

Study of Mono and Di-O-caffeoylquinic Acid Isomers in Acmella oleracea Extracts by HPLC-MS/MS and Application of Linear Equation of Deconvolution Analysis Algorithm for Their Characterization

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
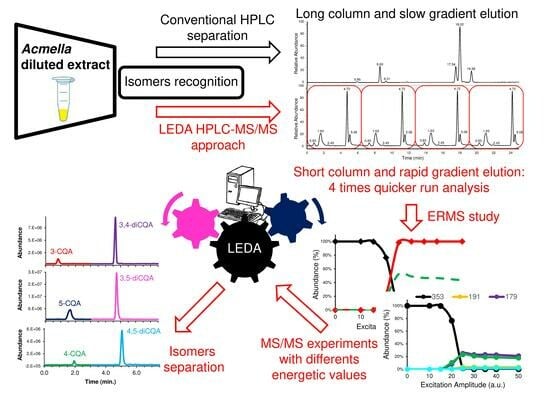
2. Results and Discussion
- Separation by a conventional chromatographic method;
- MS/MS characterization by energetic dimension of CID mechanism;
- Interpretation of obtained MS/MS spectra (CID study);
- Application of the LEDA approach in Acmella WS;
- Assessment of the LEDA quantitative performances.
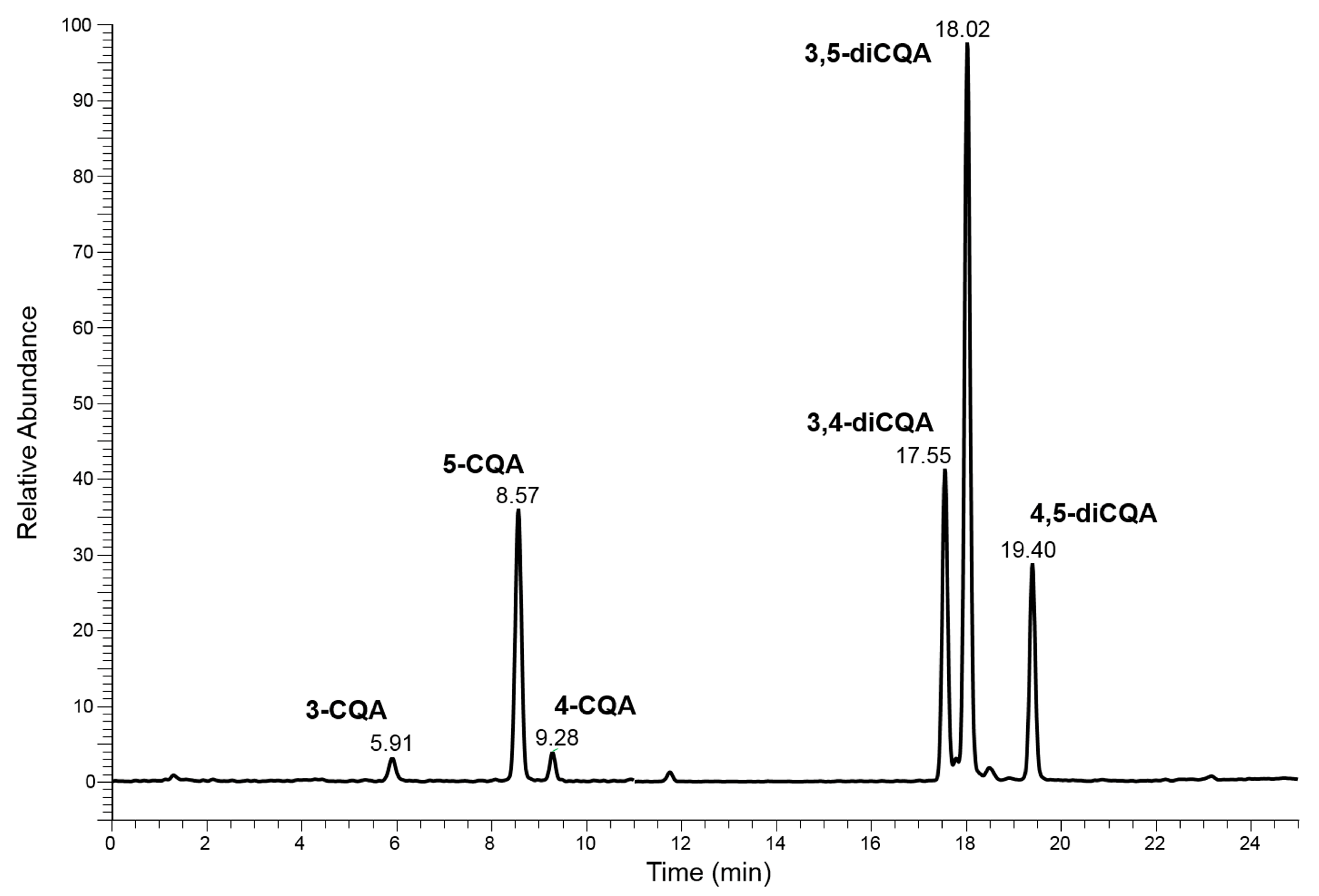
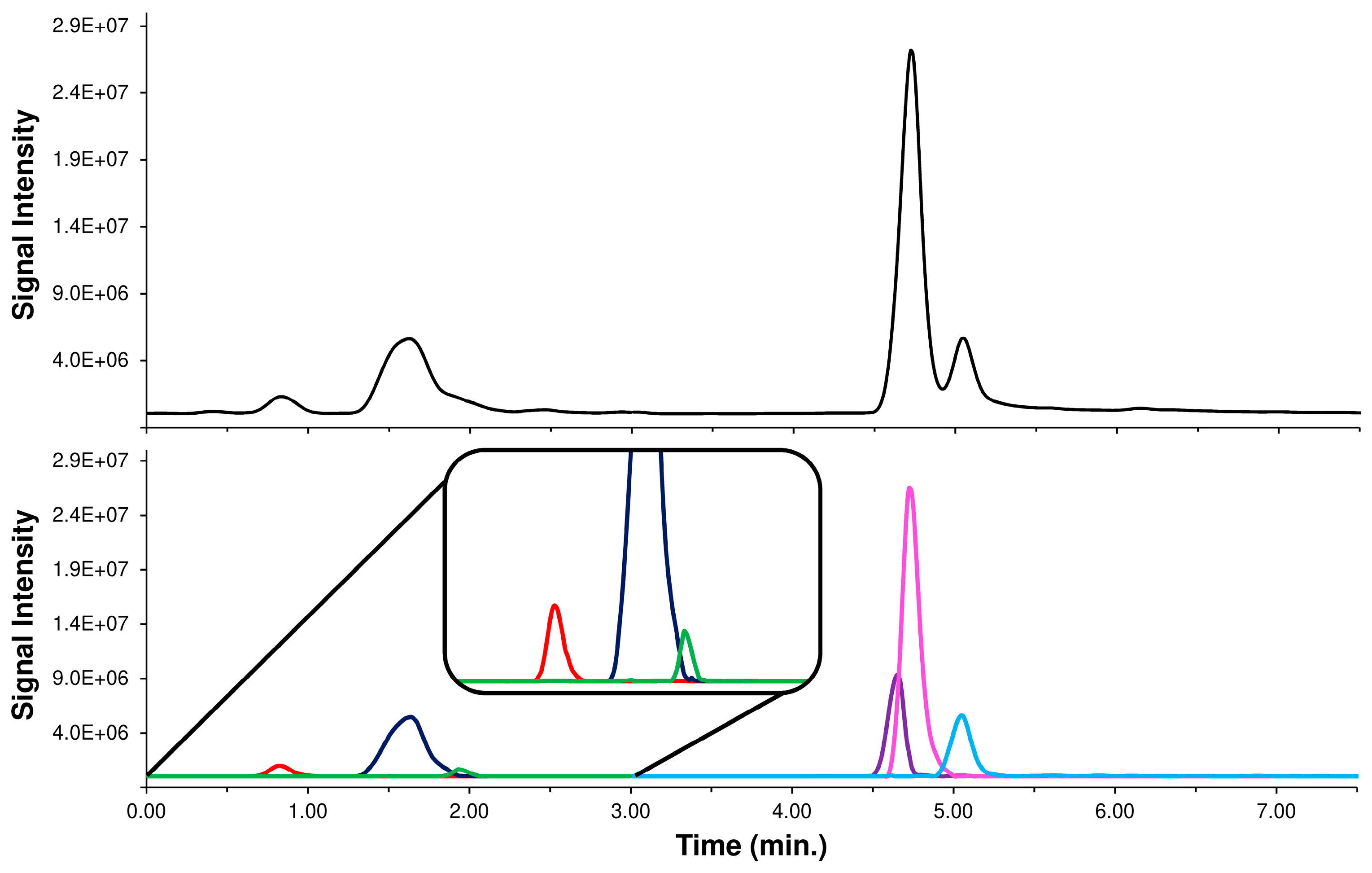
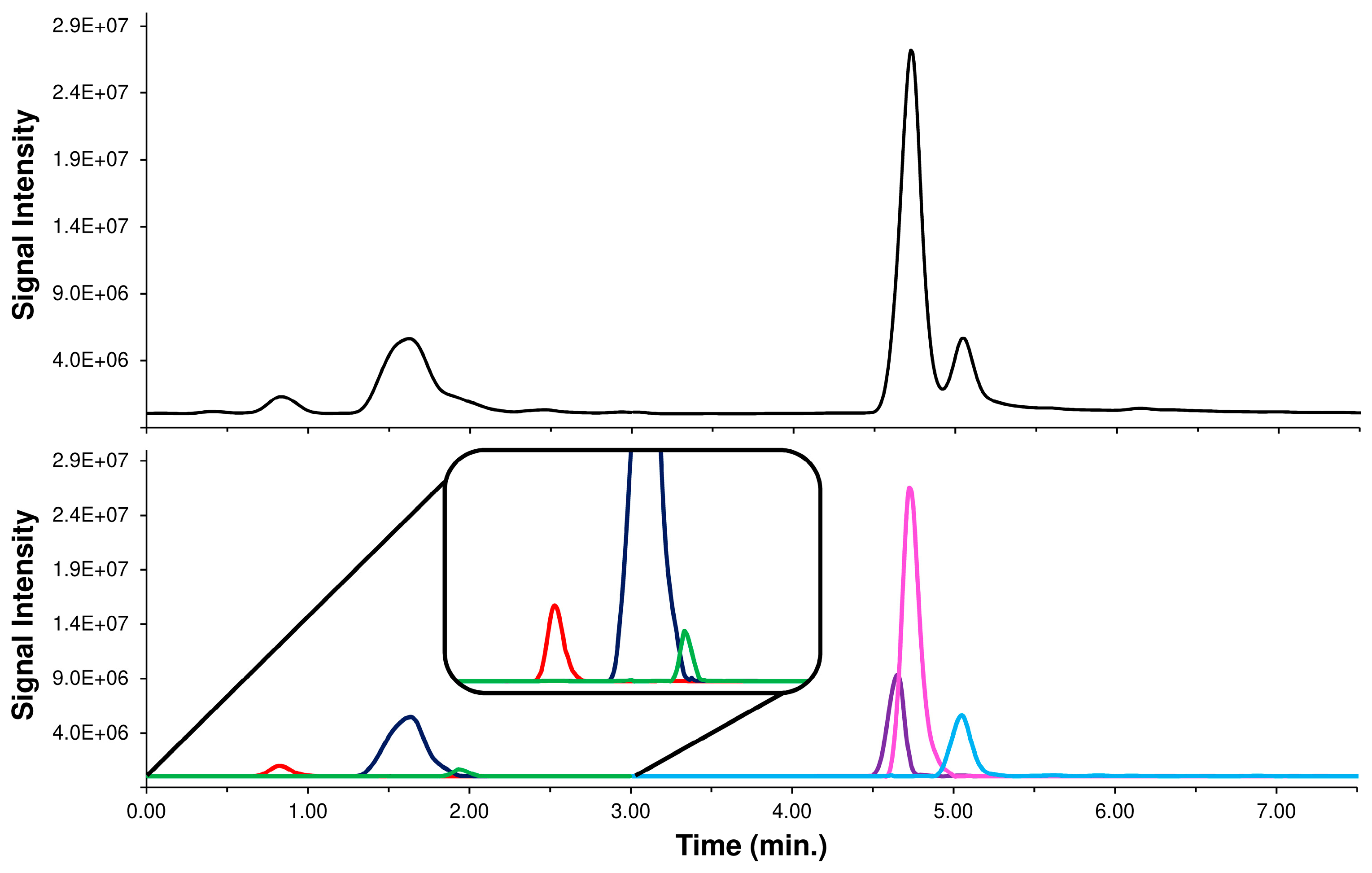
2.1. Conventional Chromatographic Separation of Caffeoylquinic-Isomers in Acmella WS
