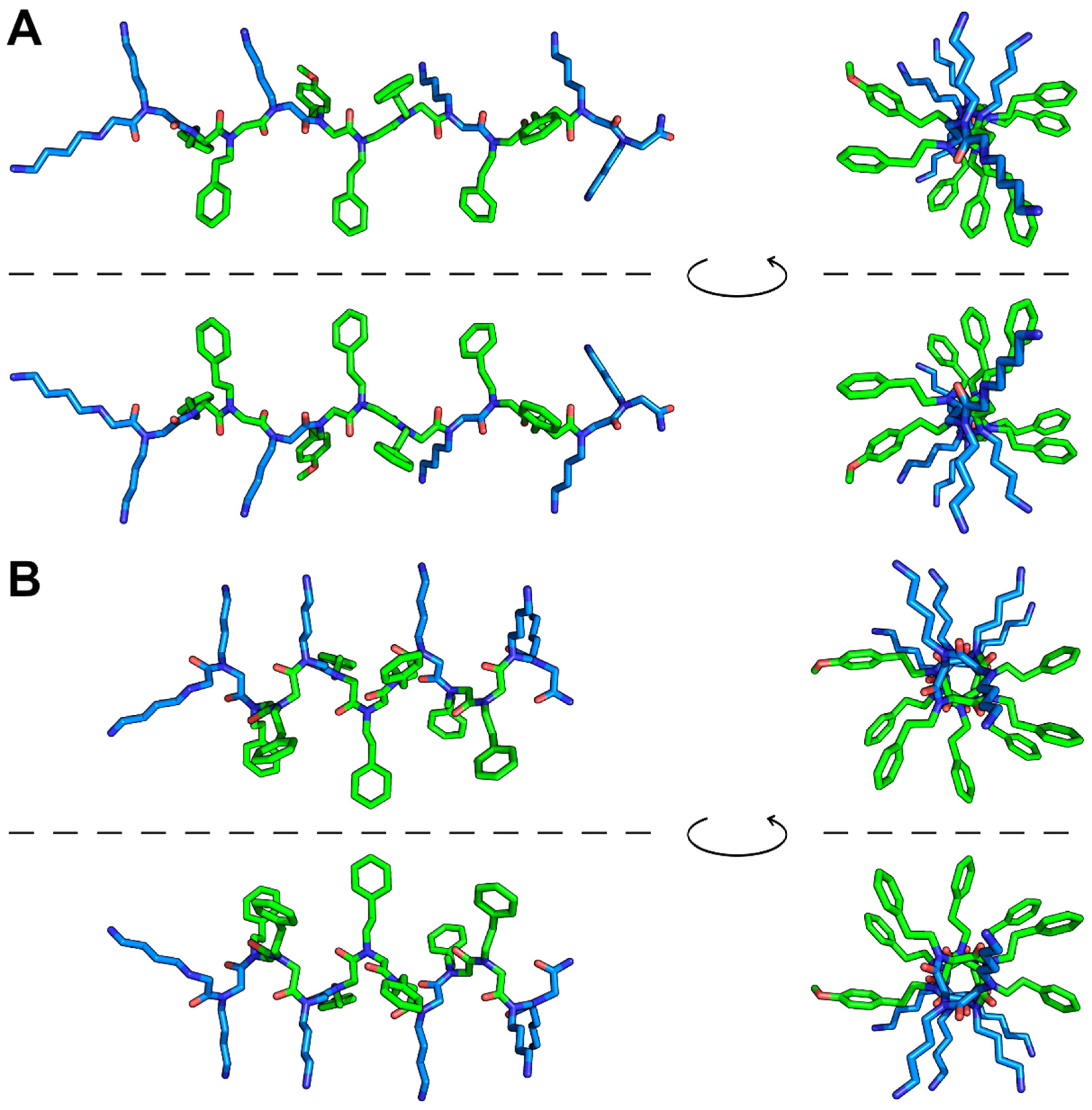
3.4. Conformational Analysis of P13#1
3.5. The Purification of P13#1
3.6. The Preparation of the Labeled Peptoids
3.7. The Production and Labelling of the Recombinant Peptides
3.8. Antimicrobial Assay
3.9. Anti-Biofilm Assay
3.10. The Interaction of Labeled Species with LPSs and SDS
3.11. The Interaction of the Labeled Species with Bacterial Cells
3.12. KD Calues and Binding Stoichiometries toward LPS
3.13. MTT Assay
3.14. The Immune-Modulatory Activity of P13#1
3.15. In Vivo Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E.W. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; De Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; Van Der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef]
- Narayana, J.L.; Huang, H.N.; Wu, C.J.; Chen, J.Y. Efficacy of the antimicrobial peptide TP4 against Helicobacter pylori infection: In vitro membrane perturbation via micellization and in vivo suppression of host immune responses in a mouse model. Oncotarget 2015, 6, 12936–12954. [Google Scholar] [CrossRef]
- Barreto-Santamaría, A.; Rivera, Z.J.; García, J.E.; Curtidor, H.; Patarroyo, M.E.; Patarroyo, M.A.; Arévalo-Pinzón, G. Shorter antibacterial peptide having high selectivity for E. coli membranes and low potential for inducing resistance. Microorganisms 2020, 8, 867. [Google Scholar] [CrossRef]
- Chatupheeraphat, C.; Peamchai, J.; Luk-in, S.; Eiamphungporn, W. Synergistic effect and antibiofilm activity of the antimicrobial peptide K11 with conventional antibiotics against multidrug-resistant and extensively drug-resistant Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2023, 13, 1153868. [Google Scholar] [CrossRef]
- Chou, S.; Wang, J.; Shang, L.; Akhtar, M.U.; Wang, Z.; Shi, B.; Feng, X.; Shan, A. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci. 2019, 7, 2394–2409. [Google Scholar] [CrossRef]
- Pamp, S.J.; Gjermansen, M.; Johansen, H.K.; Tolker-Nielsen, T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008, 68, 223–240. [Google Scholar] [CrossRef]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-Spectrum Anti-biofilm Peptide That Targets a Cellular Stress Response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef]
- Jacobsen, A.S.; Jenssen, H. Human cathelicidin LL-37 prevents bacterial biofilm formation. Future Med. Chem. 2012, 4, 1587–1599. [Google Scholar] [CrossRef]
- Martin, L.; van Meegern, A.; Doemming, S.; Schuerholz, T. Antimicrobial peptides in human sepsis. Front. Immunol. 2015, 6, 404. [Google Scholar] [CrossRef]
- Di Grazia, A.; Cappiello, F.; Cohen, H.; Casciaro, B.; Luca, V.; Pini, A.; Di, Y.P.; Shai, Y.; Mangoni, M.L. D-Amino acids incorporation in the frog skin-derived peptide esculentin-1a(1-21)NH2 is beneficial for its multiple functions. Amino Acids 2015, 47, 2505–2519. [Google Scholar] [CrossRef]
