
Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies
 , , ,
, , ,  ,
,  and
and 
Abstract
:1. Introduction
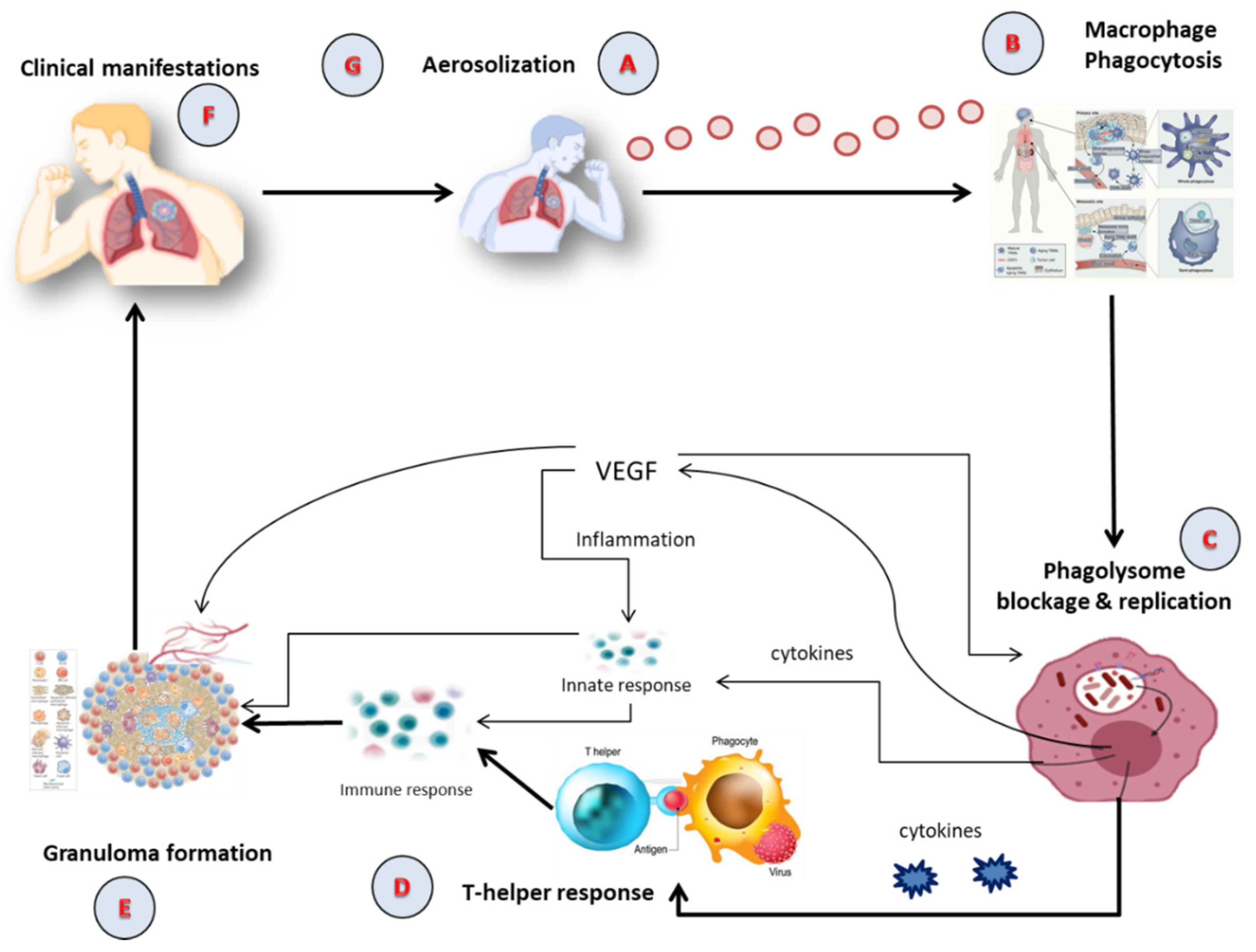
2. TB Pathophysiology
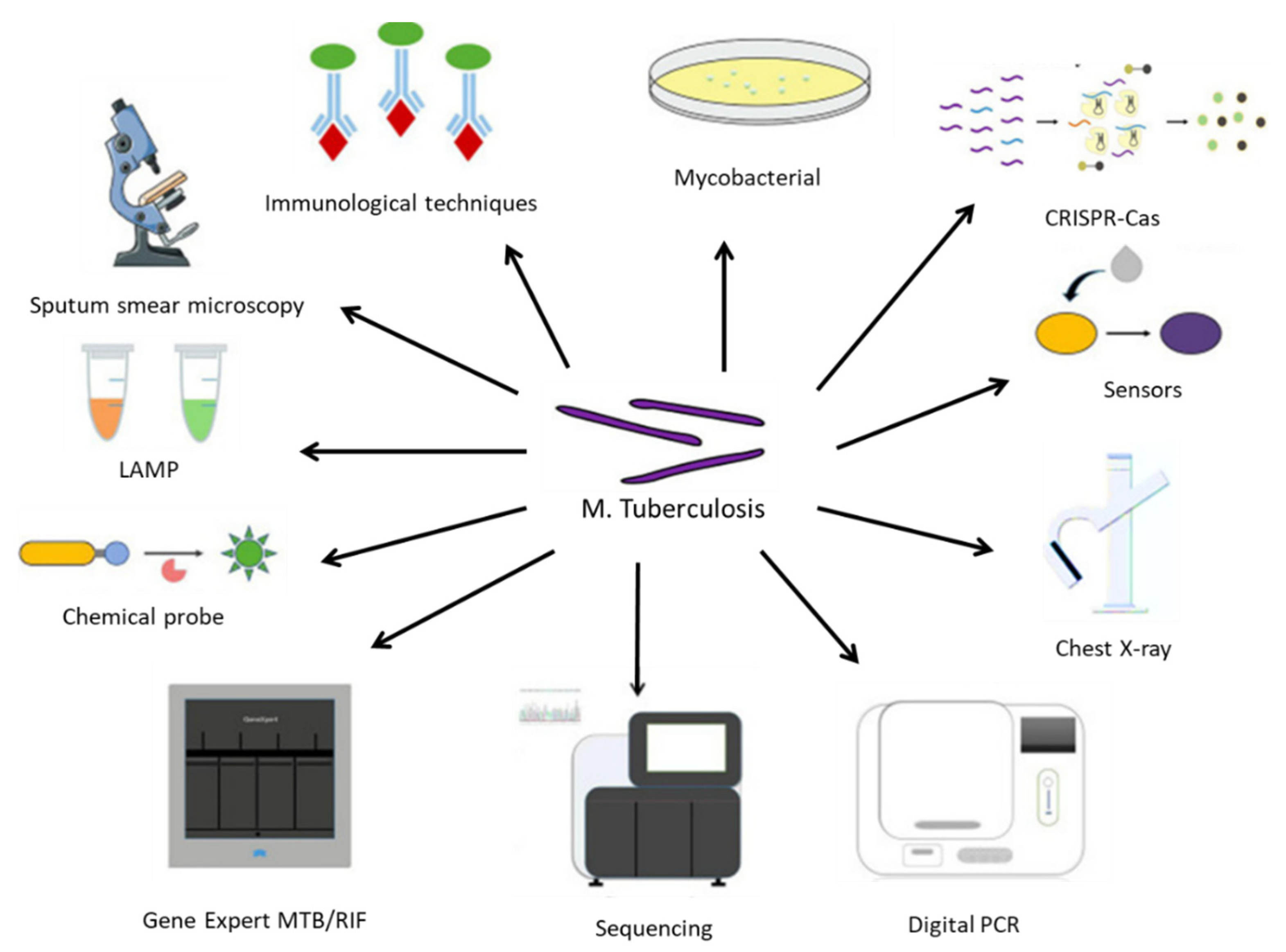
3. TB Diagnostic
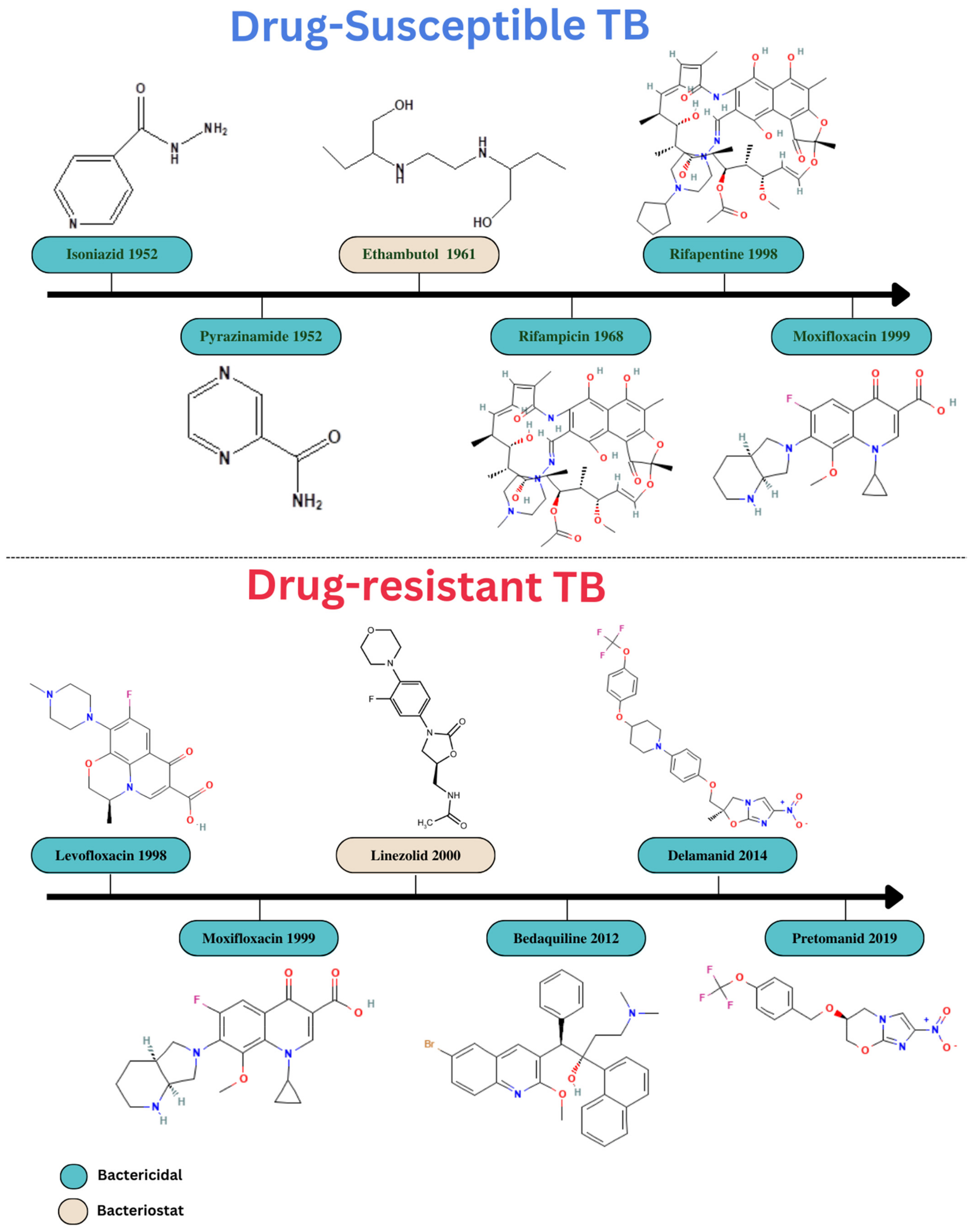
4. Conventional Treatment Options and Its Limitations
5. Nano-Based Drug Delivery in TB
- Targeted Drug Delivery: Nanocarriers can deliver drugs directly to the infected site, ensuring that the therapeutic agents reach the desired location in the body, thus increasing the efficacy and reducing potential side effects [48].
- Reduced Drug Dosage: Due to their efficient delivery and release mechanisms, nanosized drug delivery systems can often achieve therapeutic effects with reduced drug doses, minimizing potential side effects and toxicity [16].
- Controlled Release: The drugs encapsulated in nanocarriers can be released in a sustained manner over time, ensuring consistent drug levels and potentially reducing the frequency of dosing [47].