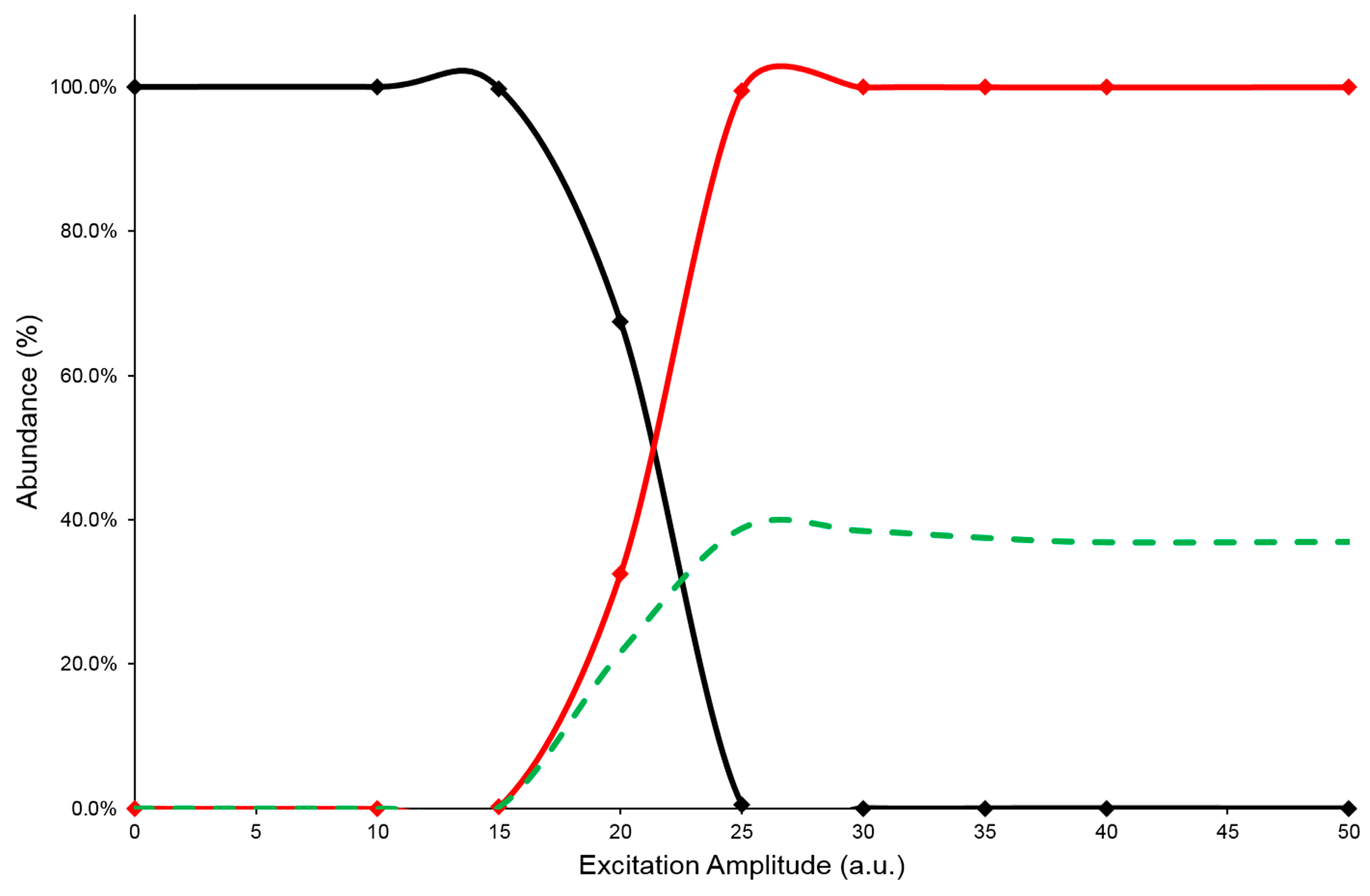
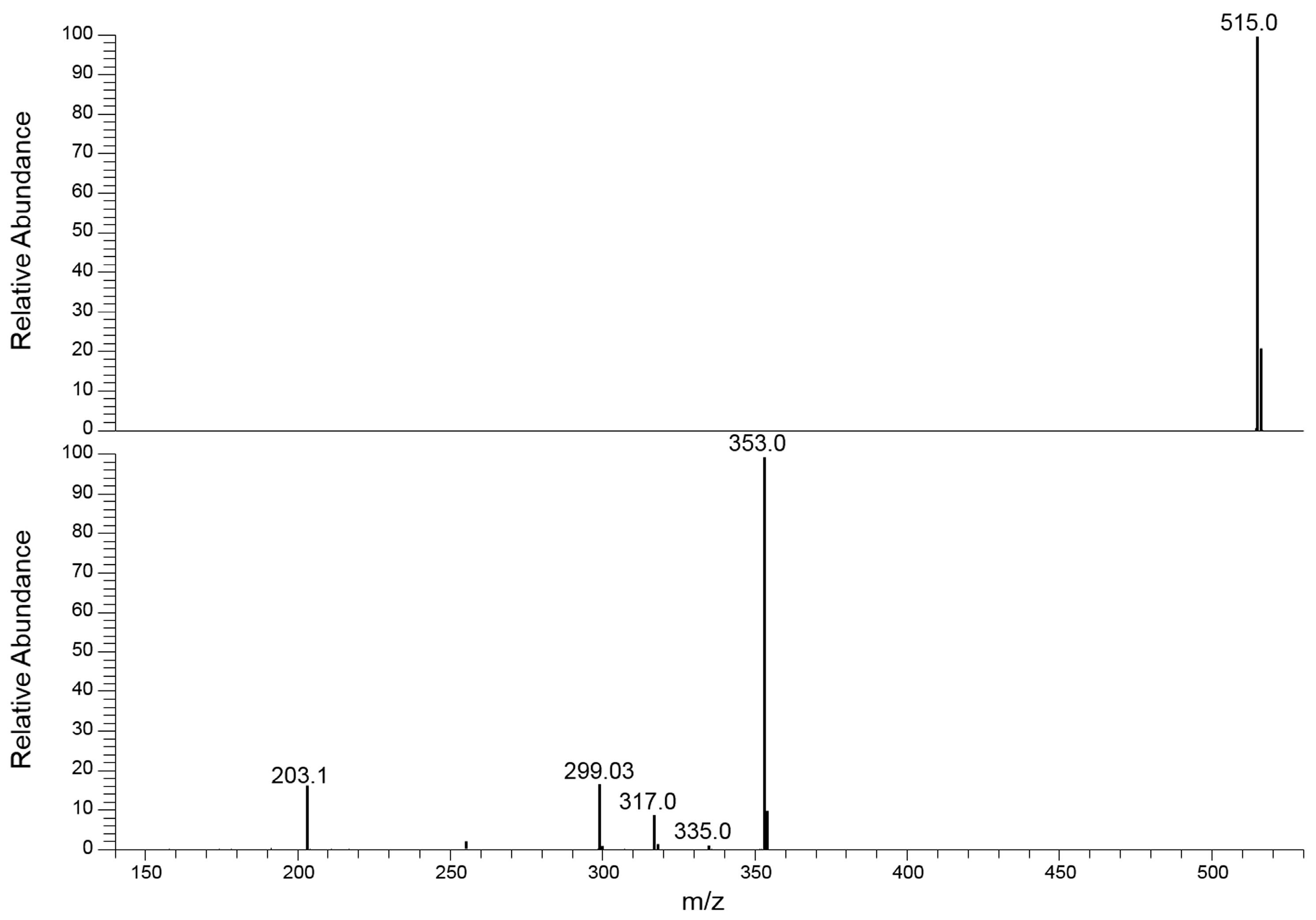
2.2. MS/MS Characterization by Energetic Dimension of CID Mechanism
2.3. Collision-Induced Dissociation Study
2.4. HPLC-MS/MS Supported by LEDA Approach
2.5. Evaluation of Quali-Quantitative Performance of LEDA Approach
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Standard and Calibration Solutions
3.3. Preparation of Phenolic Extracts
3.4. MS and ERMS Experiments
3.5. HPLC-MS/MS Methods
3.6. The LEDA Alghorithm
- (Pi/Ri)m is the abundance ratio between the product ion (Pi) vs. reference ion (Ri) measured (m) in the sample;
- (Pi/Ri)x is the characteristic abundance ratio between the Pi vs. Ri of pure isomer;
- [%]x is the concentration (%) of the isomer in the sample.