- Quercini, L.; Brunetti, J.; Riolo, G.; Bindi, S.; Scali, S.; Lampronti, I.; D’Aversa, E.; Wronski, S.; Pollini, S.; Gentile, M.; et al. An antimicrobial molecule mitigates signs of sepsis in vivo and eradicates infections from lung tissue. FASEB J. 2020, 34, 192–207. [Google Scholar] [CrossRef]
- Cresti, L.; Cappello, G.; Vailati, S.; Melloni, E.; Brunetti, J.; Falciani, C.; Bracci, L.; Pini, A. In Vivo Efficacy and Toxicity of an Antimicrobial Peptide in a Model of Endotoxin-Induced Pulmonary Inflammation. Int. J. Mol. Sci. 2023, 24, 7967. [Google Scholar] [CrossRef]
- Brunetti, J.; Carnicelli, V.; Ponzi, A.; Di Giulio, A.; Lizzi, A.R.; Cristiano, L.; Cresti, L.; Cappello, G.; Pollini, S.; Mosconi, L.; et al. Antibacterial and anti-inflammatory activity of an antimicrobial peptide synthesized with D amino acids. Antibiotics 2020, 9, 840. [Google Scholar] [CrossRef]
- Mora, P.; Mas-Moruno, C.; Tamboredo, S.; Cruz, L.J.; Pérez-Payá, E.; Albericio, F. Design of a minimized cyclic tetrapeptide that neutralizes bacterial endotoxins. J. Pept. Sci. 2006, 12, 491–496. [Google Scholar] [CrossRef]
- van Os, N.; Javed, A.; Broere, F.; van Dijk, A.; Balhuizen, M.D.; van Eijk, M.; Rooijakkers, S.H.M.; Bardoel, B.W.; Heesterbeek, D.A.C.; Haagsman, H.P.; et al. Novel insights in antimicrobial and immunomodulatory mechanisms of action of PepBiotics CR-163 and CR-172. J. Glob. Antimicrob. Resist. 2022, 30, 406–413. [Google Scholar] [CrossRef]
- Javed, A.; Slingerland, C.J.; Wood, T.M.; Martin, N.I.; Broere, F.; Weingarth, M.H.; Veldhuizen, E.J.A. Chimeric Peptidomimetic Antibiotic Efficiently Neutralizes Lipopolysaccharides (LPS) and Bacteria-Induced Activation of RAW Macrophages. ACS Infect. Dis. 2023, 9, 518–526. [Google Scholar] [CrossRef]
- van Harten, R.M.; Veldhuizen, E.J.A.; Haagsman, H.P.; Scheenstra, M.R. The cathelicidin CATH-2 efficiently neutralizes LPS- and E. coli-induced activation of porcine bone marrow derived macrophages. Vet. Immunol. Immunopathol. 2022, 244, 110369. [Google Scholar] [CrossRef]
- Ibrahim, H.R.; Hamasaki, K.; Miyata, T. Novel peptide motifs from lysozyme suppress pro-inflammatory cytokines in macrophages by antagonizing toll-like receptor and LPS-scavenging action. Eur. J. Pharm. Sci. 2017, 107, 240–248. [Google Scholar] [CrossRef]
- Tran, T.B.; Velkov, T.; Nation, R.L.; Forrest, A.; Tsuji, B.T.; Bergen, P.J.; Li, J. Pharmacokinetics/pharmacodynamics of colistin and polymyxin B: Are we there yet? Int. J. Antimicrob. Agents 2016, 48, 592–597. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef]
- Dohm, M.T.; Kapoor, R.; Barron, A.E. Peptoids: Bio-Inspired Polymers as Potential Pharmaceuticals. Curr. Pharm. Des. 2012, 17, 2732–2747. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg. Med. Chem. Lett. 1994, 4, 2657–2662. [Google Scholar] [CrossRef]
