

Adenylosuccinic Acid Is a Non-Toxic Small Molecule In Vitro and In Vivo



Abstract
:1. Introduction
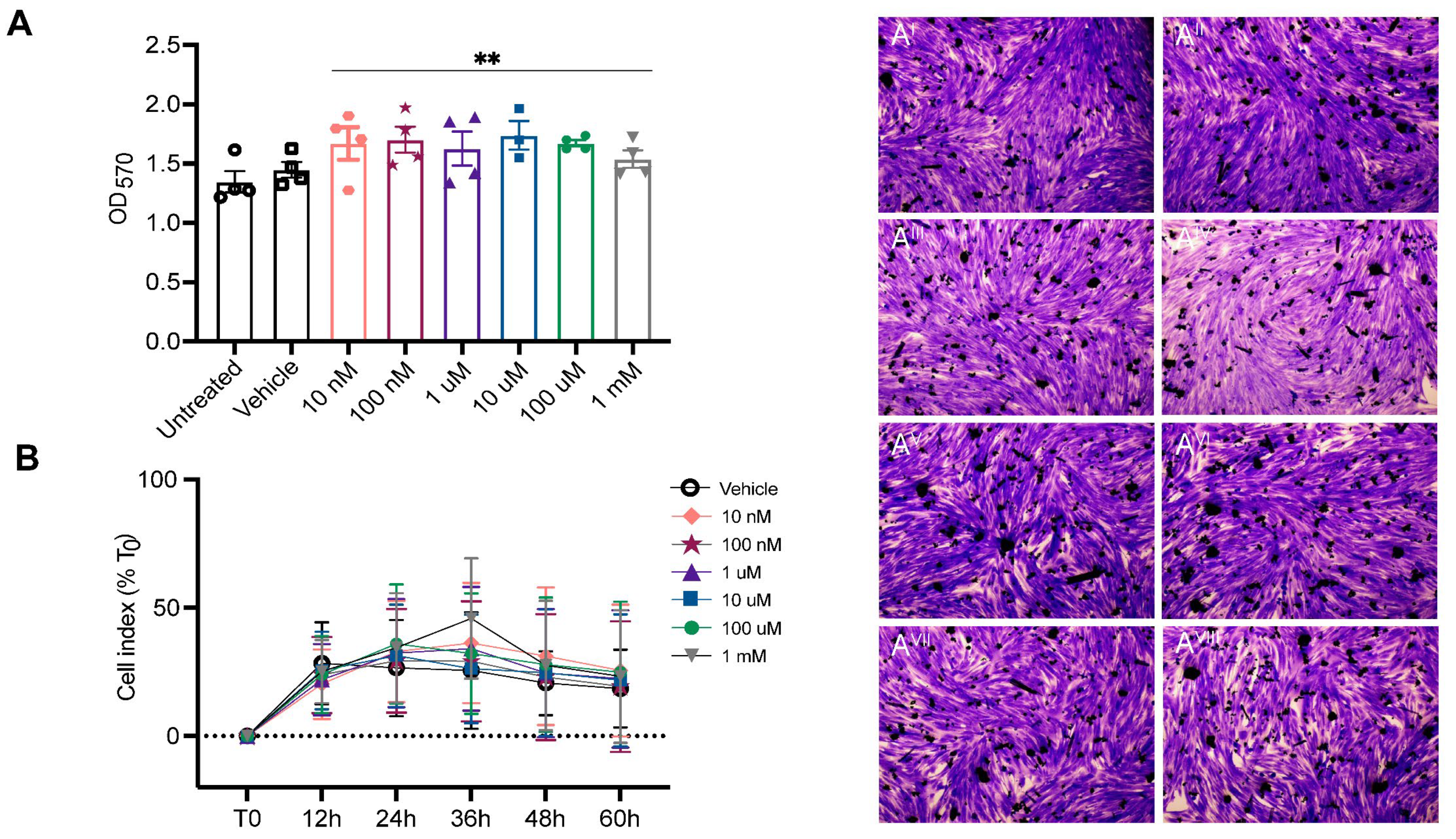
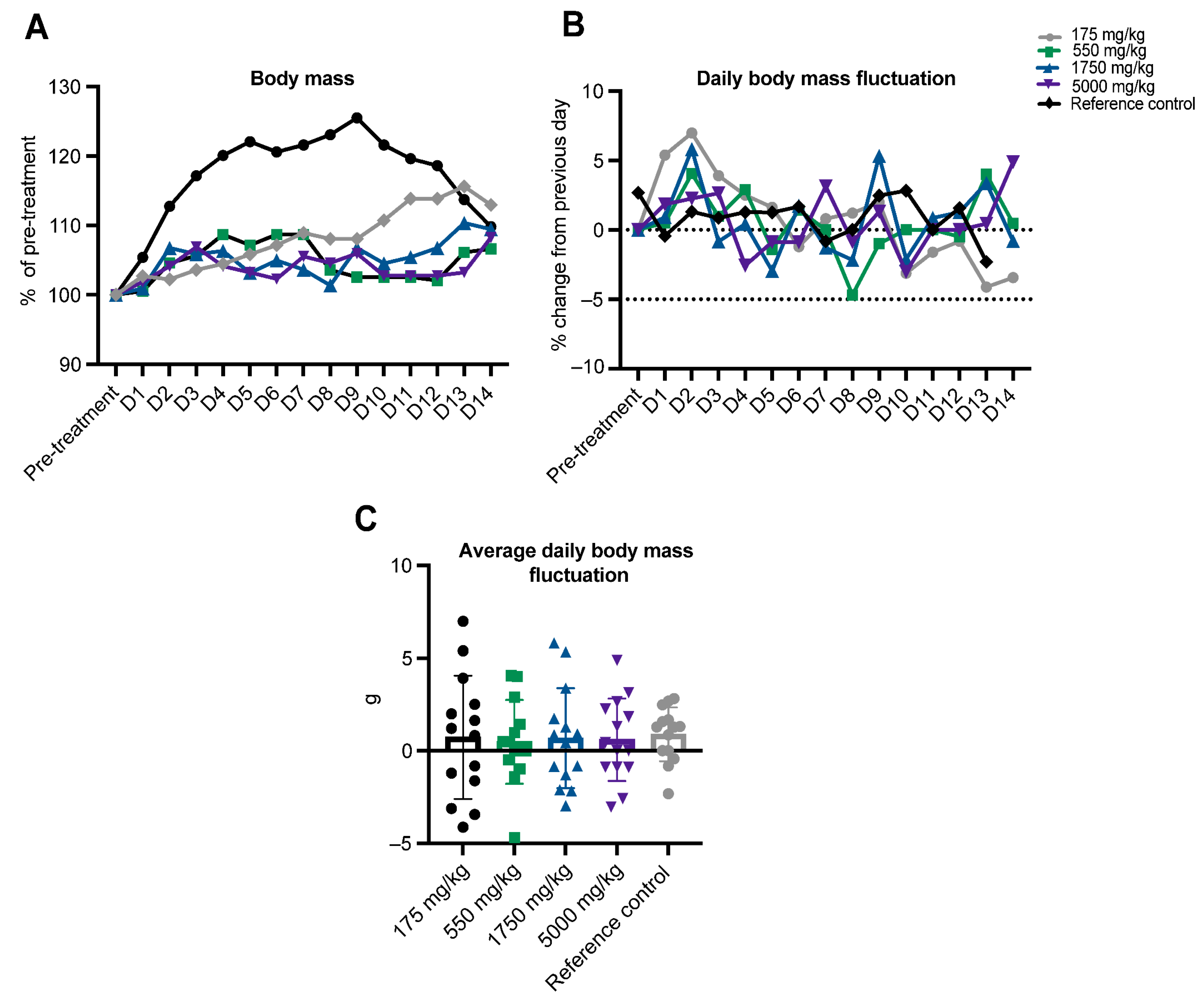
2. Results
3. Discussion
4. Materials and Methods
4.1. In Vitro Toxicity Testing
4.2. In Vivo Toxicity Testing
4.2.1. Animals
4.2.2. Drug Details
4.2.3. Acute Oral Toxicity Procedure
4.2.4. Haematology
4.2.5. Biochemistry
4.2.6. Histopathology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonsett, B.; Rudman, A. The dystrophin connection—ATP? Med. Hypotheses 1992, 38, 139–154. [Google Scholar] [CrossRef]
- Timpani, A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Budzinska, M.; Zimna, A.; Kurpisz, M. The role of mitochondria in Duchenne muscular dystrophy. J. Physiol. Pharmacol. 2021, 72, 157–166. [Google Scholar]
- Bonsett, C.; Rudman, A. Duchenne’s muscular dystrophy: A tissue culture perspective. Indiana Med. J. Indiana State Med. Assoc. 1984, 77, 446. [Google Scholar]
- Chandel, N.S. Nucleotide metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040592. [Google Scholar] [CrossRef] [PubMed]
- Arinze, I.J. Facilitating understanding of the purine nucleotide cycle and the one-carbon pool: Part I: The purine nucleotide cycle. Biochem. Mol. Biol. Educ. 2005, 33, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.L.; Hines, J.J.; Sabina, R.L.; Harbury, O.L.; Holmes, E.W. Disruption of the purine nucleotide cycle by inhibition of adenylosuccinate lyase produces skeletal muscle dysfunction. J. Clin. Investig. 1984, 74, 1422–1427. [Google Scholar] [CrossRef]
- Lowenstein, J.M.; Goodman, M.N. The purine nucleotide cycle in skeletal muscle. Fed. Proc. 1978, 37, 2308–2312. [Google Scholar]
- Rybalka, E.; Kourkais, S.; Bonsett, C.A.; Moghadaszadeh, B.; Beggs, A.H.; Timpani, C.A. Adenylosuccinic Acid: An Orphan Drug with Untapped Potential. Pharmaceuticals 2023, 16, 822. [Google Scholar] [CrossRef] [PubMed]