3.7. Calibration Curve of HPLC-MS/MS Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Uthpala, T.G.G.; Navaratne, S.B. Acmella oleracea plant; identification, applications and use as an emerging food source—Review. Food Rev. Int. 2020, 37, 399–414. [Google Scholar] [CrossRef]
- Pinheiro, M.S.S.; Moysés, D.A.; Galucio, N.C.R.; Santos, W.O.; Pina, J.R.S.; Oliveira, L.C.; Silva, S.Y.S.; Silva, S.C.; Frazão, N.F.; Marinho, P.S.B.; et al. Cytotoxic and molecular evaluation of spilanthol obtained from Acmella oleracea (L.) R. K. Jansen (jambu) in human gastric cancer cells. Nat. Prod. Res. 2023, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lobina, C.; Sau, R.; Fara, F.; Maccioni, P.; Carai, M.A.M.; Colombo, G. Analgesic properties of a food grade lecithin delivery system of Zingiber officinale and Acmella oleracea standardized extracts in rats. Nat. Prod. Res. 2021, 35, 3078–3082. [Google Scholar] [CrossRef] [PubMed]
- Yien, R.M.K.; Gomes, A.C.C.; Fiorot, R.G.; Miranda, A.L.P.; Neves, G.A.; Andrade, B.S.A.; Costa, F.C.; Tributino, J.L.M.; Simas, N.K. Alkylamides from Acmella oleracea: Antinociceptive effect and molecular docking with cannabinoid and TRPV1 receptors. Nat. Prod. Res. 2023, 37, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Lalthanpuii, P.B.; Lalchhandama, K. Chemical composition and broad-spectrum anthelmintic activity of a cultivar of toothache plant, Acmella oleracea, from Mizoram, India. Pharm. Biol. 2020, 58, 393–399. [Google Scholar] [CrossRef]
- Rondanelli, M.; Riva, A.; Allegrini, P.; Faliva, M.A.; Naso, M.; Peroni, G.; Nichetti, M.; Gasparri, C.; Spadaccini, D.; Iannello, G.; et al. The Use of a New Food-Grade Lecithin Formulation of Highly Standardized Ginger (Zingiber officinale) and Acmella oleracea Extracts for the Treatment of Pain and Inflammation in a Group of Subjects with Moderate Knee Osteoarthritis. J. Pain Res. 2020, 13, 761–770. [Google Scholar] [CrossRef]
- Spinozzi, E.; Ferrati, M.; Baldassarri, C.; Cappellacci, L.; Marmugi, M.; Caselli, A.; Benelli, G.; Maggi, F.; Petrelli, R. A review of the chemistry and biological activities of Acmella oleracea (“jambù”, Asteraceae), with a view to the development of bioinsecticides and acaricides. Plants 2022, 11, 2721. [Google Scholar] [CrossRef]
- Nascimento, L.E.S.; Arriola, N.D.A.; da Silva, L.A.L.; Faqueti, L.G.; Sandjo, L.P.; de Araújo, C.E.S.; Biavatti, M.W.; Barcelos-Oliveira, J.L.; Dias de Mello Castanho Amboni, R. Phytochemical profile of different anatomical parts of jambu (Acmella oleracea (L.) R.K. Jansen): A comparison between hydroponic and conventional cultivation using PCA and cluster analysis. Food Chem. 2020, 332, 127393. [Google Scholar] [CrossRef]
- Bellumori, M.; Zonfrillo, B.; Maggini, V.; Bogani, P.; Gallo, E.; Firenzuoli, F.; Mulinacci, N.; Innocenti, M. Acmella oleracea (L.) R.K. Jansen: Alkylamides and phenolic compounds in aerial parts and roots of in vitro seedlings. J. Pharm. Biomed. Anal. 2022, 220, 11499. [Google Scholar] [CrossRef]
- Li, A.N.; Li, S.; Zhang, Y.J.; Xu, X.-R.; Chen, Y.M.; Li, H.B. Resources and Biological Activities of Natural Polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef]