- Weiser, L.J.; Santiso, E.E. Molecular modeling studies of peptoid polymers. AIMS Mater. Sci. 2017, 4, 1029–1051. [Google Scholar] [CrossRef]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef]
- Lee, J.; Kang, D.; Choi, J.; Huang, W.; Wadman, M.; Barron, A.E.; Seo, J. Effect of side chain hydrophobicity and cationic charge on antimicrobial activity and cytotoxicity of helical peptoids. Bioorg. Med. Chem. Lett. 2018, 28, 170–173. [Google Scholar] [CrossRef]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef]
- Bolt, H.L.; Eggimann, G.A.; Jahoda, C.A.B.; Zuckermann, R.N.; Sharples, G.J.; Cobb, S.L. Exploring the links between peptoid antibacterial activity and toxicity. Medchemcomm 2017, 8, 886–896. [Google Scholar] [CrossRef]
- De Riccardis, F. The Challenge of Conformational Isomerism in Cyclic Peptoids. Eur. J. Org. Chem. 2020, 2020, 2981–2994. [Google Scholar] [CrossRef]
- Nam, H.Y.; Choi, J.; Kumar, S.D.; Nielsen, J.E.; Kyeong, M.; Wang, S.; Kang, D.; Lee, Y.; Lee, J.; Yoon, M.H.; et al. Helicity Modulation Improves the Selectivity of Antimicrobial Peptoids. ACS Infect. Dis. 2020, 6, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Shagaghi, N.; Palombo, E.A.; Clayton, A.H.A.; Bhave, M. Archetypal tryptophan-rich antimicrobial peptides: Properties and applications. World J. Microbiol. Biotechnol. 2016, 32, 31. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, M.R.; Ghosh, A.; Harmouche, N.; Casciaro, B.; Luca, V.; Bortolotti, A.; Cappiello, F.; Stella, L.; Bhunia, A.; Bechinger, B.; et al. Membrane perturbing activities and structural properties of the frog-skin derived peptide Esculentin-1a(1-21)NH2 and its Diastereomer Esc(1-21)-1c: Correlation with their antipseudomonal and cytotoxic activity. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 2327–2339. [Google Scholar] [CrossRef]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. 2017, 49, 916–925. [Google Scholar] [CrossRef]
- Shai, Y.; Oren, Z. Diastereomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 1996, 271, 7305–7308. [Google Scholar] [CrossRef]
- Bosi, S.; Fabbro, A.; Ballerini, L.; Prato, M. Carbon nanotubes: A promise for nerve tissue engineering? Nanotechnol. Rev. 2013, 2, 47–57. [Google Scholar] [CrossRef]
- Dürr, U.H.N.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Pane, K.; Durante, L.; Crescenzi, O.; Cafaro, V.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Izzo, V.; Di Donato, A.; Notomista, E. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: Application to the detection of “cryptic” antimicrobial peptides. J. Theor. Biol. 2017, 419, 254–265. [Google Scholar] [CrossRef]