- Gooding, J.R.; Jensen, M.V.; Dai, X.; Wenner, B.R.; Lu, D.; Arumugam, M.; Ferdaoussi, M.; MacDonald, F.E.; Newgard, C.B. Adenylosuccinate is an insulin secretagogue derived from glucose-induced purine metabolism. Cell Rep. 2015, 13, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Bonsett, C.A.; Rudman, A. ‘Oil globules’ in Duchenne muscular dystrophy—History, demonstration, metabolic significance. Med. Hypotheses 1994, 43, 327–338. [Google Scholar] [CrossRef]
- Lipsky, M.S.; Sharp, L.K. From idea to market: The drug approval process. J. Am. Board. Fam. Pract. 2001, 14, 362–367. [Google Scholar] [PubMed]
- Van Norman, G.A. Drugs and devices: Comparison of European and US approval processes. JACC Basic. Transl. Sci. 2016, 1, 399–412. [Google Scholar] [PubMed]
- Clarridge, K.E.; Chin, S.J.; Stone, K.D. Overview of FDA drug approval and labeling. J. Allergy Clin. Immunol. Pract. 2022, 10, 3051–3056. [Google Scholar] [CrossRef] [PubMed]
- OECD. Test No. 425: Acute Oral Toxicity: Up-and-Down Procedure; OECD: Paris, France, 2022. [Google Scholar]
- Mazzaccara, C.; Labruna, G.; Cito, G.; Scarfo, M.; De Felice, M.; Pastore, L.; Sacchetti, L. Age-Related Reference Intervals of the Main Biochemical and Hematological Parameters in C57BL/6J, 129SV/EV and C3H/HeJ Mouse Strains. PLoS ONE 2008, 3, e3772. [Google Scholar] [CrossRef]
- Zaias, J.; Mineau, M.; Cray, C.; Yoon, D.; Altman, N.H. Reference values for serum proteins of common laboratory rodent strains. J. Am. Assoc. Lab. Anim. Sci. 2009, 48, 387–390. [Google Scholar]
- Silva-Santana, G.; Bax, J.C.; Fernandez, D.C.S.; Bacellar, D.T.L.; Hooper, C.; Dias, A.A.S.O.; Silva, C.B.; de Souza, A.M.; Ramos, S.; Santos, R.A.; et al. Clinical hematological and biochemical parameters in Swiss, BALB/c, C57BL/6 and B6D2F1 Mus musculus. Anim. Models Exp. Med. 2020, 3, 304–315. [Google Scholar] [CrossRef]
- Nemzek, J.A.; Bolgos, G.L.; Williams, B.A.; Remick, D.G. Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm. Res. 2001, 50, 523–527. [Google Scholar] [CrossRef]