- Huang, W.Y.; Yi-Zhong Cai, Y.Z.; Zhang, Y. Natural Phenolic Compounds From Medicinal Herbs and Dietary Plants: Potential Use for Cancer Prevention. Nutr. Cancer 2009, 62, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Alcázar Magaña, A.; Kamimura, N.; Soumyanath, A.; Stevens, J.F.; Maier, C.S. Caffeoylquinic acids: Chemistry, biosynthesis, occurrence, analytical challenges, and bioactivity. Plant J. 2021, 107, 1299–1319. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tian, Z.; Cui, Y.; Liu, Z.; Ma, X. Chlorogenic acid: A comprehensive review of the dietary sources, processing effects, bioavailability, beneficial properties, mechanisms of action, and future directions. Compr. Rev. Food Sci. Food Saf. 2020, 19, 3130–3158. [Google Scholar] [CrossRef] [PubMed]
- Wianowska, D.; Gil, M. Recent advances in extraction and analysis procedures of natural chlorogenic acids. Phytochem. Rev. 2019, 18, 73–302. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, P.; Cheng, G.; Zhang, Y. A Brief Review of Phenolic Compounds Identified from Plants: Their Extraction, Analysis, and Biological Activity. Nat. Prod. Commun. 2022, 17. [Google Scholar] [CrossRef]
- Khoddami, A.; Wilkes, M.A.; Roberts, T.H. Techniques for Analysis of Plant Phenolic Compounds. Molecules 2013, 18, 2328. [Google Scholar] [CrossRef]
- Wianowska, D.; Typek, R.; Dawidowicz, A.L. Chlorogenic Acid Stability in Pressurized Liquid Extraction Conditions. J. AOAC Int. 2015, 98, 415–421. [Google Scholar] [CrossRef]
- Crotti, S.; Menicatti, M.; Pallecchi, M.; Bartolucci, G. Tandem mass spectrometry approaches for recognition of isomeric compounds mixtures. Mass Spectrom. Rev. 2021, 42, e21757. [Google Scholar] [CrossRef]
- Kurimoto, A.; Daikoku, S.; Mutsuga, S.; Kanie, O. Analysis of energy-resolved mass spectra at MSn in a Pursuit to characterize structural isomers of oligosaccharides. Anal. Chem. 2006, 78, 3461–3466. [Google Scholar] [CrossRef]
- Kertesz, T.M.; Hall, L.H.; Hill, D.W.; Grant, D.F. CE50: Quantifying collision induced dissociation energy for small molecule characterization and identification. J. Am. Soc. Mass Spec. 2009, 20, 1759–1767. [Google Scholar] [CrossRef]
- Memboeuf, A.; Jullien, L.; Lartia, R.; Brasme, B.; Gimbert, Y. Tandem mass spectrometric analysis of a mixture of isobars using the survival yield technique. J. Am. Soc. Mass Spec. 2011, 22, 1744–1752. [Google Scholar] [CrossRef]
- Dit Fouque, J.D.; Maroto, A.; Memboeuf, A. Purification and Quantification of an Isomeric Compound in a Mixture by Collisional Excitation in Multistage Mass Spectrometry Experiments. Anal. Chem. 2016, 88, 10821–10825. [Google Scholar] [CrossRef] [PubMed]
- Dit Fouque, J.D.; Lartia, R.; Maroto, A.; Memboeuf, A. Quantification of intramolecular click chemistry modified synthetic peptide isomers in mixtures using tandem mass spectrometry and the survival yield technique. Anal. Bioanal. Chem. 2018, 410, 5765–5777. [Google Scholar] [CrossRef] [PubMed]
- Maroto, A.; Dit Fouque, D.J.; Memboeuf, A. Ion trap MS using high trapping gas pressure enables unequivocal structural analysis of three isobaric compounds in a mixture by using energy-resolved mass spectrometry and the survival yield technique. J Mass Spectrom. 2020, 55, e4478. [Google Scholar] [CrossRef]
- Dit Fouque, J.D.; Maroto, A.; Memboeuf, A. Structural analysis of a compound despite the presence of an isobaric interference by using in-source Collision Induced Dissociation and tandem mass spectrometry. J. Mass Spectrom. 2021, 56, e4698. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.T.; Chen, D.; Glish, G.L. Identifying the D-Pentoses Using Water Adduction to Lithium Cationized Molecule. J. Am. Soc. Mass Spectrom. 2017, 28, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.T.; Chen, D.; Wallbillich, N.J.; Glish, G.L. Distinguishing Biologically Relevant Hexoses by Water Adduction to the Lithium-Cationized Molecule. Anal. Chem. 2017, 89, 10504–10510. [Google Scholar] [CrossRef]