- Pane, K.; Sgambati, V.; Zanfardino, A.; Smaldone, G.; Cafaro, V.; Angrisano, T.; Pedone, E.; Di Gaetano, S.; Capasso, D.; Haney, E.F.; et al. A new cryptic cationic antimicrobial peptide from human apolipoprotein E with antibacterial activity and immunomodulatory effects on human cells. FEBS J. 2016, 283, 2115–2131. [Google Scholar] [CrossRef]
- Bosso, A.; Pirone, L.; Gaglione, R.; Pane, K.; Del Gatto, A.; Zaccaro, L.; Di Gaetano, S.; Diana, D.; Fattorusso, R.; Pedone, E.; et al. A new cryptic host defense peptide identified in human 11-hydroxysteroid dehydrogenase-1 β-like: From in silico identification to experimental evidence. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Pane, K.; Cafaro, V.; Avitabile, A.; Torres, M.D.T.; Vollaro, A.; De Gregorio, E.; Catania, M.R.; Di Maro, A.; Bosso, A.; Gallo, G.; et al. Identification of Novel Cryptic Multifunctional Antimicrobial Peptides from the Human Stomach Enabled by a Computational-Experimental Platform. ACS Synth. Biol. 2018, 7, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Gaglione, R.; Dell’Olmo, E.; Bosso, A.; Chino, M.; Pane, K.; Ascione, F.; Itri, F.; Caserta, S.; Amoresano, A.; Lombardi, A.; et al. Novel human bioactive peptides identified in Apolipoprotein B: Evaluation of their therapeutic potential. Biochem. Pharmacol. 2017, 130, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.T.; Melo, M.C.R.; Crescenzi, O.; Notomista, E.; de la Fuente-Nunez, C. Mining for encrypted peptide antibiotics in the human proteome. Nat. Biomed. Eng. 2022, 6, 67–75. [Google Scholar] [CrossRef]
- Lone, A.; Arnous, A.; Hansen, P.R.; Mojsoska, B.; Jenssen, H. Synthesis of Peptoids Containing Multiple Nhtrp and Ntrp Residues: A Comparative Study of Resin, Cleavage Conditions and Submonomer Protection. Front. Chem. 2020, 8, 370. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the Bovine Antimicrobial Peptide Indolicidin Bound to Dodecylphosphocholine and Sodium Dodecyl Sulfate Micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef]
- Maayan, G.; Ward, M.D.; Kirshenbaum, K. Folded biomimetic oligomers for enantioselective catalysis. Proc. Natl. Acad. Sci. USA 2009, 106, 13679–13684. [Google Scholar] [CrossRef]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef]
- Mikkelsen, H.; McMullan, R.; Filloux, A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in ladS. PLoS ONE 2011, 6, e29113. [Google Scholar] [CrossRef]
- Bragonzi, A.; Paroni, M.; Nonis, A.; Cramer, N.; Montanari, S.; Rejman, J.; Di Serio, C.; Döring, G.; Tümmler, B. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 2009, 180, 138–145. [Google Scholar] [CrossRef]