5.1. Polymeric Nanoparticles (PNPs)
5.2. Solid Lipid Nanoparticles (SLNs)
5.3. Nanostructured Lipid Carriers (NLCs)
5.4. Liposomes
5.5. Nanoemulsions (NEs)
5.6. Polymeric Micelles (PMs)
5.7. Dendrimers
5.8. Carbon Nanotubes (CNTs)
5.9. Metallic Nanoparticles (MNPs)
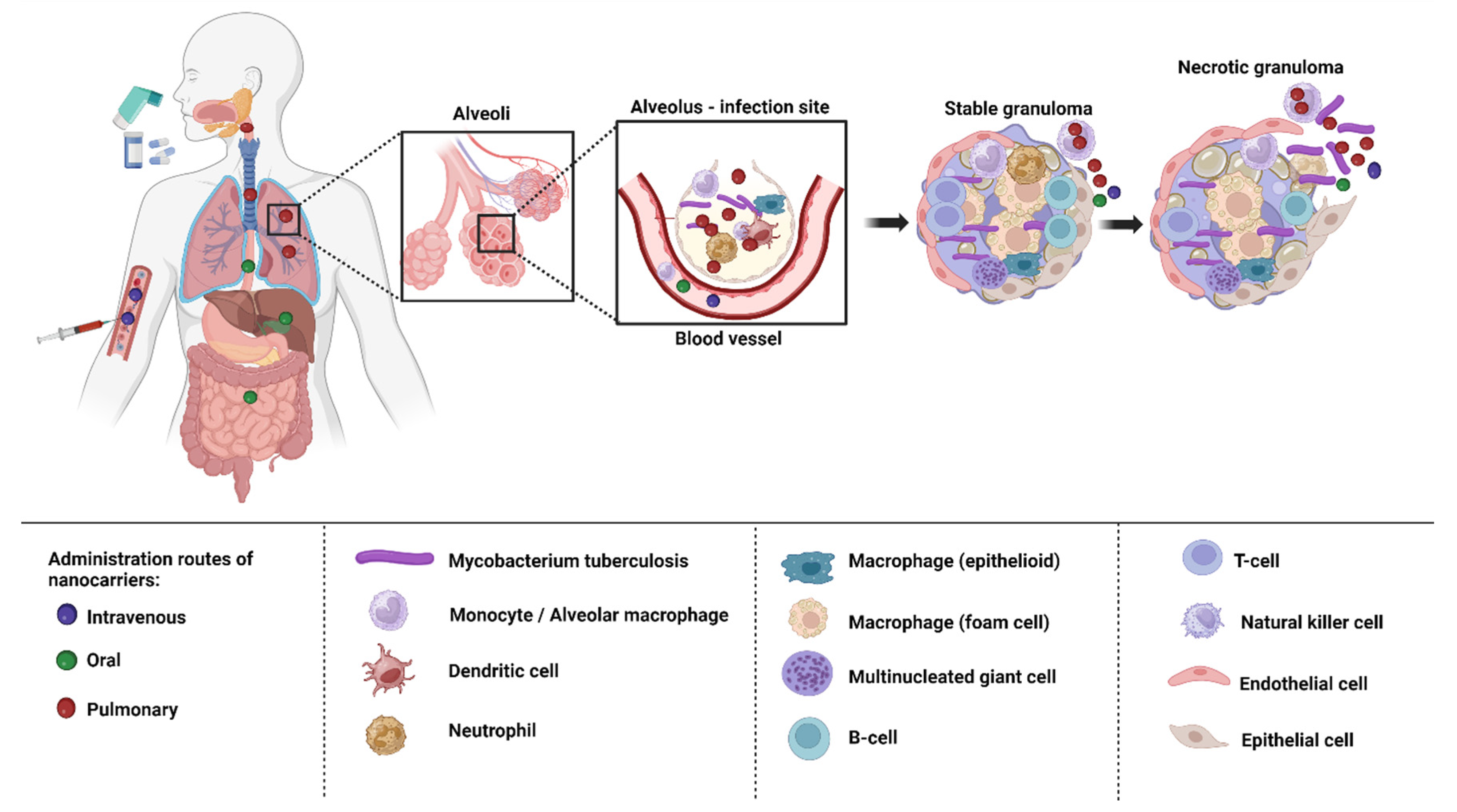
6. Delivery of Nanoformulations for TB Treatment
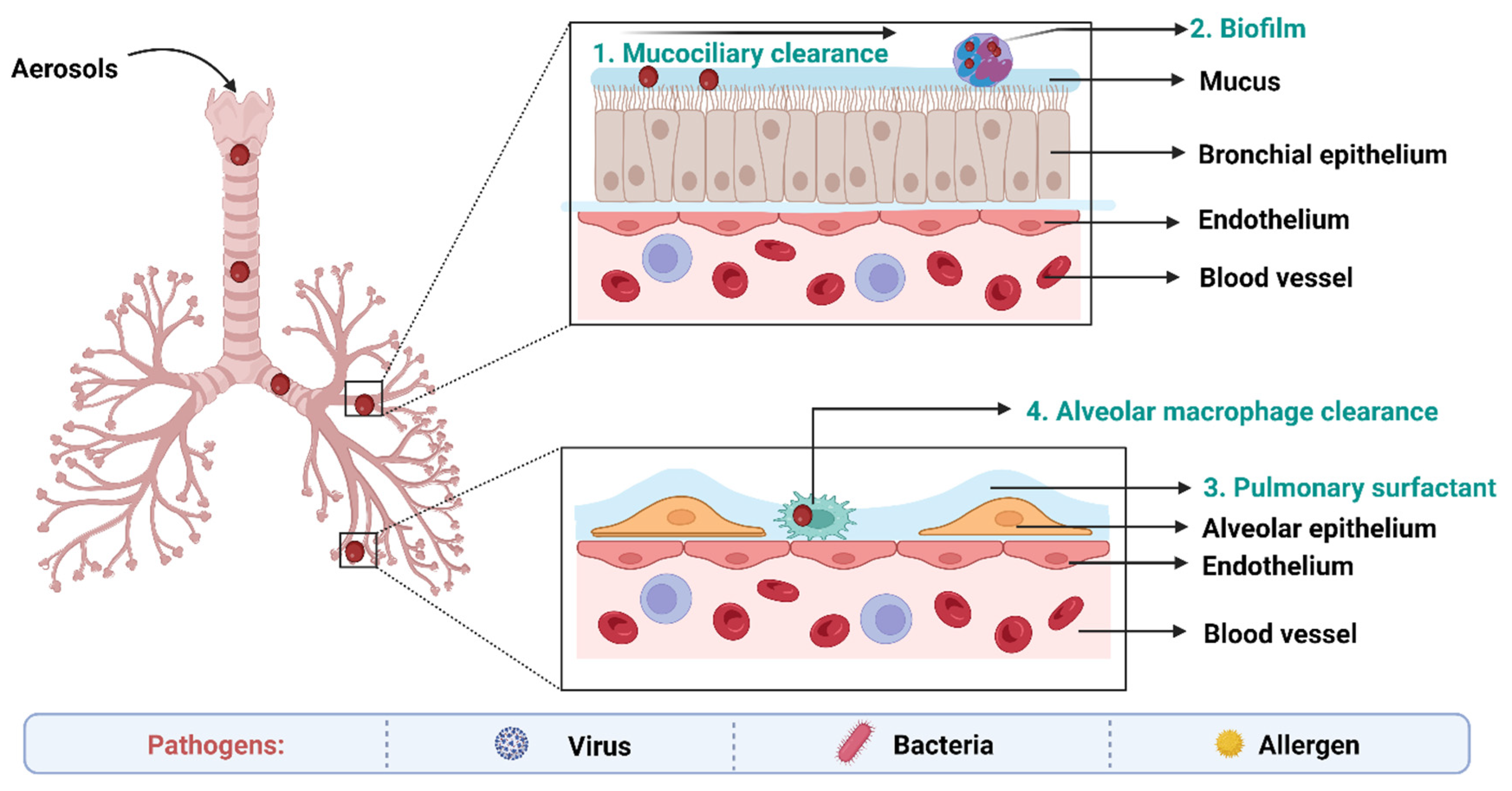
7. Biological Barriers to Delivery of Anti-TB Drugs
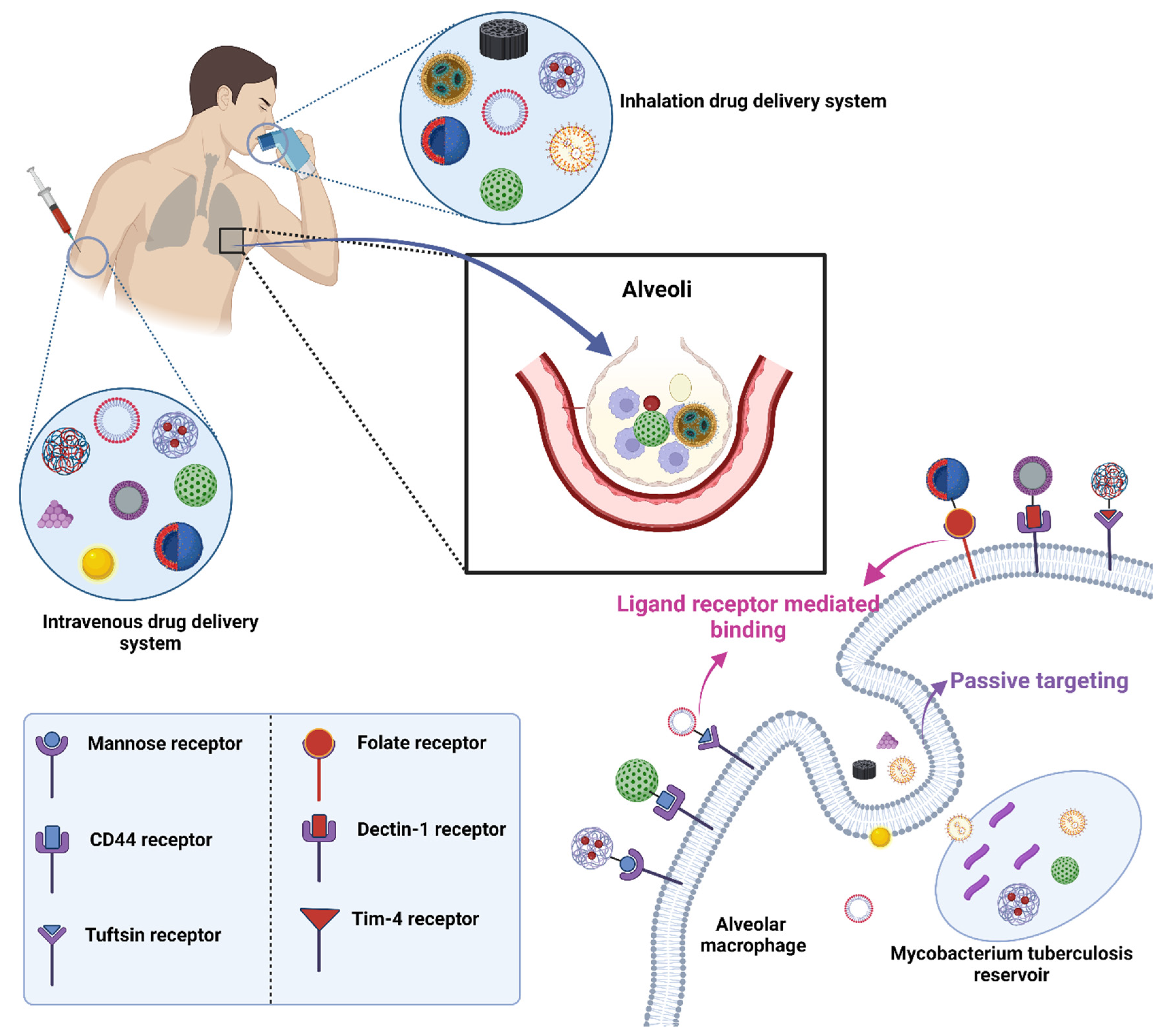
8. Ligand Conjugated Nanoformulations for Circumvention of Pulmonary Barriers
8.1. Mannose Targeting
8.2. Folic Acid Targeting
8.3. Hyaluronic Acid (HA) Targeting
8.4. Tuftsin Receptor Targeting
8.5. Mycolic Acid Targeting
9. Patents
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singh, H.; Jindal, S.; Singh, M.; Sharma, G.; Kaur, I.P. Nano-Formulation of Rifampicin with Enhanced Bioavailability: Development, Characterization and in-Vivo Safety. Int. J. Pharm. 2015, 485, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.-C.; Xu, L.-R.; Zhao, C.-J.; Zhang, H.-Y.; Li, Q.-Y.; Liu, M.-J.; Zhang, Y.-X.; Tang, Z.; Ma, X.-X. Prevalence and Risk Factors of Tuberculosis among People Living with HIV/AIDS in China: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2023, 23, 584. [Google Scholar] [CrossRef] [PubMed]
- Adigun, R.; Singh, R. Tuberculosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chan, J.G.Y.; Tyne, A.S.; Pang, A.; McLachlan, A.J.; Perera, V.; Chan, J.C.Y.; Britton, W.J.; Chan, H.K.; Duke, C.C.; Young, P.M.; et al. Murine Pharmacokinetics of Rifapentine Delivered as an Inhalable Dry Powder. Int. J. Antimicrob. Agents 2015, 45, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Nasiruddin, M.; Neyaz, M.K.; Das, S. Nanotechnology-Based Approach in Tuberculosis Treatment. Tuberc. Res. Treat. 2017, 2017, 4920209. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.R.; Muñoz-Torrico, M.; Duarte, R.; Galvão, T.; Bonini, E.H.; Arbex, F.F.; Arbex, M.A.; Augusto, V.M.; Rabahi, M.F.; de Queiroz Mello, F.C. Risk Factors for Tuberculosis: Diabetes, Smoking, Alcohol Use, and the Use of Other Drugs. J. Bras. Pneumol. 2018, 44, 145–152. [Google Scholar] [CrossRef]
- Global Tuberculosis Report 2022. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 14 September 2023).