- McInnes, E.F. Background Lesions in Laboratory Animals: A Color Atlas; Saunders/Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Rybalka, E.; Goodman, C.A.; Campejli, D.G.; Hayes, A.; Timpani, C.A. Adenylosuccinic acid: A novel inducer of the cytoprotectant Nrf2 with efficacy in Duchenne muscular dystrophy. Curr. Med. Res. Opin. 2021, 37, 465–467. [Google Scholar] [CrossRef]
- Zou, G.L.; Zhang, X.R.; Ma, Y.L.; Lu, Q.; Zhao, R.; Zhu, Y.Z.; Wang, Y.Y. The role of Nrf2/PIWIL2/purine metabolism axis in controlling radiation-induced lung fibrosis. Am. J. Cancer Res. 2020, 10, 2752–2767. [Google Scholar]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; de Almeida Rodriguez, R.P.; West, J.A.; Sewell, G.W.; Md-Ibrahim, M.N.; Reikine, S.; Sirago, G.; Unger, L.W.; Iglesias-Romero, A.B.; Ramshorn, K.; et al. FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell 2020, 180, 278–295.e23. [Google Scholar] [CrossRef] [PubMed]
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals 2020, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, A.D.; Mrowietz, U. Fumaric acid esters, their place in the treatment of psoriasis. Br. J. Dermatol. 2004, 150, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.J.; Chalmers, R.J.; Griffiths, C.E. Fumaric acid esters for severe psoriasis: A retrospective review of 58 cases. Br. J. Dermatol. 2005, 153, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. Fumaric acid esters for psoriasis: A systematic review. Ir. J. Med. Sci. 2017, 186, 161–177. [Google Scholar] [CrossRef]
- Palte, M.J.; Wehr, A.; Tawa, M.; Perkin, K.; Leigh-Pemberton, R.; Hanna, J.; Miller, C.; Penner, N. Improving the Gastrointestinal Tolerability of Fumaric Acid Esters: Early Findings on Gastrointestinal Events with Diroximel Fumarate in Patients with Relapsing-Remitting Multiple Sclerosis from the Phase 3, Open-Label EVOLVE-MS-1 Study. Adv. Ther. 2019, 36, 3154–3165. [Google Scholar] [CrossRef]
- Fox, E.J.; Vasquez, A.; Grainger, W.; Ma, T.S.; von Hehn, C.; Walsh, J.; Li, J.; Zambrano, J. Gastrointestinal tolerability of delayed-release dimethyl fumarate in a multicenter, open-label study of patients with relapsing forms of multiple sclerosis (MANAGE). Int. J. MS Care 2016, 18, 9–18. [Google Scholar] [CrossRef]