- Bragonzi, A.; Wiehlmann, L.; Klockgether, J.; Cramer, N.; Worlitzsch, D.; Döning, G.; Tümmler, B. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 2006, 152, 3261–3269. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Silipo, A.; Bianconi, I.; Lore’, N.I.; Scamporrino, A.; Sturiale, L.; Garozzo, D.; Lanzetta, R.; Parrilli, M.; Bragonzi, A.; et al. Persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73 exhibits an under-acylated LPS structure responsible of its low inflammatory activity. Mol. Immunol. 2015, 63, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; Jeukens, J.; Freschi, L.; Alcalá-Franco, B.; Facchini, M.; Boyle, B.; Molinaro, A.; Kukavica-Ibrulj, I.; Tümmler, B.; Levesque, R.C.; et al. Comparative genomics and biological characterization of sequential Pseudomonas aeruginosa isolates from persistent airways infection. BMC Genom. 2015, 16, 1105. [Google Scholar] [CrossRef] [PubMed]
- Bloemendaal, A.L.A.; Brouwer, E.C.; Fluit, A.C. Methicillin resistance transfer from Staphylocccus epidermidis to methicillin-susceptible Staphylococcus aureus in a patient during antibiotic therapy. PLoS ONE 2010, 5, e11841. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, L.L.; Duthie, E.S. Staphylococcal Coagulase: Mode of Action and Antigenicity. J. Gen. Microbiol. 1952, 6, 95–107. [Google Scholar] [CrossRef]
- Kasetty, G.; Papareddy, P.; Kalle, M.; Rydengård, V.; Mörgelin, M.; Albiger, B.; Malmsten, M.; Schmidtchen, A. Structure-activity studies and therapeutic potential of host defense peptides of human thrombin. Antimicrob. Agents Chemother. 2011, 55, 2880–2890. [Google Scholar] [CrossRef]
- Pane, K.; Durante, L.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Sgambati, V.; Di Maro, A.; Carpentieri, A.; Izzo, V.; Di Donato, A.; et al. Rational design of a carrier protein for the production of recombinant toxic peptides in Escherichia coli. PLoS ONE 2016, 11, e0146552. [Google Scholar] [CrossRef]
- Loutet, S.A.; Valvano, M.A. Extreme antimicrobial peptide and polymyxin B resistance in the genus Burkholderia. Front. Cell. Infect. Microbiol. 2011, 1, 6. [Google Scholar] [CrossRef]
- Ghimire, J.; Guha, S.; Nelson, B.J.; Morici, L.A.; Wimley, W.C. The Remarkable Innate Resistance of Burkholderia bacteria to Cationic Antimicrobial Peptides: Insights into the Mechanism of AMP Resistance. J. Membr. Biol. 2022, 255, 503–511. [Google Scholar] [CrossRef]
- Mhlongo, J.T.; Waddad, A.Y.; Albericio, F.; de la Torre, B.G. Antimicrobial Peptide Synergies for Fighting Infectious Diseases. Adv. Sci. 2023, 10, 2300472. [Google Scholar] [CrossRef]
- Zharkova, M.S.; Orlov, D.S.; Golubeva, O.Y.; Chakchir, O.B.; Eliseev, I.E.; Grinchuk, T.M.; Shamova, O.V. Application of antimicrobial peptides of the innate immune system in combination with conventional antibiotics-a novel way to combat antibiotic resistance? Front. Cell. Infect. Microbiol. 2019, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Terminology relating to methods for the determination of susceptibility of bacteria to antimicrobial agents. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2000, 6, 503–508. [Google Scholar] [CrossRef]