- Barrenechea-Pulache, A.; Portocarrero-Bonifaz, A.; Rojas-Roque, C.; Gamboa-Unsihuay, J.E.; Hernández-Vásquez, A. Forgetting Other Communicable Diseases during the COVID-19 Pandemic: Tuberculosis Mortality in Peru. Lancet Reg. Health–Am. 2022, 9, 100226. [Google Scholar] [CrossRef] [PubMed]
- Merk, H.; Ködmön, C.; Werf, M.J. van der Will We Reach the Sustainable Development Goals Target for Tuberculosis in the European Union/European Economic Area by 2030? Eurosurveillance 2019, 24, 1900153. [Google Scholar] [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-Tuberculosis Treatment Strategies and Drug Development: Challenges and Priorities. Nat. Rev. Microbiol. 2022, 20, 685–701. [Google Scholar] [CrossRef]
- Buya, A.B.; Witika, B.A.; Bapolisi, A.M.; Mwila, C.; Mukubwa, G.K.; Memvanga, P.B.; Makoni, P.A.; Nkanga, C.I. Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review. Pharmaceutics 2021, 13, 2041. [Google Scholar] [CrossRef]
- Kumar, G.; Virmani, T.; Pathak, K.; Alhalmi, A. A Revolutionary Blueprint for Mitigation of Hypertension via Nanoemulsion. BioMed Res. Int. 2022, 2022, e4109874. [Google Scholar] [CrossRef]
- Verma, N.; Arora, V.; Awasthi, R.; Chan, Y.; Jha, N.K.; Thapa, K.; Jawaid, T.; Kamal, M.; Gupta, G.; Liu, G.; et al. Recent Developments, Challenges and Future Prospects in Advanced Drug Delivery Systems in the Management of Tuberculosis. J. Drug Deliv. Sci. Technol. 2022, 75, 103690. [Google Scholar] [CrossRef]
- Alshawwa, S.Z.; Kassem, A.A.; Farid, R.M.; Mostafa, S.K.; Labib, G.S. Nanocarrier Drug Delivery Systems: Characterization, Limitations, Future Perspectives and Implementation of Artificial Intelligence. Pharmaceutics 2022, 14, 883. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Sharma, A.; Sharma, A.; Kumar, G.; Virmani, T.; Mukherjee, S. 17—Nanotechnology in Pulmonary Tissue Engineering. In Nanostructured Materials for Tissue Engineering; Mondal, A., Nayak, A.K., Chakraborty, P., Eds.; Nanotechnology in Biomedicine; Elsevier: Amsterdam, The Netherlands, 2023; pp. 537–556. ISBN 978-0-323-95134-0. [Google Scholar]
- Virmani, T.; Kumar, G.; Virmani, R.; Sharma, A.; Pathak, K. Nanocarrier-Based Approaches to Combat Chronic Obstructive Pulmonary Disease. Nanomedicine 2022, 17, 1833–1854. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Mandlik, S.; Pokharkar, V. Nanocarrier-Based Approaches for the Efficient Delivery of Anti-Tubercular Drugs and Vaccines for Management of Tuberculosis. Front. Pharmacol. 2021, 12, 749945. [Google Scholar] [CrossRef] [PubMed]
- Bouzeyen, R.; Javid, B. Therapeutic Vaccines for Tuberculosis: An Overview. Front. Immunol. 2022, 13, 878471. [Google Scholar] [CrossRef] [PubMed]
- Covián, C.; Fernández-Fierro, A.; Retamal-Díaz, A.; Díaz, F.E.; Vasquez, A.E.; Lay, M.K.; Riedel, C.A.; González, P.A.; Bueno, S.M.; Kalergis, A.M. BCG-Induced Cross-Protection and Development of Trained Immunity: Implication for Vaccine Design. Front. Immunol. 2019, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Kia, P.; Ruman, U.; Pratiwi, A.R.; Hussein, M.Z. Innovative Therapeutic Approaches Based on Nanotechnology for the Treatment and Management of Tuberculosis. IJN 2023, 18, 1159–1191. [Google Scholar] [CrossRef] [PubMed]
- Fries, C.N.; Curvino, E.J.; Chen, J.-L.; Permar, S.R.; Fouda, G.G.; Collier, J.H. Advances in Nanomaterial Vaccine Strategies to Address Infectious Diseases Impacting Global Health. Nat. Nanotechnol. 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Churchyard, G.; Kim, P.; Shah, N.S.; Rustomjee, R.; Gandhi, N.; Mathema, B.; Dowdy, D.; Kasmar, A.; Cardenas, V. What We Know about Tuberculosis Transmission: An Overview. J. Infect. Dis. 2017, 216, S629–S635. [Google Scholar] [CrossRef]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef]
- Doyle, R.M.; Burgess, C.; Williams, R.; Gorton, R.; Booth, H.; Brown, J.; Bryant, J.M.; Chan, J.; Creer, D.; Holdstock, J.; et al. Direct Whole-Genome Sequencing of Sputum Accurately Identifies Drug-Resistant Mycobacterium Tuberculosis Faster than MGIT Culture Sequencing. J. Clin. Microbiol. 2018, 56, e00666-18. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, B.B.; Fernandez-Suarez, M.; Heller, D.; Ambravaneswaran, V.; Irimia, D.; Toner, M.; Fortune, S.M. Asymmetry and Aging of Mycobacterial Cells Lead to Variable Growth and Antibiotic Susceptibility. Science 2012, 335, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Pai, M. Tuberculosis: The Story after the Primer. Nat. Rev. Dis. Primers 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Koduri, L.V.S.K.; Bhatt, T.D.; Jhamb, S.S.; Mishra, V.; Gill, M.S.; Suresh, S. In Vitro-In Vivo Evaluation of Novel Co-Spray Dried Rifampicin Phospholipid Lipospheres for Oral Delivery. AAPS PharmSciTech 2017, 18, 138–146. [Google Scholar] [CrossRef]
- Erokhina, M.; Lepekha, L.; Voronezhskaya, E.; Nezlin, L.; Avdienko, V.; Ergeshov, A. Application of Laser Scanning Confocal Microscopy for the Visualization of M. Tuberculosis in Lung Tissue Samples with Weak Ziehl–Neelsen Staining. J. Clin. Med. 2019, 8, 1185. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.; Irfan, M.; Jabeen, K.; Iqbal, N.; Hasan, R.; Migliori, G.B.; Zumla, A.; Visca, D.; Centis, R.; Tiberi, S. Post Tuberculosis Treatment Infectious Complications. Int. J. Infect. Dis. 2020, 92, S41–S45. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, J.; Jerome, K.R. Applications of Digital PCR for Clinical Microbiology. J. Clin. Microbiol. 2017, 55, 1621–1628. [Google Scholar] [CrossRef]
- Cao, D.-H.; Wei, T.-Q.; Wei, Q.; Liu, L.-R. Reply: Challenges in the Diagnosis of Renal Tuberculosis. Kaohsiung J. Med. Sci. 2015, 31, 494–495. [Google Scholar] [CrossRef]
- Zhou, G.; Luo, Q.; Luo, S.; Teng, Z.; Ji, Z.; Yang, J.; Wang, F.; Wen, S.; Ding, Z.; Li, L.; et al. Interferon-γ Release Assays or Tuberculin Skin Test for Detection and Management of Latent Tuberculosis Infection: A Systematic Review and Meta-Analysis. Lancet Infect. Dis. 2020, 20, 1457–1469. [Google Scholar] [CrossRef]
- Mehta, K.; Spaink, H.P.; Ottenhoff, T.H.M.; van der Graaf, P.H.; van Hasselt, J.G.C. Host-Directed Therapies for Tuberculosis: Quantitative Systems Pharmacology Approaches. Trends Pharmacol. Sci. 2022, 43, 293–304. [Google Scholar] [CrossRef]
- Dong, B.; He, Z.; Li, Y.; Xu, X.; Wang, C.; Zeng, J. Improved Conventional and New Approaches in the Diagnosis of Tuberculosis. Front. Microbiol. 2022, 13, 924410. [Google Scholar] [CrossRef] [PubMed]
- Luca, S.; Mihaescu, T. History of BCG Vaccine. Maedica 2013, 8, 53–58. [Google Scholar] [PubMed]
- Syggelou, A.; Spyridis, N.; Benetatou, K.; Kourkouni, E.; Kourlaba, G.; Tsagaraki, M.; Maritsi, D.; Eleftheriou, I.; Tsolia, M. BCG Vaccine Protection against TB Infection among Children Older than 5 Years in Close Contact with an Infectious Adult TB Case. J. Clin. Med. 2020, 9, 3224. [Google Scholar] [CrossRef] [PubMed]
- Davenne, T.; McShane, H. Why Don’t We Have an Effective Tuberculosis Vaccine yet? Expert. Rev. Vaccines 2016, 15, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Borah Slater, K.; Kim, D.; Chand, P.; Xu, Y.; Shaikh, H.; Undale, V. A Current Perspective on the Potential of Nanomedicine for Anti-Tuberculosis Therapy. Trop. Med. Infect. Dis. 2023, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Meeting Report of a Technical Expert Consultation: Non-Inferiority Analysis of Xpert MTB/RIF Ultra Compared to Xpert MTB/RIF; WHO: Geneva, Switzerland, 2017; Volume 10. [Google Scholar]
- Jang, J.G.; Chung, J.H. Diagnosis and Treatment of Multidrug-Resistant Tuberculosis. Yeungnam Univ. J. Med. 2020, 37, 277–285. [Google Scholar] [CrossRef]
- World Health Organization. Rapid Communication: Key Changes to Treatment of Multidrug- and Rifampicin-Resistant Tuberculosis; WHO: Geneva, Switzerland, 2018; pp. 1–7. [Google Scholar]
- Chopra, H.; Mohanta, Y.K.; Rauta, P.R.; Ahmed, R.; Mahanta, S.; Mishra, P.K.; Panda, P.; Rabaan, A.A.; Alshehri, A.A.; Othman, B.; et al. An Insight into Advances in Developing Nanotechnology Based Therapeutics, Drug Delivery, Diagnostics and Vaccines: Multidimensional Applications in Tuberculosis Disease Management. Pharmaceuticals 2023, 16, 581. [Google Scholar] [CrossRef]
- Parveen, S.; Sur, T.; Sarkar, S.; Roy, R. Antagonist Impact of Selenium-Based Nanoparticles Against Mycobacterium Tuberculosis. Appl. Biochem. Biotechnol. 2023, 195, 3606–3614. [Google Scholar] [CrossRef]
- Virmani, R.; Virmani, T.; Pathak, K. Chapter 17—Nanovesicles for Delivery of Central Nervous System Drugs. In Applications of Nanovesicular Drug Delivery; Nayak, A.K., Hasnain, M.S., Aminabhavi, T.M., Torchilin, V.P., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 315–339. ISBN 978-0-323-91865-7. [Google Scholar]
- Singla, R.K.; Sai, C.S.; Chopra, H.; Behzad, S.; Bansal, H.; Goyal, R.; Gautam, R.K.; Tsagkaris, C.; Joon, S.; Singla, S.; et al. Natural Products for the Management of Castration-Resistant Prostate Cancer: Special Focus on Nanoparticles Based Studies. Front. Cell Dev. Biol. 2021, 9, 745177. [Google Scholar] [CrossRef]