- Russo, C.D.; Scott, K.A.; Pirmohamed, M. Dimethyl fumarate induced lymphopenia in multiple sclerosis: A review of the literature. Pharmacol. Ther. 2021, 219, 107710. [Google Scholar] [CrossRef]
- Colas, C.; Ung, P.M.; Schlessinger, A. SLC Transporters: Structure, Function, Drug Discovery. Medchemcomm 2016, 7, 1069–1081. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.J.; Pontefract, S.K. Adverse drug reactions. Clin. Med. 2016, 16, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Goodman, C.A.; Stathis, C.G.; White, J.D.; Mamchaoui, K.; Butler-Browne, G.; Gueven, N.; Hayes, A.; Rybalka, E. Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci. Rep. 2020, 10, 1125. [Google Scholar] [CrossRef] [PubMed]
- Longbrake, E.E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult. Scler. J.–Exp. Transl. Clin. 2015, 1, 2055217315596994. [Google Scholar] [CrossRef]
- Timpani, C.A.; Rybalka, E. Calming the (Cytokine) Storm: Dimethyl Fumarate as a Therapeutic Candidate for COVID-19. Pharmaceuticals 2021, 14, 15. [Google Scholar] [CrossRef]
- Timpani, C.A.; Mamchaoui, K.; Butler-Browne, G.; Rybalka, E. Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants 2020, 9, 1268. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cheregi, B.D.; Sorensen, J.C.; Nurgali, K.; Hayes, A. Chemotherapeutic agents induce mitochondrial superoxide production and toxicity but do not alter respiration in skeletal muscle in vitro. Mitochondrion 2018, 42, 33–49. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Dose | Post-Fasted Body Weight (g) | ASA Mass (mg) | Volume H2O/Dose (mL) |
|---|---|---|---|
| 175 mg/kg | 20.4 | 3.57 | 0.204 |
| 550 mg/kg | 19.6 | 10.78 | 0.196 |
| 1750 mg/kg | 22.2 | 38.85 | 0.222 |
| 5000 mg/kg | 21.8 | 109 | 0.218 |
| Observations | ||||||
|---|---|---|---|---|---|---|
| Observations | Symptoms | 175 mg/kg ASA n = 1 | 550 mg/kg ASA n = 1 | 1750 mg/kg ASA n = 1 | 5000 mg/kg ASA n = 1 | Untreated |
| Body weight | >5% loss of body weight | - | - | - | - | - |
| Food and water consumption | - | - | - | - | - | - |