- Campbell, M.T.; Chen, D.; Glish, G.L. Distinguishing Linkage Position and Anomeric Configuration of Glucose-Glucose Disaccharides by Water Adduction to Lithiated Molecules. Anal. Chem. 2018, 90, 2048–2054. [Google Scholar] [CrossRef]
- Menicatti, M.; Guandalini, L.; Dei, S.; Floriddia, E.; Teodori, E.; Traldi, P.; Bartolucci, G. The power of energy resolved tandem mass spectrometry experiments for resolution of isomers: The case of drug plasma stability investigation of multidrug resistance inhibitors. Rapid Commun. Mass Spectrom. 2016, 30, 423–432. [Google Scholar] [CrossRef]
- Menicatti, M.; Pallecchi, M.; Bua, S.; Vullo, D.; Di Cesare Mannelli, L.; Ghelardini, C.; Carta, F.; Supuran, C.T.; Bartolucci, G. Resolution of co-eluting isomers of anti-inflammatory drugs conjugated to carbonic anhydrase inhibitors from plasma in liquid chromatography by energy-resolved tandem mass spectrometry. J. Enzyme Inhib. Med. Chem. 2018, 33, 671–679. [Google Scholar] [CrossRef]
- Menicatti, M.; Pallecchi, M.; Ricciutelli, M.; Galarini, R.; Moretti, S.; Sagratini, G.; Vittori, S.; Lucarini, S.; Caprioli, G.; Bartolucci, G. Determination of coeluted isomers in wine samples by application of MS/MS deconvolution analysis. J. Mass Spectrom. 2020, 55, e4607. [Google Scholar] [CrossRef] [PubMed]
- Pallecchi, M.; Braconi, L.; Menicatti, M.; Giachetti, S.; Dei, S.; Teodori, E.; Bartolucci, G. Simultaneous Degradation Study of Isomers in Human Plasma by HPLC-MS/MS and Application of LEDA Algorithm for Their Characterization. Int. J. Mol. Sci. 2022, 23, 13139. [Google Scholar] [CrossRef] [PubMed]
- March, R.E.; Todd, J.F. Quadrupole Ion Trap Mass Spectrometry, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 2–416. [Google Scholar]
- Cooks, R.G.; Glish, G.L.; McLuckey, S.A.; Kaiser, R.E. Ion Trap Mass Spectrometry. Chem. Eng. News 1991, 69, 26–41. [Google Scholar] [CrossRef]
- Johnson, J.V.; Yost, R.A.; Kelley, P.E.; Bradford, D.C. Tandem-in-Space and Tandem-in-Time Mass Spectrometry: Triple Quadrupoles and Quadrupole Ion Traps. Anal. Chem. 1990, 62, 2162–2172. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Yates, J.R. The Quadrupole Ion Trap Mass Spectrometer—A Small Solution to a Big Challenge. Anal. Biochem. 1997, 244, 1–15. [Google Scholar] [CrossRef]
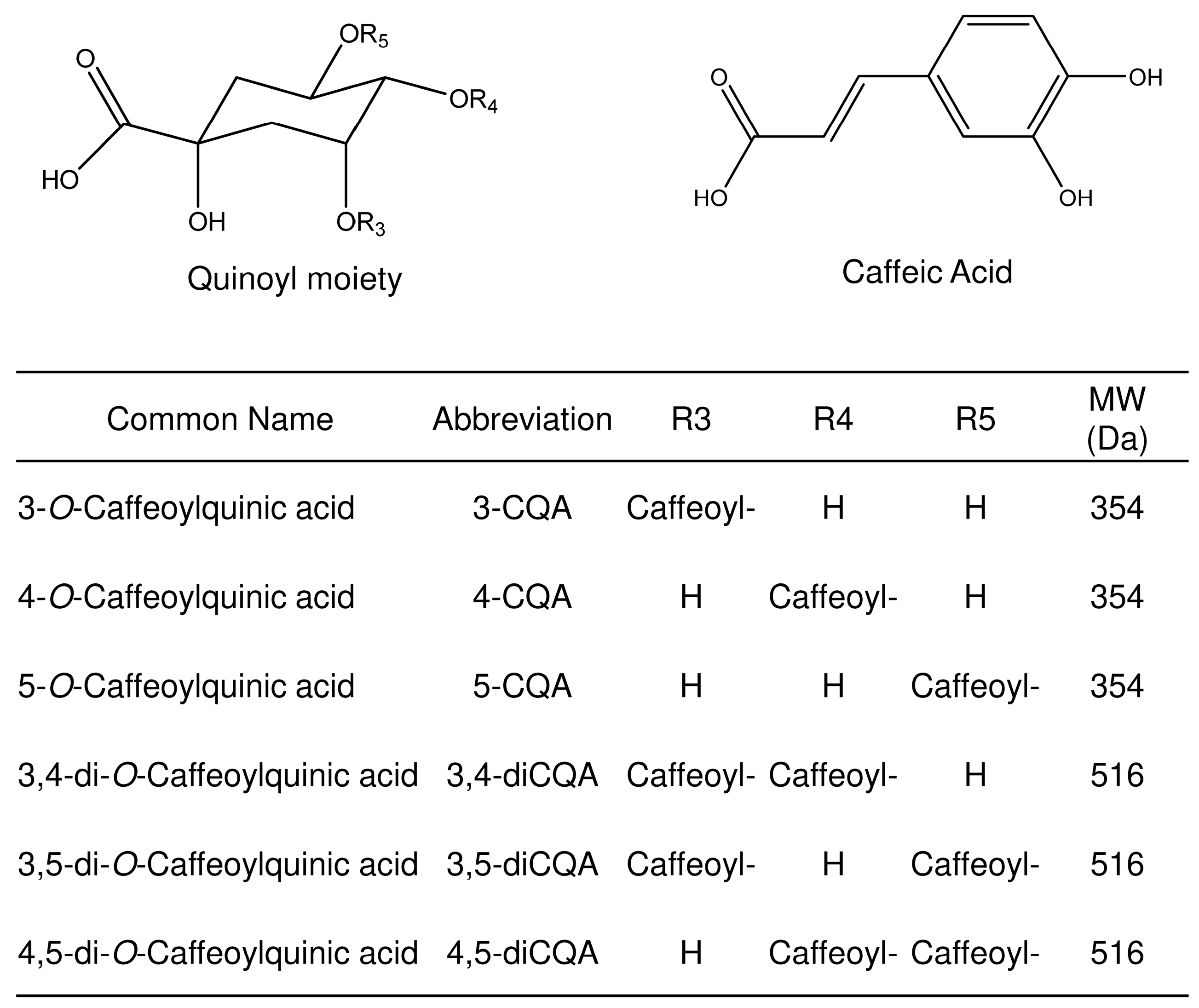
- Clifford, M.N.; Johnston, K.L.; Knight, S.; Kuhnert, N. Hierarchical Scheme for LC-MSn Identification of Chlorogenic Acids. J. Agric. Food Chem. 2003, 51, 2900–2911. [Google Scholar] [CrossRef]
- Clifford, M.N.; Knight, S.; Kuhnert, N. Discriminating between the six isomers of dicaffeoylquinic acid by LC-MSn. J Agric Food Chem. 2005, 53, 3821–3832. [Google Scholar] [CrossRef]
- Pallecchi, M.; Lucio, L.; Braconi, L.; Menicatti, M.; Dei, S.; Teodori, E.; Bartolucci, G. Isomers Recognition in HPLC-MS/MS Analysis of Human Plasma Samples by Using an Ion Trap Supported by a Linear Equations-Based Algorithm. Int. J. Mol. Sci. 2023, 24, 11155. [Google Scholar] [CrossRef]
- ICH Q2B, Validation of Analytical Procedure: Methodology, International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. 1996. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 27 March 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SYExA50 (a.u.) | ExAmax (a.u.) | PiYmax (%) |
|---|---|---|---|
| 3-CQA | 21.0 | 25 | 56 |
| 5-CQA | 20.5 | 25 | 53 |
| 4-CQA | 19.9 | 25 | 39 |
| 3,4-diCQA | 24.4 | 30 | 41 |
| 3,5-diCQA | 18.1 | 25 | 39 |