- Fratini, F.; Mancini, S.; Turchi, B.; Friscia, E.; Pistelli, L.; Giusti, G.; Cerri, D. A novel interpretation of the Fractional Inhibitory Concentration Index: The case Origanum vulgare L. and Leptospermum scoparium J. R. et G. Forst essential oils against Staphylococcus aureus strains. Microbiol. Res. 2017, 195, 11–17. [Google Scholar] [CrossRef]
- Witherell, K.S.; Price, J.; Bandaranayake, A.D.; Olson, J.; Call, D.R. Circumventing colistin resistance by combining colistin and antimicrobial peptides to kill colistin-resistant and multidrug-resistant Gram-negative bacteria. J. Glob. Antimicrob. Resist. 2020, 22, 706–712. [Google Scholar] [CrossRef]
- Sacco, F.; Bitossi, C.; Casciaro, B.; Loffredo, M.R.; Fabiano, G.; Torrini, L.; Raponi, F.; Raponi, G.; Mangoni, M.L. The Antimicrobial Peptide Esc(1-21) Synergizes with Colistin in Inhibiting the Growth and in Killing Multidrug Resistant Acinetobacter baumannii Strains. Antibiotics 2022, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm activity of host defence peptides: Complexity provides opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, A.; Moretta, A.; Canè, C.; Cirillo, A.; Duilio, A. Antimicrobial and antibiofilm peptides. Biomolecules 2020, 10, 652. [Google Scholar] [CrossRef]
- Ridyard, K.E.; Overhage, J. The potential of human peptide ll-37 as an antimicrobial and anti-biofilm agent. Antibiotics 2021, 10, 650. [Google Scholar] [CrossRef]
- Siepi, M.; Oliva, R.; Masino, A.; Gaglione, R.; Arciello, A.; Russo, R.; Di Maro, A.; Zanfardino, A.; Varcamonti, M.; Petraccone, L.; et al. Environment-sensitive fluorescent labelling of peptides by luciferin analogues. Int. J. Mol. Sci. 2021, 22, 13312. [Google Scholar] [CrossRef]
- Facchin, B.M.; dos Reis, G.O.; Vieira, G.N.; Mohr, E.T.B.; da Rosa, J.S.; Kretzer, I.F.; Demarchi, I.G.; Dalmarco, E.M. Inflammatory biomarkers on an LPS-induced RAW 264.7 cell model: A systematic review and meta-analysis. Inflamm. Res. 2022, 71, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Torre, A.; Bacconi, M.; Sammicheli, C.; Galletti, B.; Laera, D.; Fontana, M.R.; Grandi, G.; De Gregorio, E.; Bagnoli, F.; Nuti, S.; et al. Four-component Staphylococcus aureus vaccine 4C-staph enhances Fcγ receptor expression in neutrophils and monocytes and mitigates S. aureus infection in neutropenic mice. Infect. Immun. 2015, 83, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Bae, T.; Schneewind, O.; Takeuchi, F.; Hiramatsu, K. Genome sequence of Staphylococcus aureus strain newman and comparative analysis of staphylococcal genomes: Polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008, 190, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Bregestovski, P.D. Chloride channels of glycine and GABA receptors with blockers: Monte Carlo minimization and structure-activity relationships. Biophys. J. 2000, 78, 1786–1803. [Google Scholar] [CrossRef]