| Motor activity | Home-cage activity | - | - | - | + (<30 m) | - |
| Neurological system | Tremors, limb tone, ataxia | - | - | - | - | - |
| Respiratory system | Gasping, heaving, cyanosis | - | - | - | - | - |
| Gastrointestinal function | Abdominal griping, vomiting, diarrhoea | - | - | - | + (diarrhoea) | - |
| Skin and mucous membranes | Secretions, excretions | - | - | - | - | - |
| Pain | Grimacing, altered social activity | - | - | - | - | |
| Parameters | Reference Range | ASA Dose | Reference Control | |||
|---|---|---|---|---|---|---|
| 175 mg/kg | 550 mg/kg | 1750 mg/kg | 5000 mg/kg | |||
| Haematological | ||||||
| Erythrocytes (×1012/L) | 3.47–11.73 a | 10.5 | 11.06 | 10.66 | 11.0 | 11.02 |
| Haematocrit (L/L) | 0.16–0.58 a | 0.48 | 0.54 | 0.54 | 0.54 | 0.54 |
| Haemoglobin (g/L) | 57.0–170.0 a | 156 | 162 | 153 | 157 | 158 |
| MCV (fL) | 25.7–59.7 b | 48 | 49 | 50 | 49 | 49 |
| MCH (pg) | 9.0–19.2 b | 16 | 15 | 14 | 14 | 14 |
| MCHC (g/L) | 264.6–314.2 b | 323 * | 300 | 286 | 289 | 290 |
| Platelet (×109/L) | 520–1880 b | 942 | 954 | 1040 | 1077 | 1188 |
| WCC (×109/L) | 2.2–11.53 a | 8.9 | 5.5 | 3.8 | 4.6 | 5.8 |
| Segmented Neutrophils (×109/L) | 0.12–0.30 b | 0.3 | 0.7 * | 0.2 | 0.4 * | 0.5 * |
| Lymphocytes (×109/L) | 4.36–5.68 b | 8.5 * | 4.6 | 3.4 * | 3.4 * | 4.8 |
| Monocytes (×109/L) | 0–0.3 b | 0.18 | 0.06 | 0.23 | 0 | 0 |
| Eosinophils (×109/L) | 0–5.1 d | 0 | 2 | 0 | 7* | 0.1 |
| Basophils (×109/L) | 0–0.1 d | 0 | 0 | 0 | 0.1 | 0 |
| Biochemical | ||||||
| Total Protein (g/L) | 45–83 a | 48 | 42 * | 53 | 40 * | 53 |
| Albumin (g/L) | 20–47 a | 29 | 24 | 30 | 21 | 29 |
| Total globulin (g/L) | 18–21 c | 19 | 18 | 23 * | 19 | 24 |
| Glucose (mmol/L) | 5.2–12.2 a | 18.1 * | 13.8 * | 15 * | IS | IS |
| Sodium (mmol/L) | 149.0–281.4 a | 142 * | IS | 145 * | IS | IS |
| Potassium (mmol/L) | 4.0–14.0 a | 10.8 | IS | 7.5 | IS | IS |
| Chloride (mmol/L) | 110.0–204.4 a | 98 * | IS | 101 * | IS | IS |
| Calcium (mmol/L) | 2.3–3.5 a | 2.64 | 2.15 * | 2.57 | 1.94 * | 2.66 |
| Phosphorous (mmol/L) | 2.0–3.1 a | 3.32 * | 2.23 | 3.95 * | 2.83 | 3.69 |