- Dhir, S.; Dutt, R.; Singh, R.P.; Chauhan, M.; Virmani, T.; Kumar, G.; Alhalmi, A.; Aleissa, M.S.; Rudayni, H.A.; Al-Zahrani, M. Amomum Subulatum Fruit Extract Mediated Green Synthesis of Silver and Copper Oxide Nanoparticles: Synthesis, Characterization, Antibacterial and Anticancer Activities. Processes 2023, 11, 2698. [Google Scholar] [CrossRef]
- Lin, W.; Fan, S.; Liao, K.; Huang, Y.; Cong, Y.; Zhang, J.; Jin, H.; Zhao, Y.; Ruan, Y.; Lu, H.; et al. Engineering Zinc Oxide Hybrid Selenium Nanoparticles for Synergetic Anti-Tuberculosis Treatment by Combining Mycobacterium Tuberculosis Killings and Host Cell Immunological Inhibition. Front. Cell. Infect. Microbiol. 2023, 12, 1074533. [Google Scholar] [CrossRef] [PubMed]
- Costa-Gouveia, J.; Aínsa, J.A.; Brodin, P.; Lucía, A. How Can Nanoparticles Contribute to Antituberculosis Therapy? Drug Discov. Today 2017, 22, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Sharma, A.K.; Kasu, R.; Gupta, U. Polymeric Nanoparticles: A Holistic Approach to Combat Tuberculosis. Crit. Rev. Ther. Drug Carr. Syst. 2022, 39, 83–115. [Google Scholar] [CrossRef] [PubMed]
- Varma, J.N.R.; Kumar, T.S.; Prasanthi, B.; Ratna, J.V. Formulation and Characterization of Pyrazinamide Polymeric Nanoparticles for Pulmonary Tuberculosis: Efficiency for Alveolar Macrophage Targeting. Indian J. Pharm. Sci. 2015, 77, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef]
- Xia, W.; Tao, Z.; Zhu, B.; Zhang, W.; Liu, C.; Chen, S.; Song, M. Targeted Delivery of Drugs and Genes Using Polymer Nanocarriers for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 9118. [Google Scholar] [CrossRef] [PubMed]
- Virmani, T.; Kumar, G.; Virmani, R.; Sharma, A.; Pathak, K. Xanthan Gum-Based Drug Delivery Systems for Respiratory Diseases. In Natural Polymeric Materials based Drug Delivery Systems in Lung Diseases; Dureja, H., Adams, J., Löbenberg, R., Andreoli Pinto, T.d.J., Dua, K., Eds.; Springer Nature: Singapore, 2023; pp. 279–295. ISBN 978-981-19765-6-8. [Google Scholar]
- Colone, M.; Calcabrini, A.; Stringaro, A. Drug Delivery Systems of Natural Products in Oncology. Molecules 2020, 25, 4560. [Google Scholar] [CrossRef]
- Virmani, T.; Kumar, G.; Sharma, A.; Pathak, K.; Akhtar, M.S.; Afzal, O.; Altamimi, A.S.A. Amelioration of Cancer Employing Chitosan, Its Derivatives, and Chitosan-Based Nanoparticles: Recent Updates. Polymers 2023, 15, 2928. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Taheri, M.; Nouri, F.; Farmani, A.; Moez, N.M.; Arabestani, M.R. Nano Drug Delivery in Intracellular Bacterial Infection Treatments. Biomed. Pharmacother. 2022, 146, 112609. [Google Scholar] [CrossRef]
- Pandey, R.; Sharma, A.; Zahoor, A.; Sharma, S.; Khuller, G.K.; Prasad, B. Poly (DL-Lactide-Co-Glycolide) Nanoparticle-Based Inhalable Sustained Drug Delivery System for Experimental Tuberculosis. J. Antimicrob. Chemother. 2003, 52, 981–986. [Google Scholar] [CrossRef]
- Araujo, V.H.S.; Delello Di Filippo, L.; Duarte, J.L.; Spósito, L.; de Camargo, B.A.F.; da Silva, P.B.; Chorilli, M. Exploiting Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery against Cutaneous Fungal Infections. Crit. Rev. Microbiol. 2021, 47, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Lucks, S.; Müller, R. Medication Vehicles Made of Solid Lipid Particles (Solid Lipid Nanospheres—SLN). European Patent Office 0605497B1, 14 April 1996. [Google Scholar]
- Gasco, M.R. Method for Producing Solid Lipid Microspheres Having a Narrow Size Distribution. U.S. Patent 5250236A, 5 October 1993. [Google Scholar]
- Liu, D.; Chen, L.; Jiang, S.; Zhu, S.; Qian, Y.; Wang, F.; Li, R.; Xu, Q. Formulation and Characterization of Hydrophilic Drug Diclofenac Sodium-Loaded Solid Lipid Nanoparticles Based on Phospholipid Complexes Technology. J. Liposome Res. 2014, 24, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Khan, M.A.; Khan, W.S.; Shafique, M.; Khan, M. Fabrication of Niclosamide Loaded Solid Lipid Nanoparticles: In Vitro Characterization and Comparative in Vivo Evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Sumera; Anwar, A.; Ovais, M.; Khan, A.; Raza, A. Docetaxel-loaded Solid Lipid Nanoparticles: A Novel Drug Delivery System. IET Nanobiotechnol. 2017, 11, 621–629. [Google Scholar] [CrossRef]
- Virmani, T.; Kumar, G.; Sharma, A.; Pathak, K. An Overview of Ocular Drug Delivery Systems. In Nanotechnology in Ophthalmology; Elsevier: Amsterdam, The Netherlands, 2023; pp. 23–48. ISBN 978-0-443-15264-1. [Google Scholar]
- Kumar, G.; Virmani, T.; Sharma, A.; Pathak, K. Codelivery of Phytochemicals with Conventional Anticancer Drugs in Form of Nanocarriers. Pharmaceutics 2023, 15, 889. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.-S.; Na, Y.-G.; Cho, C.-W. Sustained Cytotoxicity of Wogonin on Breast Cancer Cells by Encapsulation in Solid Lipid Nanoparticles. Nanomaterials 2018, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Khatak, S.; Mehta, M.; Awasthi, R.; Paudel, K.R.; Singh, S.K.; Gulati, M.; Hansbro, N.G.; Hansbro, P.M.; Dua, K.; Dureja, H. Solid Lipid Nanoparticles Containing Anti-Tubercular Drugs Attenuate the Mycobacterium Marinum Infection. Tuberculosis 2020, 125, 102008. [Google Scholar] [CrossRef]
- Obinu, A.; Porcu, E.P.; Piras, S.; Ibba, R.; Carta, A.; Molicotti, P.; Migheli, R.; Dalpiaz, A.; Ferraro, L.; Rassu, G.; et al. Solid Lipid Nanoparticles as Formulative Strategy to Increase Oral Permeation of a Molecule Active in Multidrug-Resistant Tuberculosis Management. Pharmaceutics 2020, 12, 1132. [Google Scholar] [CrossRef]
- Talluri, S.V.; Kuppusamy, G.; Karri, V.V.S.R.; Tummala, S.; Madhunapantula, S.V. Lipid-Based Nanocarriers for Breast Cancer Treatment—Comprehensive Review. Drug Deliv. 2015, 23, 1291–1305. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Nanostructured Lipid Matrices for Improved Microencapsulation of Drugs. Int. J. Pharm. 2002, 242, 121–128. [Google Scholar] [CrossRef]
- Pinheiro, M.; Ribeiro, R.; Vieira, A.; Andrade, F.; Reis, S. Design of a Nanostructured Lipid Carrier Intended to Improve the Treatment of Tuberculosis. Drug Des. Devel Ther. 2016, 10, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Bharti Sharma, J.; Bhatt, S.; Tiwari, A.; Tiwari, V.; Kumar, M.; Verma, R.; Kaushik, D.; Virmani, T.; Kumar, G.; Al Kamaly, O.; et al. Statistical Optimization of Tetrahydrocurcumin Loaded Solid Lipid Nanoparticles Using Box Behnken Design in the Management of Streptozotocin-Induced Diabetes Mellitus. Saudi Pharm. J. 2023, 31, 101727. [Google Scholar] [CrossRef] [PubMed]
- Nabi, B.; Rehman, S.; Aggarwal, S.; Baboota, S.; Ali, J. Nano-Based Anti-Tubercular Drug Delivery: An Emerging Paradigm for Improved Therapeutic Intervention. Drug Deliv. Transl. Res. 2020, 10, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Ahalwat, S.; Bhatt, D.C. Development of Novel Lipid Matrix for Improved Sustained Release Effect of a Hydrophilic Drug via Response Surface Methodology. J. Drug Deliv. Sci. Technol. 2022, 67, 102993. [Google Scholar] [CrossRef]
- Patil, T.S.; Deshpande, A.S. Nanostructured Lipid Carrier-Mediated Lung Targeted Drug Delivery System to Enhance the Safety and Bioavailability of Clofazimine. Drug Dev. Ind. Pharm. 2021, 47, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.S.; Figueiredo, C.; Azevedo, N.F.; Braeckmans, K.; De Smedt, S.C. Nanomaterials and Molecular Transporters to Overcome the Bacterial Envelope Barrier: Towards Advanced Delivery of Antibiotics. Adv. Drug Deliv. Rev. 2018, 136–137, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Hatae, A.C.; Roque-Borda, C.A.; Pavan, F.R. Strategies for Lipid-Based Nanocomposites with Potential Activity against Mycobacterium Tuberculosis: Microbial Resistance Challenge and Drug Delivery Trends. OpenNano 2023, 13, 100171. [Google Scholar] [CrossRef]
- Pham, D.-D.; Fattal, E.; Tsapis, N. Pulmonary Drug Delivery Systems for Tuberculosis Treatment. Int. J. Pharm. 2015, 478, 517–529. [Google Scholar] [CrossRef]
- Rinaldi, F.; Hanieh, P.N.; Sennato, S.; De Santis, F.; Forte, J.; Fraziano, M.; Casciardi, S.; Marianecci, C.; Bordi, F.; Carafa, M. Rifampicin-Liposomes for Mycobacterium Abscessus Infection Treatment: Intracellular Uptake and Antibacterial Activity Evaluation. Pharmaceutics 2021, 13, 1070. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Krause, R.W.; Noundou, X.S.; Walker, R.B. Preparation and Characterization of Isoniazid-Loaded Crude Soybean Lecithin Liposomes. Int. J. Pharm. 2017, 526, 466–473. [Google Scholar] [CrossRef]
- Singh, S.; Virmani, T.; Kohli, K. Nanoemulsion System for Improvement of Raspberry Ketone Oral Bioavailability. IGJPS 2020, 10, 33–42. [Google Scholar] [CrossRef]
- Virmani, T.; Kumar, G.; Pathak, K. Non-Aqueous Nanoemulsions: An Innovative Lipid-Based Drug Carrier. Available online: https://www.igi-global.com/chapter/non-aqueous-nanoemulsions/www.igi-global.com/chapter/non-aqueous-nanoemulsions/300404 (accessed on 19 April 2022).