- Siepi, M.; Morales-Narváez, E.; Domingo, N.; Monti, D.M.; Notomista, E.; Merkoçi, A. Production of biofunctionalized MoS2 flakes with rationally modified lysozyme: A biocompatible 2D hybrid material. 2D Mater. 2017, 4, 035007. [Google Scholar] [CrossRef]
- Siepi, M.; Donadio, G.; Dardano, P.; De Stefano, L.; Monti, D.M.; Notomista, E. Denatured lysozyme-coated carbon nanotubes: A versatile biohybrid material. Sci. Rep. 2019, 9, 16643. [Google Scholar] [CrossRef]
- Siepi, M.; Oliva, R.; Battista, F.; Petraccone, L.; Del Vecchio, P.; Izzo, V.; Dal Piaz, F.; Isticato, R.; Notomista, E.; Donadio, G. Molecular dissection of dh3w, a fluorescent peptidyl sensor for zinc and mercury. Sensors 2020, 20, 598. [Google Scholar] [CrossRef]
- Wetlaufer, D.B.; Edsall, J.T.; Hollingworth, B.R. Ultraviolet difference spectra of tyrosine groups in proteins and amino acids. J. Biol. Chem. 1958, 233, 1421–1428. [Google Scholar] [CrossRef]
- Pane, K.; Verrillo, M.; Avitabile, A.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Di Maro, A.; Rega, C.; Amoresano, A.; Izzo, V.; et al. Chemical Cleavage of an Asp-Cys Sequence Allows Efficient Production of Recombinant Peptides with an N-Terminal Cysteine Residue. Bioconjug. Chem. 2018, 29, 1373–1383. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Li, J.; Kleintschek, T.; Rieder, A.; Cheng, Y.; Baumbach, T.; Obst, U.; Schwartz, T.; Levkin, P.A. Hydrophobic liquid-infused porous polymer surfaces for antibacterial applications. ACS Appl. Mater. Interfaces 2013, 5, 6704–6711. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Tan, M.; Ho, B.; Ding, J.L.; Wohland, T. Determination of critical micelle concentrations and aggregation numbers by fluorescence correlation spectroscopy: Aggregation of a lipopolysaccharide. Anal. Chim. Acta 2006, 556, 216–225. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (MBC) 1 | MIC | |||
|---|---|---|---|---|
| (μM) | (μM) | |||
| Bacterial Strain | P13#1 | (P)GKY20 | Vanc 2 | Pol B 2 |
| Gram(−) | ||||
| Pseudomonas aeruginosa PAO1 | 1.56 (1.56) | 3.12 (3.12) | 0.18 | |
| Pseudomonas aeruginosa KK27 | 1.56 (1.56) | nd 3 | 0.36 | |
| Pseudomonas aeruginosa RP73 | 1.56 (1.56) | 1.56 (1.56) | 0.09 | |
| Pseudomonas aeruginosa AA2 | 1.56 (1.56) | nd | 0.36 | |
| Pseudomonas aeruginosa PA14 | 1.56 (1.56) | 6.25 (6.25) | 0.18 | |
| Klebsiella pneumoniae ATCC 700603 | 1.56 (1.56) | 6.25 (6.25) | 0.36 | |
| Acinetobacter baumanii ATCC 17878 | 1.56 (1.56) | 3.12 (3.12) | 0.18 | |
| Burkholderia cenocepacea LMG 18863 | >50 | >50 | >46.2 | |
| Escherichia coli ATCC 25922 | 1.56 (1.56) | 6.25 (6.25) | 0.72 | |