| Urea (mmol/L) | 14.5–21.4 a | 4.1 * | 5.3 * | 5.5 * | 4.9 * | 5.4 * |
| Total bilirubin (µmol/L) | 3.4–14.3 a | 9 | 1 * | 4 | IS | IS |
| Alanine aminotransferase (ALT; U/L) | 42–73 a | 39 * | 50 | 75 * | 25 * | 42 |
| Aspartate aminotransferase (AST; U/L) | 51–122 a | 97 | 76 | 220 * | 62 | 86 |
| Alkaline phosphatase (ALP; U/L) | 103–217 a | 140 | 155 | 193 | 137 | 178 |
| GammaGT (U/L) | 6.0–8.0 a | 7 | <5 * | <5 * | IS | IS |
| Creatine kinase (U/L) | 105–649 a | 100 * | 43 * | 290 | IS | IS |
| Total cholesterol (mmol/L) | 1.3–3.4 a | 2.02 | 1.77 | 2.29 | 2.02 | 2.27 |
| Pathology | Symptoms | ASA Dose | ||||
|---|---|---|---|---|---|---|
| 175 mg/kg | 550 mg/kg | 1750 mg/kg | 5000 mg/kg | Reference Control | ||
| Macropathology | Splenic melanosis (background lesion) | - | + | + | - | - |
| Kidney | Rare interstitial lymphoplasmacytic infiltrate with fibrosis | - | + | + | + | - |
| Liver | Multiple random foci of necrosis with neutrophil aggregates | +++ | +++ | + | + | +++ |
| Mild anisocytosis with scant megalocytosis | + | + | + | ++ | - | |
| Stomach | Focal neutrophilic infiltrate into gastric mucosa; | - | - | + | ++ | + |
| Low-grade focal granulocytic gastritis | - | - | + | +++ | +++ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timpani, C.A.; Rasmussen, L.; Rybalka, E. Adenylosuccinic Acid Is a Non-Toxic Small Molecule In Vitro and In Vivo. Pharmaceuticals 2023, 16, 1458. https://doi.org/10.3390/ph16101458
Timpani CA, Rasmussen L, Rybalka E. Adenylosuccinic Acid Is a Non-Toxic Small Molecule In Vitro and In Vivo. Pharmaceuticals. 2023; 16(10):1458. https://doi.org/10.3390/ph16101458
Chicago/Turabian StyleTimpani, Cara A., Lorna Rasmussen, and Emma Rybalka. 2023. "Adenylosuccinic Acid Is a Non-Toxic Small Molecule In Vitro and In Vivo" Pharmaceuticals 16, no. 10: 1458. https://doi.org/10.3390/ph16101458
APA StyleTimpani, C. A., Rasmussen, L., & Rybalka, E. (2023). Adenylosuccinic Acid Is a Non-Toxic Small Molecule In Vitro and In Vivo. Pharmaceuticals, 16(10), 1458. https://doi.org/10.3390/ph16101458