- Kumar, G.; Virmani, T.; Pathak, K.; Kamaly, O.A.; Saleh, A. Central Composite Design Implemented Azilsartan Medoxomil Loaded Nanoemulsion to Improve Its Aqueous Solubility and Intestinal Permeability: In Vitro and Ex Vivo Evaluation. Pharmaceuticals 2022, 15, 1343. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Altamimi, M.A.; Alshehri, S.; Imam, S.S.; Shakeel, F.; Singh, S.K. Novel Approach for Transdermal Delivery of Rifampicin to Induce Synergistic Antimycobacterial Effects Against Cutaneous and Systemic Tuberculosis Using a Cationic Nanoemulsion Gel. Int. J. Nanomed. 2020, 15, 1073–1094. [Google Scholar] [CrossRef] [PubMed]
- Gothwal, A.; Khan, I.; Gupta, U. Polymeric Micelles: Recent Advancements in the Delivery of Anticancer Drugs. Pharm. Res. 2016, 33, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Gorain, B.; Choudhury, H.; Patro Sisinthy, S.; Kesharwani, P. Polymeric Micelle-Based Drug Delivery Systems for Tuberculosis Treatment. In Nanotechnology Based Approaches for Tuberculosis Treatment; Elsevier: Amsterdam, The Netherlands, 2020; pp. 175–191. ISBN 978-0-12-819811-7. [Google Scholar]
- Virmani, R.; Pathak, K. Targeted Polymeric Micellar Systems for Respiratory Diseases. In Targeting Chronic Inflammatory Lung Diseases Using Advanced Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2020; pp. 411–439. ISBN 978-0-12-820658-4. [Google Scholar]
- Ebrahim Attia, A.B.; Ong, Z.Y.; Hedrick, J.L.; Lee, P.P.; Ee, P.L.R.; Hammond, P.T.; Yang, Y.-Y. Mixed Micelles Self-Assembled from Block Copolymers for Drug Delivery. Curr. Opin. Colloid. Interface Sci. 2011, 16, 182–194. [Google Scholar] [CrossRef]
- Varela-Moreira, A.; Shi, Y.; Fens, M.H.A.M.; Lammers, T.; Hennink, W.E.; Schiffelers, R.M. Clinical Application of Polymeric Micelles for the Treatment of Cancer. Mater. Chem. Front. 2017, 1, 1485–1501. [Google Scholar] [CrossRef]
- Amarnath Praphakar, R.; Sam Ebenezer, R.; Vignesh, S.; Shakila, H.; Rajan, M. Versatile pH-Responsive Chitosan-g-Polycaprolactone/Maleic Anhydride–Isoniazid Polymeric Micelle To Improve the Bioavailability of Tuberculosis Multidrugs. ACS Appl. Bio Mater. 2019, 2, 1931–1943. [Google Scholar] [CrossRef]
- An, H.; Deng, X.; Wang, F.; Xu, P.; Wang, N. Dendrimers as Nanocarriers for the Delivery of Drugs Obtained from Natural Products. Polymers 2023, 15, 2292. [Google Scholar] [CrossRef]
- Fana, M.; Gallien, J.; Srinageshwar, B.; Dunbar, G.L.; Rossignol, J. PAMAM Dendrimer Nanomolecules Utilized as Drug Delivery Systems for Potential Treatment of Glioblastoma: A Systematic Review. Int. J. Nanomed. 2020, 15, 2789–2808. [Google Scholar] [CrossRef]
- Shukla, R.; Sethi, A.; Handa, M.; Mohan, M.; Tripathi, P.K.; Kesharwani, P. Dendrimer-Based Drug Delivery Systems for Tuberculosis Treatment. In Nanotechnology Based Approaches for Tuberculosis Treatment; Elsevier: Amsterdam, The Netherlands, 2020; pp. 163–174. ISBN 978-0-12-819811-7. [Google Scholar]
- Ahmed, R.; Aucamp, M.; Ebrahim, N.; Samsodien, H. Supramolecular Assembly of Rifampicin and PEGylated PAMAM Dendrimer as a Novel Conjugate for Tuberculosis. J. Drug Deliv. Sci. Technol. 2021, 66, 102773. [Google Scholar] [CrossRef]
- Zomorodbakhsh, S.; Abbasian, Y.; Naghinejad, M.; Sheikhpour, M. The Effects Study of Isoniazid Conjugated Multi-Wall Carbon Nanotubes Nanofluid on Mycobacterium Tuberculosis. Int. J. Nanomed. 2020, 15, 5901–5909. [Google Scholar] [CrossRef] [PubMed]
- Sheikhpour, M.; Delorme, V.; Kasaeian, A.; Amiri, V.; Masoumi, M.; Sadeghinia, M.; Ebrahimzadeh, N.; Maleki, M.; Pourazar, S. An Effective Nano Drug Delivery and Combination Therapy for the Treatment of Tuberculosis. Sci. Rep. 2022, 12, 9591. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Paz, C.; Fernández-Paz, E.; Salcedo-Abraira, P.; Rojas, S.; Barrios-Esteban, S.; Csaba, N.; Horcajada, P.; Remuñán-López, C. Microencapsulated Isoniazid-Loaded Metal-Organic Frameworks for Pulmonary Administration of Antituberculosis Drugs. Molecules 2021, 26, 6408. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, N.V.; Khatri, H.N.; Patel, M.M. Formulation, Optimization, and Characterization of Rifampicin-Loaded Solid Lipid Nanoparticles for the Treatment of Tuberculosis. Drug Dev. Ind. Pharm. 2018, 44, 1975–1989. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Mokhtarzadeh, A.; Panahi-Azar, V.; Mohammadi, A.; Hamishehkar, H.; Mesgari-Abbasi, M.; Ezzati Nazhad Dolatabadi, J.; De La Guardia, M. Ethambutol-Loaded Solid Lipid Nanoparticles as Dry Powder Inhalable Formulation for Tuberculosis Therapy. AAPS PharmSciTech 2019, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Azami, A.; Mokhtarzadeh, A.; Rahbar Saadat, Y.; Omidi, Y.; Ezzati Nazhad Dolatabadi, J. Formulation and Characterization of Ethambutol Loaded Nanostructured Lipid Carrier. Lat. Am. J. Pharm. 2017, 36, 247–252. [Google Scholar]
- Makled, S.; Boraie, N.; Nafee, N. Nanoparticle-Mediated Macrophage Targeting-a New Inhalation Therapy Tackling Tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1037–1055. [Google Scholar] [CrossRef]
- Singh, A.; Das, S.S.; Ruokolainen, J.; Kesari, K.K.; Singh, S.K. Biopolymer-Capped Pyrazinamide-Loaded Colloidosomes: In Vitro Characterization and Bioavailability Studies. ACS Omega 2023, 8, 25515–25524. [Google Scholar] [CrossRef]
- Vieira, A.C.C.; Chaves, L.L.; Pinheiro, S.; Pinto, S.; Pinheiro, M.; Lima, S.C.; Ferreira, D.; Sarmento, B.; Reis, S. Mucoadhesive Chitosan-Coated Solid Lipid Nanoparticles for Better Management of Tuberculosis. Int. J. Pharm. 2018, 536, 478–485. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Krause, R.W.M. Conjugation of Isoniazid to a Zinc Phthalocyanine via Hydrazone Linkage for pH-Dependent Liposomal Controlled Release. Appl. Nanosci. 2018, 8, 1313–1323. [Google Scholar] [CrossRef]
- Patil, J.S.; Devi, V.K.; Devi, K.; Sarasija, S. A Novel Approach for Lung Delivery of Rifampicin-Loaded Liposomes in Dry Powder Form for the Treatment of Tuberculosis. Lung India 2015, 32, 331–338. [Google Scholar] [CrossRef] [PubMed]
- El-Ridy, M.S.; Mostafa, D.M.; Shehab, A.; Nasr, E.A.; Abd El-Alim, S. Biological Evaluation of Pyrazinamide Liposomes for Treatment of Mycobacterium Tuberculosis. Int. J. Pharm. 2007, 330, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lou, C.; Wang, X.; Wang, C.; Shi, Z.; Niu, N. Preparation, Characterization, and in-Vitro Cytotoxicity of Nanoliposomes Loaded with Anti-Tubercular Drugs and TGF-Β1 siRNA for Improving Spinal Tuberculosis Therapy. BMC Infect. Dis. 2022, 22, 824. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Buttaro, B.A.; Xue, H.Y.; Tran, N.T.; Wong, H.L. Lipid-Polymer Hybrid Nanoparticles Carrying Linezolid Improve Treatment of Methicillin-Resistant Staphylococcus Aureus (MRSA) Harbored inside Bone Cells and Biofilms. Eur. J. Pharm. Biopharm. 2020, 151, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Xiang, H.; Li, X.; Luo, C.; Ma, X.; Zhao, W.; Chen, J.; Tian, Z.; Li, X.; Song, X. Development of Rifapentine-Loaded PLGA-Based Nanoparticles: In Vitro Characterisation and in Vivo Study in Mice. Int. J. Nanomed. 2020, 15, 7491–7507. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Gothwal, A.; Pandey, P.K.; Chauhan, D.S.; Pachouri, P.K.; Gupta, U.D.; Gupta, U. HPMA-PLGA Based Nanoparticles for Effective In Vitro Delivery of Rifampicin. Pharm. Res. 2018, 36, 19. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.; Raizaday, A.; Hussain, T.; Nagshubha, B. Development and Optimization of Dual Drugs (Isoniazid and Moxiflox-Acin) Loaded Functional PLGA Nanoparticles for the Synergistic Treatment of Tuberculosis. Curr. Drug Deliv. 2016, 13, 1034–1052. [Google Scholar] [CrossRef] [PubMed]
- Gajendiran, M.; Gopi, V.; Elangovan, V.; Murali, R.V.; Balasubramanian, S. Isoniazid Loaded Core Shell Nanoparticles Derived from PLGA–PEG–PLGA Tri-Block Copolymers: In Vitro and in Vivo Drug Release. Colloids Surf. B: Biointerfaces 2013, 104, 107–115. [Google Scholar] [CrossRef]
- Hakkimane, S.S.; Shenoy, V.P.; Gaonkar, S.L.; Bairy, I.; Guru, B.R. Antimycobacterial Susceptibility Evaluation of Rifampicin and Isoniazid Benz-Hydrazone in Biodegradable Polymeric Nanoparticles against Mycobacterium Tuberculosis H37Rv Strain. IJN 2018, 13, 4303–4318. [Google Scholar] [CrossRef]
- Abdelghany, S.; Parumasivam, T.; Pang, A.; Roediger, B.; Tang, P.; Jahn, K.; Britton, W.J.; Chan, H.-K. Alginate Modified-PLGA Nanoparticles Entrapping Amikacin and Moxifloxacin as a Novel Host-Directed Therapy for Multidrug-Resistant Tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 52, 642–651. [Google Scholar] [CrossRef]
- Pham, D.-D.; Fattal, E.; Tsapis, N. Pyrazinamide-Loaded Poly(Lactide-Co-Glycolide) Nanoparticles: Optimization by Experimental Design. J. Drug Deliv. Sci. Technol. 2015, 30, 384–390. [Google Scholar] [CrossRef]
- Shah, S.; Cristopher, D.; Sharma, S.; Soniwala, M.; Chavda, J. Inhalable Linezolid Loaded PLGA Nanoparticles for Treatment of Tuberculosis: Design, Development and in Vitro Evaluation. J. Drug Deliv. Sci. Technol. 2020, 60, 102013. [Google Scholar] [CrossRef]
- Oliva, R.; Ginestra, G.; Piperno, A.; Mazzaglia, A.; Nostro, A.; Scala, A. Harnessing the Power of PLA-PEG Nanoparticles for Linezolid Delivery against Methicillin-Resistant Staphylococcus Aureus. Int. J. Pharm. 2023, 642, 123067. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, N.; Khuller, G.K.; Prabhakar, T.; Pal, N.; Gupta, P.; Gupta, U. Nanoformulations of Moxifloxacin, Econozole and Ethionamide as Novel Treatment Regimens Against MDR TB—An Experimental Study. Curr. Nanosci. 2016, 12, 110–117. [Google Scholar] [CrossRef]
- Gajendiran, M.; Jo, H.; Kim, K.; Balasubramanian, S. In Vitro Controlled Release of Tuberculosis Drugs by Amphiphilic Branched Copolymer Nanoparticles. J. Ind. Eng. Chem. 2019, 77, 181–188. [Google Scholar] [CrossRef]
- Debnath, S.K.; Saisivam, S.; Omri, A. PLGA Ethionamide Nanoparticles for Pulmonary Delivery: Development and in Vivo Evaluation of Dry Powder Inhaler. J. Pharm. Biomed. Anal. 2017, 145, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Malhotra, S.; Shafiq, N.; Pandhi, P.; Khuller, G.K.; Sharma, S. In Vitro Physicochemical Characterization and Short Term In Vivo Tolerability Study of Ethionamide Loaded PLGA Nanoparticles: Potentially Effective Agent for Multidrug Resistant Tuberculosis. J. Microencapsul. 2011, 28, 717–728. [Google Scholar] [CrossRef]
- Dineshkumar, P.; Panneerselvam, T.; Brundavani, K.D.; Selvaraj, K.; Kumar, P.V. Formulation of Rifampicin Loaded PEGylated 5.0G EDA-PAMAM Dendrimers as Effective Long-Duration Release Drug Carriers. Curr. Drug Ther. 2017, 12, 115–126. [Google Scholar] [CrossRef]
- Rodrigues, B.; Shende, P. Monodispersed Metal-Based Dendrimeric Nanoclusters for Potentiation of Anti-Tuberculosis Action. J. Mol. Liq. 2020, 304, 112731. [Google Scholar] [CrossRef]
- Bazán Henostroza, M.A.; Curo Melo, K.J.; Nishitani Yukuyama, M.; Löbenberg, R.; Araci Bou-Chacra, N. Cationic Rifampicin Nanoemulsion for the Treatment of Ocular Tuberculosis. Colloids Surf. A Physicochem. Eng. Asp. 2020, 597, 124755. [Google Scholar] [CrossRef]
- Choudhary, A.; Jain, P.; Mohapatra, S.; Mustafa, G.; Ansari, M.J.; Aldawsari, M.F.; Alalaiwe, A.S.; Mirza, M.A.; Iqbal, Z. A Novel Approach of Targeting Linezolid Nanoemulsion for the Management of Lymph Node Tuberculosis. ACS Omega 2022, 7, 15688–15694. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Chan, L.W.; Wong, T.W. Critical Physicochemical and Biological Attributes of Nanoemulsions for Pulmonary Delivery of Rifampicin by Nebulization Technique in Tuberculosis Treatment. Drug Deliv. 2017, 24, 1631–1647. [Google Scholar] [CrossRef] [PubMed]