| Salmonella typhimurium ATCC 14028 | 1.56 (1.56) | 3.12 (3.12) | 0.36 | |
| Salmonella enteriditis 706 RIVM | 1.56 (1.56) | 6.25 (6.25) | 0.72 | |
| Gram(+) | ||||
| Staphylococcus aureus ATCC 6538P | 0.78 (0.78) | 1.56 (1.56) | 0.34 | |
| Staphylococcus aureus WKZ2 (MRSA) | 1.56 (1.56) | nd | 0.34 | |
| Staphylococcus aureus Newman | 3.12 (3.12) | 6.25 (6.25) | 0.17 | |
| Enterococcus faecalis ATCC 29212 | 1.56 (1.56) | 1.56 (1.56) | 0.34 | |
| Bacterial Strains | Antibiotic | MIC (μg/mL) | ΣFICi 1 |
|---|---|---|---|
| P13#1 | |||
| Gram(−) | |||
| Pseudomonas aeruginosa PAO1 | Tobramycin | 0.0078 | 0.626 |
| Ciprofloxacin | 0.125 | 0.750 | |
| Colistin | 0.25 | 0.376 | |
| Meropenem | 0.25 | 2 | |
| Gram(+) | |||
| Staphylococcus aureus ATCC 6538P | Tobramycin | 0.00097 | 0.750 |
| Ciprofloxacin | 0.125 | 1 | |
| Meropenem | 0.0625 | 2 |
| Peptide/ Peptoid | LPSEc 1 (µg/mL) | KD (µM) | [Binding Sites] (µM) | Stoichiometry 2 (Peptoid:LPS) |
|---|---|---|---|---|
| Luc-P13#1 | 40 | 0.121 (±0.032) | 4.53 (±0.123) | 1.13:1 |
| Luc-P13#1 | 5 | 0.085 (±0.021) | 2.21 (±0.107) | 4.42:1 |
| Luc-GKY20 | 40 | 0.382 (±0.030) | 6.12 (±0.109) | 1.53:1 |
| Luc-GKY20 | 5 | 0.109 (±0.022) | 2.62 (±0.153) | 5.24:1 |
| LPSPa 3 (µg/mL) | ||||
| Luc-P13#1 | 5 | 0.032 (±0.0085) | 1.91 (±0.049) | - |
| Luc-GKY20 | 5 | 0.054 (±0.013) | 2.1 (±0.08) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cafaro, V.; Bosso, A.; Di Nardo, I.; D’Amato, A.; Izzo, I.; De Riccardis, F.; Siepi, M.; Culurciello, R.; D’Urzo, N.; Chiarot, E.; et al. The Antimicrobial, Antibiofilm and Anti-Inflammatory Activities of P13#1, a Cathelicidin-like Achiral Peptoid. Pharmaceuticals 2023, 16, 1386. https://doi.org/10.3390/ph16101386
Cafaro V, Bosso A, Di Nardo I, D’Amato A, Izzo I, De Riccardis F, Siepi M, Culurciello R, D’Urzo N, Chiarot E, et al. The Antimicrobial, Antibiofilm and Anti-Inflammatory Activities of P13#1, a Cathelicidin-like Achiral Peptoid. Pharmaceuticals. 2023; 16(10):1386. https://doi.org/10.3390/ph16101386
Chicago/Turabian StyleCafaro, Valeria, Andrea Bosso, Ilaria Di Nardo, Assunta D’Amato, Irene Izzo, Francesco De Riccardis, Marialuisa Siepi, Rosanna Culurciello, Nunzia D’Urzo, Emiliano Chiarot, and et al. 2023. "The Antimicrobial, Antibiofilm and Anti-Inflammatory Activities of P13#1, a Cathelicidin-like Achiral Peptoid" Pharmaceuticals 16, no. 10: 1386. https://doi.org/10.3390/ph16101386
APA StyleCafaro, V., Bosso, A., Di Nardo, I., D’Amato, A., Izzo, I., De Riccardis, F., Siepi, M., Culurciello, R., D’Urzo, N., Chiarot, E., Torre, A., Pizzo, E., Merola, M., & Notomista, E. (2023). The Antimicrobial, Antibiofilm and Anti-Inflammatory Activities of P13#1, a Cathelicidin-like Achiral Peptoid. Pharmaceuticals, 16(10), 1386. https://doi.org/10.3390/ph16101386