| 4,5-diCQA | 22.1 | 25 | 43 |
| Isomers | ChromSys 1 Acmella WS ± SD (mg L−1) | ChromSys 1 Acmella ES ± SD (mg L−1) | ChromSys 2 Acmella WS ± SD (mg L−1) | ChromSys 2 Acmella ES ± SD (mg L−1) |
|---|---|---|---|---|
| 3-CQA | 0.6 ± 0.1 | 12 ± 2 | 0.8 ± 0.1 | 15 ± 2 |
| 5-CQA | 6.0 ± 0.5 | 120 ± 10 | 5.5 ± 0.2 | 110 ± 4 |
| 4-CQA | 0.4 ± 0.1 | 8 ± 2 | 0.4 ± 0.1 | 8 ± 2 |
| 3,4-diCQA | 6.5 ± 0.5 | 130 ± 10 | 7.0 ± 0.5 | 140 ± 10 |
| 3,5-diCQA | 16.5 ± 1.0 | 330 ± 20 | 18.0 ± 0.5 | 360 ± 10 |
| 4,5-diCQA | 4.0 ± 0.5 | 80 ± 10 | 4.5 ± 0.5 | 90 ± 10 |
| Time Segment | Time Segment (min) | Precursor Ion (m/z) | MS/MS Event | Pis Scan Range (m/z) | ExA (a.u.) | |
|---|---|---|---|---|---|---|
| ChromSys 1 | ||||||
| CQAs | 1 | 0.0–11.0 | 353 | Ri | 300–365 | 15 |
| Pis | 95–250 | 25 | ||||
| diCQAs | 2 | 11.0–25.0 | 515 | Ri | 450–530 | 10 |
| Pis | 140–365 | 30 | ||||
| ChromSys 2 | ||||||
| CQAs | 1 | 0.0–3.0 | 353 | Ri | 300–365 | 15 |
| Pis | 95–250 | 25 | ||||
| diCQAs | 2 | 3.0–7.5 | 515 | Ri | 450–530 | 10 |
| Pis | 140–365 | 30 |
| Isomers Group | Ratio Pi/Ri (m/z) | 3-CQA Ratio Value ± SD | 5-CQA Ratio Value ± SD | 4-CQA Ratio Value ± SD |
|---|---|---|---|---|
| CQAs | 191/353 | 0.23 ± 0.01 | 0.30 ± 0.01 | 0.02 ± 0.01 |
| 179/353 | 0.20 ± 0.01 | 0.02 ± 0.01 | 0.20 ± 0.01 | |
| 173/353 | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.17 ± 0.01 | |
| 135/353 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.01 ±0.01 | |
| Ratio Pi/Ri (m/z) | 3,4-diCQA Ratio Value ± SD | 3,5-diCQA Ratio Value ± SD | 4,5-diCQA Ratio Value ± SD | |
| diCQAs | 353/515 | 0.28 ± 0.01 | 0.37 ± 0.01 | 0.26 ± 0.01 |
| 335/515 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.01 ± 0.01 | |
| 317/515 | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 | |
| 299/515 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.04 ± 0.01 | |
| 203/515 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.04 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellumori, M.; Pallecchi, M.; Zonfrillo, B.; Lucio, L.; Menicatti, M.; Innocenti, M.; Mulinacci, N.; Bartolucci, G. Study of Mono and Di-O-caffeoylquinic Acid Isomers in Acmella oleracea Extracts by HPLC-MS/MS and Application of Linear Equation of Deconvolution Analysis Algorithm for Their Characterization. Pharmaceuticals 2023, 16, 1375. https://doi.org/10.3390/ph16101375
Bellumori M, Pallecchi M, Zonfrillo B, Lucio L, Menicatti M, Innocenti M, Mulinacci N, Bartolucci G. Study of Mono and Di-O-caffeoylquinic Acid Isomers in Acmella oleracea Extracts by HPLC-MS/MS and Application of Linear Equation of Deconvolution Analysis Algorithm for Their Characterization. Pharmaceuticals. 2023; 16(10):1375. https://doi.org/10.3390/ph16101375
Chicago/Turabian StyleBellumori, Maria, Marco Pallecchi, Beatrice Zonfrillo, Luigi Lucio, Marta Menicatti, Marzia Innocenti, Nadia Mulinacci, and Gianluca Bartolucci. 2023. "Study of Mono and Di-O-caffeoylquinic Acid Isomers in Acmella oleracea Extracts by HPLC-MS/MS and Application of Linear Equation of Deconvolution Analysis Algorithm for Their Characterization" Pharmaceuticals 16, no. 10: 1375. https://doi.org/10.3390/ph16101375
APA StyleBellumori, M., Pallecchi, M., Zonfrillo, B., Lucio, L., Menicatti, M., Innocenti, M., Mulinacci, N., & Bartolucci, G. (2023). Study of Mono and Di-O-caffeoylquinic Acid Isomers in Acmella oleracea Extracts by HPLC-MS/MS and Application of Linear Equation of Deconvolution Analysis Algorithm for Their Characterization. Pharmaceuticals, 16(10), 1375. https://doi.org/10.3390/ph16101375