- Grotz, E.; Tateosian, N.L.; Salgueiro, J.; Bernabeu, E.; Gonzalez, L.; Manca, M.L.; Amiano, N.; Valenti, D.; Manconi, M.; García, V.; et al. Pulmonary Delivery of Rifampicin-Loaded Soluplus Micelles against Mycobacterium Tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 53, 101170. [Google Scholar] [CrossRef]
- Moretton, M.A.; Hocht, C.; Taira, C.; Sosnik, A. Rifampicin-Loaded ‘Flower-like’ Polymeric Micelles for Enhanced Oral Bioavailability in an Extemporaneous Liquid Fixed-Dose Combination with Isoniazid. Nanomedicine 2014, 9, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Gothwal, A.; Khan, I.; Pachouri, P.K.; Bhaskar, N.; Gupta, U.D.; Chauhan, D.S.; Gupta, U. Smartly Engineered PEGylated Di-Block Nanopolymeric Micelles: Duo Delivery of Isoniazid and Rifampicin Against Mycobacterium Tuberculosis. AAPS PharmSciTech 2018, 19, 3237–3248. [Google Scholar] [CrossRef] [PubMed]
- Veeren, A.; Bhaw-Luximon, A.; Jhurry, D. Polyvinylpyrrolidone–Polycaprolactone Block Copolymer Micelles as Nanocarriers of Anti-TB Drugs. Eur. Polym. J. 2013, 49, 3034–3045. [Google Scholar] [CrossRef]
- Chen, G.; Wu, Y.; Yu, D.; Li, R.; Luo, W.; Ma, G.; Zhang, C. Isoniazid-Loaded Chitosan/Carbon Nanotubes Microspheres Promote Secondary Wound Healing of Bone Tuberculosis. J. Biomater. Appl. 2019, 33, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Jafari Nodooshan, S.; Safarkar, R.; Movahedzadeh, F.; Mosavari, N.; Novin Kashani, A.; Dehghanpour, M.; Kamalzadeh, M.; Rasouli Koohi, S.; Fathizadeh, S.; et al. Toxicity Effects of AgZnO Nanoparticles and Rifampicin on Mycobacterium Tuberculosis into the Macrophage. J. Basic. Microbiol. 2018, 58, 41–51. [Google Scholar] [CrossRef]
- Heidary, M.; Zaker Bostanabad, S.; Amini, S.M.; Jafari, A.; Ghalami Nobar, M.; Ghodousi, A.; Kamalzadeh, M.; Darban-Sarokhalil, D. The Anti-Mycobacterial Activity Of Ag, ZnO, And Ag- ZnO Nanoparticles Against MDR- And XDR-Mycobacterium Tuberculosis. Infect. Drug Resist. 2019, 12, 3425–3435. [Google Scholar] [CrossRef]
- Woźniak-Budych, M.J.; Przysiecka, Ł.; Langer, K.; Peplińska, B.; Jarek, M.; Wiesner, M.; Nowaczyk, G.; Jurga, S. Green Synthesis of Rifampicin-Loaded Copper Nanoparticles with Enhanced Antimicrobial Activity. J. Mater. Sci. Mater. Med. 2017, 28, 42. [Google Scholar] [CrossRef]
- Baranyai, Z.; Soria-Carrera, H.; Alleva, M.; Millán-Placer, A.C.; Lucía, A.; Martín-Rapún, R.; Aínsa, J.A.; de la Fuente, J.M. Nanotechnology-Based Targeted Drug Delivery: An Emerging Tool to Overcome Tuberculosis. Adv. Ther. 2021, 4, 2000113. [Google Scholar] [CrossRef]
- Kumar, G.; Khar, R.K.; Virmani, T.; Jogpal, V.; Virmani, R. Comparative Evaluation of Fast Dissolving Tablet of Atorvastatin Calcium Using Natural and Synthetic Super Disintegrating Agents. Res. J. Pharm. Technol. 2018, 11, 5001. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The Impact of Nanoparticle Protein Corona on Cytotoxicity, Immunotoxicity and Target Drug Delivery. Nanomedicine 2016, 11, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Linakis, M.W.; Roberts, J.K.; Lala, A.C.; Spigarelli, M.G.; Medlicott, N.J.; Reith, D.M.; Ward, R.M.; Sherwin, C.M.T. Challenges Associated with Route of Administration in Neonatal Drug Delivery. Clin. Pharmacokinet. 2016, 55, 185–196. [Google Scholar] [CrossRef] [PubMed]
- van Zyl, L.; du Plessis, J.; Viljoen, J. Cutaneous Tuberculosis Overview and Current Treatment Regimens. Tuberculosis 2015, 95, 629–638. [Google Scholar] [CrossRef]
- Tan, Z.M.; Lai, G.P.; Pandey, M.; Srichana, T.; Pichika, M.R.; Gorain, B.; Bhattamishra, S.K.; Choudhury, H. Novel Approaches for the Treatment of Pulmonary Tuberculosis. Pharmaceutics 2020, 12, 1196. [Google Scholar] [CrossRef]
- Labiris, N.R.; Dolovich, M.B. Pulmonary Drug Delivery. Part I: Physiological Factors Affecting Therapeutic Effectiveness of Aerosolized Medications: Physiological Factors Affecting the Effectiveness of Inhaled Drugs. Br. J. Clin. Pharmacol. 2003, 56, 588–599. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early Local Immune Defenses in the Respiratory Tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef]
- Leal, J.; Smyth, H.D.C.; Ghosh, D. Physicochemical Properties of Mucus and Their Impact on Transmucosal Drug Delivery. Int. J. Pharm. 2017, 532, 555–572. [Google Scholar] [CrossRef]
- Wang, W.; Huang, Z.; Huang, Y.; Zhang, X.; Huang, J.; Cui, Y.; Yue, X.; Ma, C.; Fu, F.; Wang, W.; et al. Pulmonary Delivery Nanomedicines towards Circumventing Physiological Barriers: Strategies and Characterization Approaches. Adv. Drug Deliv. Rev. 2022, 185, 114309. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.K.; Wang, Y.-Y.; Hida, K.; Cone, R.; Hanes, J. Nanoparticles Reveal That Human Cervicovaginal Mucus Is Riddled with Pores Larger than Viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Duncan, G.A.; Hanes, J.; Suk, J.S. Barriers to Inhaled Gene Therapy of Obstructive Lung Diseases: A Review. J. Control. Release 2016, 240, 465–488. [Google Scholar] [CrossRef] [PubMed]
- García-Mouton, C.; Parra-Ortiz, E.; Malmsten, M.; Cruz, A.; Pérez-Gil, J. Pulmonary Surfactant and Drug Delivery: Vehiculization of a Tryptophan-Tagged Antimicrobial Peptide over the Air-Liquid Interfacial Highway. Eur. J. Pharm. Biopharm. 2022, 180, 33–47. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gui, J.; Xiong, K.; Chen, M.; Gao, H.; Fu, Y. A Roadmap to Pulmonary Delivery Strategies for the Treatment of Infectious Lung Diseases. J. Nanobiotechnol. 2022, 20, 101. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.; Choi, Y.; Tanaka, M.; Choi, J. Inhalable Nanoparticles Delivery Targeting Alveolar Macrophages for the Treatment of Pulmonary Tuberculosis. J. Biosci. Bioeng. 2021, 132, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Smith, I. Mycobacterium Tuberculosis Pathogenesis and Molecular Determinants of Virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Al-Nemrawi, N.K.; Darweesh, R.S.; Al-shriem, L.A.; Al-Qawasmi, F.S.; Emran, S.O.; Khafajah, A.S.; Abu-Dalo, M.A. Polymeric Nanoparticles for Inhaled Vaccines. Polymers 2022, 14, 4450. [Google Scholar] [CrossRef]
- Blank, F.; Fytianos, K.; Seydoux, E.; Rodriguez-Lorenzo, L.; Petri-Fink, A.; von Garnier, C.; Rothen-Rutishauser, B. Interaction of Biomedical Nanoparticles with the Pulmonary Immune System. J. Nanobiotechnol. 2017, 15, 6. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Proteases and Antiproteases in Chronic Neutrophilic Lung Disease—Relevance to Drug Discovery. Br. J. Pharmacol. 2009, 158, 1048–1058. [Google Scholar] [CrossRef]
- Azad, A.K.; Rajaram, M.V.S.; Schlesinger, L.S. Exploitation of the Macrophage Mannose Receptor (CD206) in Infectious Disease Diagnostics and Therapeutics. J. Cytol. Mol. Biol. 2014, 1, 1000003. [Google Scholar] [CrossRef]
- Kang, P.B.; Azad, A.K.; Torrelles, J.B.; Kaufman, T.M.; Beharka, A.; Tibesar, E.; DesJardin, L.E.; Schlesinger, L.S. The Human Macrophage Mannose Receptor Directs Mycobacterium tuberculosis Lipoarabinomannan-Mediated Phagosome Biogenesis. J. Exp. Med. 2005, 202, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Ahalwat, S.; Bhatt, D.C.; Rohilla, S.; Jogpal, V.; Sharma, K.; Virmani, T.; Kumar, G.; Alhalmi, A.; Alqahtani, A.S.; Noman, O.M.; et al. Mannose-Functionalized Isoniazid-Loaded Nanostructured Lipid Carriers for Pulmonary Delivery: In Vitro Prospects and In Vivo Therapeutic Efficacy Assessment. Pharmaceuticals 2023, 16, 1108. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.C.; Chaves, L.L.; Pinheiro, M.; Lima, S.A.C.; Ferreira, D.; Sarmento, B.; Reis, S. Mannosylated Solid Lipid Nanoparticles for the Selective Delivery of Rifampicin to Macrophages. Artif. Cells Nanomed. Biotechnol. 2018, 46, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Galdopórpora, J.M.; Martinena, C.; Bernabeu, E.; Riedel, J.; Palmas, L.; Castangia, I.; Manca, M.L.; Garcés, M.; Lázaro-Martinez, J.; Salgueiro, M.J.; et al. Inhalable Mannosylated Rifampicin–Curcumin Co-Loaded Nanomicelles with Enhanced In Vitro Antimicrobial Efficacy for an Optimized Pulmonary Tuberculosis Therapy. Pharmaceutics 2022, 14, 959. [Google Scholar] [CrossRef]
- Khan, M.M.; Zaidi, S.S.; Siyal, F.J.; Khan, S.U.; Ishrat, G.; Batool, S.; Mustapha, O.; Khan, S.; Din, F. ud Statistical Optimization of Co-Loaded Rifampicin and Pentamidine Polymeric Nanoparticles for the Treatment of Cutaneous Leishmaniasis. J. Drug Deliv. Sci. Technol. 2023, 79, 104005. [Google Scholar] [CrossRef]
- Ebrahimnejad, P.; Sodagar Taleghani, A.; Asare-Addo, K.; Nokhodchi, A. An Updated Review of Folate-Functionalized Nanocarriers: A Promising Ligand in Cancer. Drug Discov. Today 2022, 27, 471–489. [Google Scholar] [CrossRef]
- Martín-Sabroso, C.; Torres-Suárez, A.I.; Alonso-González, M.; Fernández-Carballido, A.; Fraguas-Sánchez, A.I. Active Targeted Nanoformulations via Folate Receptors: State of the Art and Future Perspectives. Pharmaceutics 2021, 14, 14. [Google Scholar] [CrossRef]
- Morshedi, M.; Saghafi-Asl, M.; Hosseinifard, E.-S. The Potential Therapeutic Effects of the Gut Microbiome Manipulation by Synbiotic Containing-Lactobacillus Plantarum on Neuropsychological Performance of Diabetic Rats. J. Transl. Med. 2020, 18, 18. [Google Scholar] [CrossRef]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in Targeting the Folate Receptor in the Treatment/Imaging of Cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef]
- Zhu, Y.; Kruglikov, I.L.; Akgul, Y.; Scherer, P.E. Hyaluronan in Adipogenesis, Adipose Tissue Physiology and Systemic Metabolism. Matrix Biol. 2019, 78–79, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Astakhova, A.A.; Azbukina, N.V.; Goriainov, S.V.; Chistyakov, V.V.; Sergeeva, M.G. High and Low Molecular Weight Hyaluronic Acid Differentially Influences Oxylipins Synthesis in Course of Neuroinflammation. Int. J. Mol. Sci. 2019, 20, 3894. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Ruffell, B. CD44 and Its Role in Inflammation and Inflammatory Diseases. Inflamm. Allergy Drug Targets 2009, 8, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, Y.; Yoshimura, M.; Ozeki, Y.; Sugawara, I.; Udagawa, T.; Mizuno, S.; Itano, N.; Kimata, K.; Tamaru, A.; Ogura, H.; et al. Mycobacteria Exploit Host Hyaluronan for Efficient Extracellular Replication. PLoS Pathog. 2009, 5, e1000643. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.; Csaba, N.; Robla, S.; Varela-Calviño, R.; Nagy, A.; Burian, K.; Kókai, D.; Ambrus, R. Dry Powder Comprised of Isoniazid-Loaded Nanoparticles of Hyaluronic Acid in Conjugation with Mannose-Anchored Chitosan for Macrophage-Targeted Pulmonary Administration in Tuberculosis. Pharmaceutics 2022, 14, 1543. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.; Fonte, P.; Oliva, M.; Videira, M.; Ferreira, D.; Sarmento, B. Solid State Formulations Composed by Amphiphilic Polymers for Delivery of Proteins: Characterization and Stability. Int. J. Pharm. 2015, 486, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Castro, P.; Fonte, P.; Sarmento, B. How to Overcome the Limitations of Current Insulin Administration with New Non-Invasive Delivery Systems. Ther. Deliv. 2015, 6, 83–94. [Google Scholar] [CrossRef]
- Borbála Horváth, L.; Krátký, M.; Pflégr, V.; Méhes, E.; Gyulai, G.; Kohut, G.; Babiczky, Á.; Biri-Kovács, B.; Baranyai, Z.; Vinšová, J.; et al. Host Cell Targeting of Novel Antimycobacterial 4-Aminosalicylic Acid Derivatives with Tuftsin Carrier Peptides. Eur. J. Pharm. Biopharm. 2022, 174, 111–130. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Evans, I.M.; Herzog, B.; Jünemann-Ramírez, M.; Zachary, I.C. Neuropilin-1 Mediates PDGF Stimulation of Vascular Smooth Muscle Cell Migration and Signalling via p130Cas. Biochem. J. 2011, 435, 609–618. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial Cell Wall Biosynthesis: A Multifaceted Antibiotic Target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef]
- Lemmer, Y.; Kalombo, L.; Pietersen, R.-D.; Jones, A.T.; Semete-Makokotlela, B.; Van Wyngaardt, S.; Ramalapa, B.; Stoltz, A.C.; Baker, B.; Verschoor, J.A.; et al. Mycolic Acids, a Promising Mycobacterial Ligand for Targeting of Nanoencapsulated Drugs in Tuberculosis. J. Control. Release 2015, 211, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Gatfield, J.; Pieters, J. Essential Role for Cholesterol in Entry of Mycobacteria into Macrophages. Science 2000, 288, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, J.A.; Baird, M.S.; Grooten, J. Towards Understanding the Functional Diversity of Cell Wall Mycolic Acids of Mycobacterium Tuberculosis. Prog. Lipid Res. 2012, 51, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, Y.; Thanyani, S.T.; Vrey, P.J.; Driver, C.H.S.; Venter, L.; van Wyngaardt, S.; ten Bokum, A.M.C.; Ozoemena, K.I.; Pilcher, L.A.; Fernig, D.G.; et al. Chapter 5—Detection of Antimycolic Acid Antibodies by Liposomal Biosensors. Methods Enzym. 2009, 464, 79–104. [Google Scholar] [CrossRef]
- Song, X.; Lin, Q.; Guo, L.; Fu, Y.; Han, J.; Ke, H.; Sun, X.; Gong, T.; Zhang, Z. Rifampicin Loaded Mannosylated Cationic Nanostructured Lipid Carriers for Alveolar Macrophage-Specific Delivery. Pharm. Res. 2015, 32, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, S.P.; Carvalho, K.V.; de Oliveira Aguiar Soares, R.D.; Carneiro, C.M.; de Andrade, M.H.G.; Duarte, R.S.; Dos Santos, O.D.H. Functionalized Rifampicin-Loaded Nanostructured Lipid Carriers Enhance Macrophages Uptake and Antimycobacterial Activity. Colloids Surf. B Biointerfaces 2019, 175, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Maretti, E.; Costantino, L.; Rustichelli, C.; Leo, E.; Croce, M.A.; Buttini, F.; Truzzi, E.; Iannuccelli, V. Surface Engineering of Solid Lipid Nanoparticle Assemblies by Methyl α-d-Mannopyranoside for the Active Targeting to Macrophages in Anti-Tuberculosis Inhalation Therapy. Int. J. Pharm. 2017, 528, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Nimje, N.; Agarwal, A.; Saraogi, G.K.; Lariya, N.; Rai, G.; Agrawal, H.; Agrawal, G.P. Mannosylated Nanoparticulate Carriers of Rifabutin for Alveolar Targeting. J. Drug Target. 2009, 17, 777–787. [Google Scholar] [CrossRef]
- Pi, J.; Shen, L.; Shen, H.; Yang, E.; Wang, W.; Wang, R.; Huang, D.; Lee, B.-S.; Hu, C.; Chen, C.; et al. Mannosylated Graphene Oxide as Macrophage-Targeted Delivery System for Enhanced Intracellular M.Tuberculosis Killing Efficiency. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 103, 109777. [Google Scholar] [CrossRef]
- Shrivastava, P.; Gautam, L.; Sharma, R.; Dube, D.; Vyas, S.; Vyas, S.P. Dual Antitubercular Drug Loaded Liposomes for Macrophage Targeting: Development, Characterisation, Ex Vivo and in Vivo Assessment. J. Microencapsul. 2021, 38, 108–123. [Google Scholar] [CrossRef]
- Patil, K.D.; Bagade, S.B.; Bonde, S.C. Biodistribution, Pharmacokinetics and Toxicity Evaluation of Mannosylated Gelatin Nanoparticles of Linezolid for Anti-Tubercular Therapy. Mater. Technol. 2022, 37, 95–103. [Google Scholar] [CrossRef]
- Saraogi, G.K.; Sharma, B.; Joshi, B.; Gupta, P.; Gupta, U.D.; Jain, N.K.; Agrawal, G.P. Mannosylated Gelatin Nanoparticles Bearing Isoniazid for Effective Management of Tuberculosis. J. Drug Target. 2011, 19, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, P.; Fernandes, T.; Chaubey, P.; Kaur, P.; Narayanan, S.; Vk, R.; Sawarkar, S.P. Mannose-Conjugated Chitosan Nanoparticles for Delivery of Rifampicin to Osteoarticular Tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Pawde, D.M.; Viswanadh, M.K.; Mehata, A.K.; Sonkar, R.; Narendra; Poddar, S.; Burande, A.S.; Jha, A.; Vajanthri, K.Y.; Mahto, S.K.; et al. Mannose Receptor Targeted Bioadhesive Chitosan Nanoparticles of Clofazimine for Effective Therapy of Tuberculosis. Saudi Pharm. J. 2020, 28, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Agnihotri, V.V.; Patil, K.Y.; Pardeshi, S.R.; Surana, S.J. Mannose-Anchored N,N,N-Trimethyl Chitosan Nanoparticles for Pulmonary Administration of Etofylline. Int. J. Biol. Macromol. 2020, 165, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Rossi, I.; Buttini, F.; Sonvico, F.; Affaticati, F.; Martinelli, F.; Annunziato, G.; Machado, D.; Viveiros, M.; Pieroni, M.; Bettini, R. Sodium Hyaluronate Nanocomposite Respirable Microparticles to Tackle Antibiotic Resistance with Potential Application in Treatment of Mycobacterial Pulmonary Infections. Pharmaceutics 2019, 11, 203. [Google Scholar] [CrossRef] [PubMed]
- Moretton, M.A.; Chiappetta, D.A.; Andrade, F.; das Neves, J.; Ferreira, D.; Sarmento, B.; Sosnik, A. Hydrolyzed Galactomannan-Modified Nanoparticles and Flower-like Polymeric Micelles for the Active Targeting of Rifampicin to Macrophages. J. Biomed. Nanotechnol. 2013, 9, 1076–1087. [Google Scholar] [CrossRef]
- Hwang, S.M.; Kim, D.D.; Chung, S.J.; Shim, C.K. Delivery of Ofloxacin to the Lung and Alveolar Macrophages via Hyaluronan Microspheres for the Treatment of Tuberculosis. J. Control. Release 2008, 129, 100–106. [Google Scholar] [CrossRef]
- Martinelli, F.; Balducci, A.G.; Kumar, A.; Sonvico, F.; Forbes, B.; Bettini, R.; Buttini, F. Engineered Sodium Hyaluronate Respirable Dry Powders for Pulmonary Drug Delivery. Int. J. Pharm. 2017, 517, 286–295. [Google Scholar] [CrossRef]
- Horváti, K.; Gyulai, G.; Csámpai, A.; Rohonczy, J.; Kiss, E.; Bősze, S. Surface Layer Modification of Poly(d,l-Lactic- Co-Glycolic Acid) Nanoparticles with Targeting Peptide: A Convenient Synthetic Route for Pluronic F127-Tuftsin Conjugate. Bioconjugate Chem. 2018, 29, 1495–1499. [Google Scholar] [CrossRef]
- Agarwal, A.; Kandpal, H.; Gupta, H.P.; Singh, N.B.; Gupta, C.M. Tuftsin-Bearing Liposomes as Rifampin Vehicles in Treatment of Tuberculosis in Mice. Antimicrob. Agents Chemother. 1994, 38, 588–593. [Google Scholar] [CrossRef]
- Parmar, R.; Misra, R.; Mohanty, S. In Vitro Controlled Release of Rifampicin through Liquid-Crystalline Folate Nanoparticles. Colloids Surf. B: Biointerfaces 2015, 129, 198–205. [Google Scholar] [CrossRef]
- Gelperina, S.; Kisich, K.; Iseman, M.D.; Heifets, L. The Potential Advantages of Nanoparticle Drug Delivery Systems in Chemotherapy of Tuberculosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1487–1490. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Nanocarrier | Active Drug | Method of Preparation | Key Findings | Reference |
|---|---|---|---|---|
| SLNs | Rifampicin | O/W modified micro-emulsion followed by high-pressure homogenization | Provided better gastric stability which could contribute to bioavailability. | [98] |
| SLNs | Ethambutol | Hot homogenization and ultrasonication | Established that dry powder inhalation form of ethambutol-laden SLNs improved efficacy against TB. | [99] |
| SLNs | Rifampicin, isoniazid, pyrazinamide | Micro-emulsion technique | Established double the growth prevention of standard anti-TB drugs against M. marinum. | [67] |
| NLCs | Ethambutol | Hot homogenization followed by ultrasonication | Displayed improved properties on basis of in vitro evaluation testing. | [100] |
| NLCs | Isoniazid | Exhibited prolonged drug release over 24 h. | [74] | |
| NLCs | Linezolid | Spray-drying | Provided sustained drug release, mucus penetrability, possible safety at therapeutic doses, in vitro and in vivo macrophage targetability, and preferential deposition in the deep lung. | [101] |
| Colloidosomes | Pyrazinamide | In situ gelation | Provided improved drug plasma concentration and AUC. | [102] |
| Chitosan coated SLNs | Rifampicin | Ultrasonication | Chitosan-coated SLNs exhibited greater in vitro muco-adhesive characteristics and greater permeability in alveolar epithelial cells A549. | [103] |
| Liposomes | Isoniazid | Film hydration method | Provided pH-reliant drug release having greater release in acidic environment. | [104] |
| Liposomes | Isoniazid | Film hydration method | Provided improved entrapment efficiency, particle size, sustained drug release along with reduction in dosing frequency. | [80] |
| Liposomes | Rifampicin | Freeze-drying | Suggested improved delivery to macrophages than plain drug. | [105] |
| Liposomes | Pyrazinamide | Film hydration method | Exhibited significant reduction in bacterial counts in lungs. | [106] |
| Nanoliposomes | Isoniazid, rifampicin, pyrazinamide | Film hydration method | TGF-1 in human macrophages generated from THP-1 could be dramatically downregulated in vitro by nanoliposomal siTGF-1. | [107] |
| Hybrid nanoparticles | Linezolid | Nanoprecipitation method | Exhibited improved intracellular and anti-biofilm activities of nanoparticles which was mainly due to extensive build-up of nanoparticles inside the MRSA-infected biofilms and osteoblasts. | [108] |
| PLGA NPs | Rifapentine | Homogenization followed by solvent evaporation | Exhibited improved pharmacokinetic parameters as compared to free drug. | [109] |
| HPMA-PLGA based NPs | Rifampicin | Modified nanoprecipitation | Exhibited 4 times more efficacy as compared to free drug against M. tuberculosis. | [110] |
| PLGA NPs | Isoniazid and moxifloxacin | Single emulsion technique | Exhibited better activity in conjugation than individual pure drug. | [111] |
| PLGA-PEG-PLGA NPs | Isoniazid | Sonication followed by double emulsification | Provided 28 times greater bioavailability as compared to free drug. | [112] |
| PLGA NPs | Rifampicin, isoniazid | Single emulsion solvent evaporation | Exhibited improved inhibition of M. tuberculosis compared to pure drugs alone and drugs in conjugation. | [113] |
| Alginate modified PLGA NPs | Amikacin, moxifloxacin | Double emulsification | Showed a greater reduction in the number of viable bacteria when compared to formulations with just one drug loaded on a nanoparticle and untreated cells. | [114] |
| PLGA NPs | Pyrazinamide | Double emulsion | Exhibited improved properties on in vitro evaluation and could proceed to in vivo testing. | [115] |
| PLGA NPs | Linezolid | Modified emulsion solvent evaporation | Exhibited mass median aerodynamic diameter of 3.38 μm along with sustained release for 120 h in simulated lung fluid. | [116] |
| PLA-PEG NPs | Linezolid | Nanoprecipitation | Exhibited better activity against a group of Gram-positive bacteria responsible for human infections. | [117] |
| PLGA NPs | Ethionamide, moxifloxacin, econozole | Nanoprecipitation | Showed that NPs of all three drugs collectively caused reduction in CFUs in lungs as well as spleen. | [118] |
| PLGA-PEG based copolymer NPs | Rifampicin, isoniazid, pyrazinamide | Double emulsification | Exhibited more sustained release of the drug than free drug, a good indication of potential for effective treatment. | [119] |
| PLGA NPs | Rifampicin, ofloxacin, ethambutol | Spray-drying | Provided better antimicrobial efficacy on in vitro analysis along with significant synergistic effect for isoniazid vulnerable species. | [73] |
| PLGA NPs | Ethionamide | Freeze-drying | Provided improved AUC and prolonged release up to 24 h in lung fluid. | [120] |
| PLGA NPs | Ethionamide | Solvent evaporation | Demonstrated possession of excellent potential for treatment of TB. | [121] |
| PEGylated PAMAM dendrimers | Rifampicin | Dissolution solvent evaporation | Exhibited more prolonged drug release than free drug along with negligible toxicity. | [94] |
| PEGylated 5.0G PAMAM dendrimers | Rifampicin | Dissolution solvent evaporation | Provided prolonged release of drug along with reduced toxicity. | [122] |
| Metal based G4 dendrimers | Isoniazid | Solvent-free technique | Provided synergistic impact and an 85 µg/mL dose reduction when the activity was tested on M. tuberculosis H37Ra (ATCC 25177). | [123] |
| Cationic NE | Rifampicin | High-pressure homogenization | Modifications using chitosan increased permeation efficacy at diseased site. | [124] |
| NE | Linezolid | Oil phase titration | Provided lymphatic targeting of drug at the target organ only after 8 h of dose. | [125] |
| NE | Rifampicin | Spontaneous emulsification | Exhibited improved cell internalization potential and decreased plasma drug concentration along with greater quantities of drug in lungs. | [126] |
| PMs | Rifampicin | Rifampicin-loaded PMs increased the in vitro drug’s microbicidal activity against M. tuberculosis-infected THP-1 macrophages up to 2.5-fold. | [127] | |
| PMs | Rifampicin, isoniazid | Co-solvent/evaporation | Exhibited increased oral bioavailability (up to 3.3 times) of rifampicin compared to free drug in the presence of isoniazid. | [128] |
| PEGylated PMs | Rifampicin, isoniazid | Freeze-drying | Provided more efficacy against sensitive M. tuberculosis strains and found to be less hemolytic. | [129] |
| PMs | Rifampicin, isoniazid, pyrazinamide | Solvent evaporation method | Provided sustained drug release on in vitro evaluation. | [130] |
| MWCNTs | Isoniazid | Reflux system | Provided better antimicrobial potential at lower concentrations. | [95] |
| Chitosan/CNTs NPs | Isoniazid | Exhibited reduced numbers of CD3+ and CD4+ T cells in isoniazid/chitosan/carbon nanotube group. | [131] | |
| MNPs | Rifampicin | Loaded drug appeared to be more biocompatible and had stronger antimycobacterial properties. | [132] | |
| MNPs | Ag, ZnO, and Ag-ZnO | Chemical reduction and chemical synthesis | Provided improved efficacy to treat multidrug-resistant and extensively resistant M. tuberculosis. | [133] |
| MNPs | Rifampicin | Green synthesis | Provided a decrease of antibiotic dosage and inhibition of its adverse effects. | [134] |
| Nanocarrier | Ligand | Drug | Result Outcomes | Ref. |
|---|---|---|---|---|
| NLCs | Mannose | Isoniazid | Exhibited a prolonged residence time in the pulmonary region with higher pharmacokinetics than non-functionalized formulation demonstrating the improved therapeutic efficiency of the mannose functionalized NLC formulation. | [157] |
| Cationic NLCs | Mannose | Rifampicin | Demonstrated considerably better absorption efficiency in NR8383 cells and alveolar macrophages than unmodified NLCs in cell-specific targeting. | [179] |
| NLCs | Tuftsin | Rifampicin | Macrophages substantially more frequently internalized tuftsin-containing nanoparticles than tuftsin-free ones. In comparison to free rifampicin, both nanoparticles were twice as efficient against M. tuberculosis. | [180] |
| SLNs | Mannose | Rifampicin | Surface mannosylation accelerated macrophage phagocytosis, showing evidence of an active targeting promotion. | [181] |
| SLNs | Mannose | Rifampicin | The mannosylation of SLNs increased their internalization in macrophages and confirmed their biocompatibility. | [158] |
| SLNs | Mannose | Rifabutin | Revealed a nearly six-fold increase in uptake in alveolar macrophages in comparison to uncoated formulation. | [182] |
| NPs | Mannosylated and Pegylated graphene oxide | Rifampicin | Provided enhanced intracellular rifampicin delivery and pharmacokinetics dramatically improved the effectiveness of rifampicin-driven killing of intracellular BCG and M. tuberculosis bacilli in infected macrophages both in vitro and ex vivo. | [183] |
| Liposomes | Mannose | Isoniazid, rifampicin | Mannosylated liposomes had the strongest anti-TB activity when tested in Balb/C mice. The biodistribution experiments also showed increased drug concentration (accumulation) that was sustained over an extended period of time. | [184] |
| Gelatin NPs | Mannose | Linezolid | Provided reduction in the dose, frequency of administration, and side effects, resulting in increased patient compliance. | [185] |
| Gelatin NPs | Mannose | Isoniazid | Intravenous treatment of formulation significantly reduced bacterial numbers in the lungs and spleen as well as the drug’s hepatotoxicity. | [186] |
| Chitosan NPs | Mannose | Rifampicin | The drug release from conjugated nanoparticles included in in situ gel was determined to be roughly 70.3% at the end of 40 h in simulated synovial fluid. | [187] |
| Chitosan NPs | Mannose | Clofazimine | Indicated that mannosylated NPs internalized more quickly. Additionally, the H37Rv strain luciferase reporter phage (LRP) experiment demonstrated that clofazimine nanoparticles had 49.5 times greater inhibition and antimycobacterial activity than free clofazimine. | [188] |
| N,N,N-trimethyl chitosan nanoparticles | Mannose | Etofylline | Provided that therapeutic efficacy of etofylline has significantly improved according to in vivo pharmacokinetic investigations in the Wistar rat model. | [189] |
| Polymeric micelles | Mannose | Curcumin, rifampicin | Resulted in a huge (5.2-fold) improvement in the microbicidal effectiveness of co-loaded systems against M. tuberculosis H37Rv in comparison to their equivalent mannose free polymeric micelles | [159] |
| Microparticles | Sodium hyaluronate | Rifampicin, isoniazid and verapamil | Ex vivo macrophage infection studies using susceptible and drug-resistant strains were performed, as well as in vitro antimicrobial activity tests. When the powder, which did not affect Ams viability after a five-day exposure, was contrasted with the same formulation without verapamil, no appreciable differences were found. | [190] |
| NPs, PMs | Galactomannan | Rifampicin | Revealed that both nanocarriers were taken up by RAW 264.7 murine macrophages. Surface modification of nanocarriers causes a notable rise of the intracellular concentration of the drug. | [191] |
| Microspheres | Hyaluronic acid | Ofloxacin | Provided increased absorption on RAW 264.7 cells and intratracheally delivered microspheres in Sprague–Dawley rats resulted in larger lung accumulation and lower plasma levels when compared to i.v. and oral administration (OFX solution). | [192] |
| Microspheres | Sodium hyaluronate | Spray-dried microspheres reduced the viability of A549 cells because of the surfactant component | [193] | |
| PLGA NPs | Tuftsin | Coumaron derivative namely TB515 | Demonstrated that adding a Pluronic–Tuftsin conjugate coating to nanoparticles significantly enhanced the internalization rate and intracellular activity of the encapsulated therapeutic candidate against M. tuberculosis. | [194] |
| Liposomes | Tuftsin | Rifampicin | Drug-loaded liposomes were at least 2000 times more effective than the free medication at reducing the load of lung bacilli in Mtb H37Rv-infected Swiss albino mice (i.v. infection) when provided twice weekly for two weeks in i.v. administered liposomes. | [195] |
| Nano-emulsion | Folate | Rifampicin | Chitosan–folate-conjugated nano-emulsion showed a higher cell internalization capacity, a lower plasma drug concentration, and a larger lung drug content. The nano-emulsions were confirmed to be safe. | [126] |
| Liquid crystalline NPs | Folate | Rifampicin | Revealed the improved intracellular uptake and reduced cytotoxicity of NPs by alveolar macrophages. | [196] |
| PLGA NPs | Mycolic acid | Isoniazid | Provided improved enhancement in phagocytic uptake of the NPs. | [175] |
| Patent No. | Formulation/Description |
|---|---|
| US8927024B1 | For example, the copolymers of 1,8-bis-3,6-dioxaoctane anhydrides and 1,6-bis-hexane anhydrides, or any combination of these two, may serve as polyanhydride microparticles or nanoparticles. The nanoparticles or microparticles can enter in infected dendritic cells, monocytes, and other infected cells. Over time, surface erosion causes antimicrobial chemicals to escape from the particles, killing or suppressing the microorganisms and curing the infection. |
| US20090192173A1 | For the treatment of infectious disorders, substituted ethylene diamine-containing techniques and formulations are proposed. The current invention includes compositions that include new substituted ethylene diamine compounds together with antitubercular medications including rifampicin, isoniazid, pyrazinamide, and ethambutol in certain embodiments. |
| US20100310662A1 | The oral drug delivery system includes PLGA nanoparticles with an azole encapsulated therein, PLGA nanoparticles with moxifloxacin encapsulated therein, and PLGA nanoparticles with rifampicin encapsulated therein. |
| US20170044100A1 | The current invention offers brand-new indoleamine substances for treating TB, including drug-resistant M. tuberculosis, compositions containing indoleamine, and procedures for combining the indoleamine with other biologically active agents to treat TB in a subject in need of such treatment. |
| US20050084455A1 | The present invention into two anti-TB drugs and a biodegradable polymer for drug delivery in a ratio of approximately 1:2 to 2:1, wherein the anti-tubercular drugs are in the ratio of 1:2 to 2:1. Finally, it pertains to a technique of treating pulmonary TB in a subclinical context as well as a procedure for making the composition. |
| US20070128124A1 | The invention offers systems, procedures, and compositions for giving capreomycin in an aerosolized form to those who need it. Aerosol capreomycin administration may be used to lessen the intensity or length of a TB infection as well as to lessen the infectivity of TB patients. This innovation also allows for the use of capreomycin in the creation of a medication that may be administered to someone who needs it through aerosol. |
| US8697653B2 | According to PCT application publication WO2011027290, the invention is a biodegradable, inhalable microparticle formulation for drug administration that contains a drug and a lipid in a certain ratio. The present invention also covers treating pulmonary TB, MDRTB, MRSA, and MSSA pneumonia in mammals by administering a therapeutically effective amount of the formulation. The invention also involves inhaling or intratracheal instilling a microparticle formulation to an animal who needs it. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Virmani, T.; Kumar, G.; Deshmukh, R.; Sharma, A.; Duarte, S.; Brandão, P.; Fonte, P. Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals 2023, 16, 1360. https://doi.org/10.3390/ph16101360
Kumar M, Virmani T, Kumar G, Deshmukh R, Sharma A, Duarte S, Brandão P, Fonte P. Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals. 2023; 16(10):1360. https://doi.org/10.3390/ph16101360
Chicago/Turabian StyleKumar, Mahesh, Tarun Virmani, Girish Kumar, Rohitas Deshmukh, Ashwani Sharma, Sofia Duarte, Pedro Brandão, and Pedro Fonte. 2023. "Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies" Pharmaceuticals 16, no. 10: 1360. https://doi.org/10.3390/ph16101360
APA StyleKumar, M., Virmani, T., Kumar, G., Deshmukh, R., Sharma, A., Duarte, S., Brandão, P., & Fonte, P. (2023). Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals, 16(10), 1360. https://doi.org/10.3390/ph16101360