

Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Etiopathogenesis of PKAN
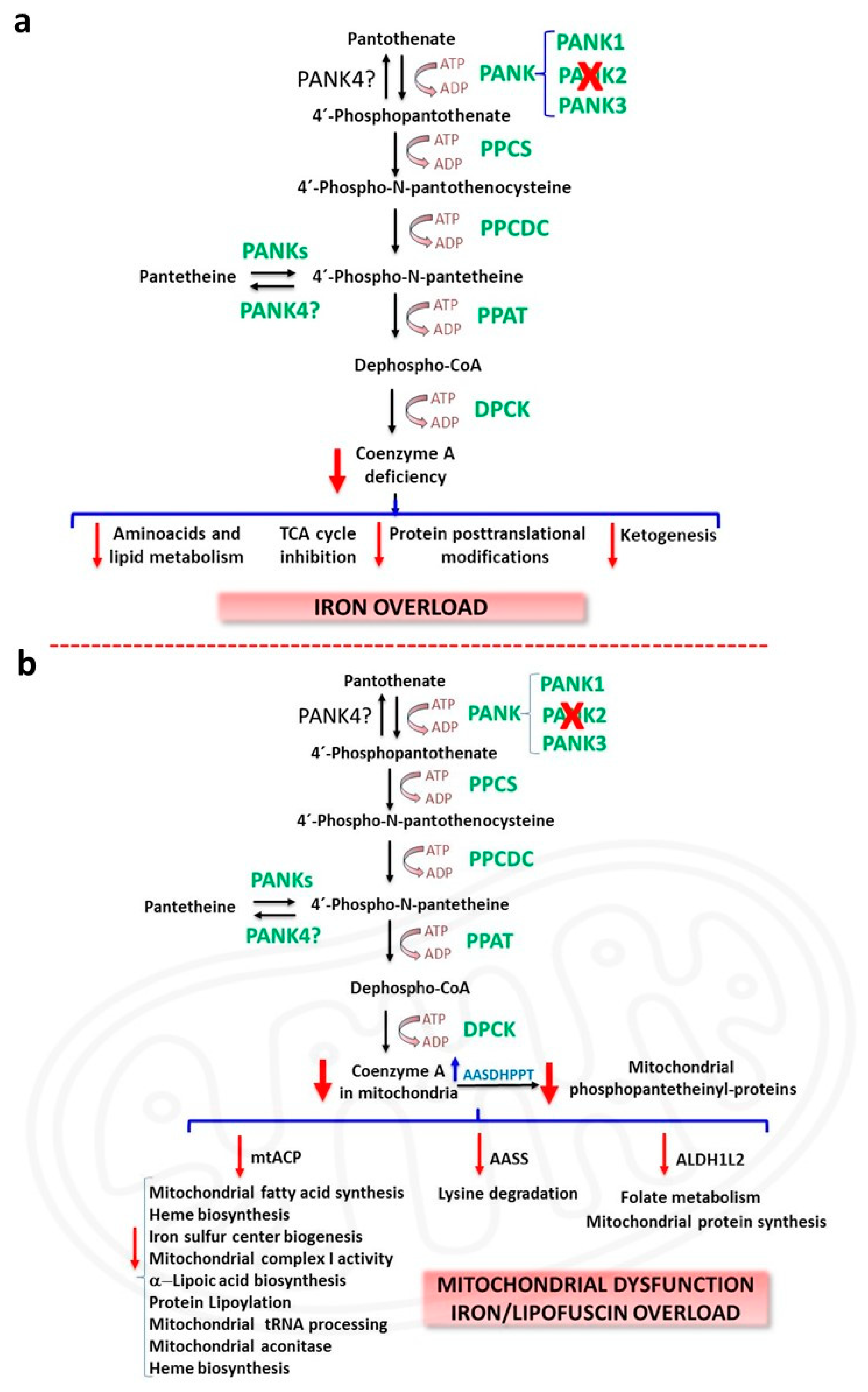
2.1. CoA Deficiency in PKAN
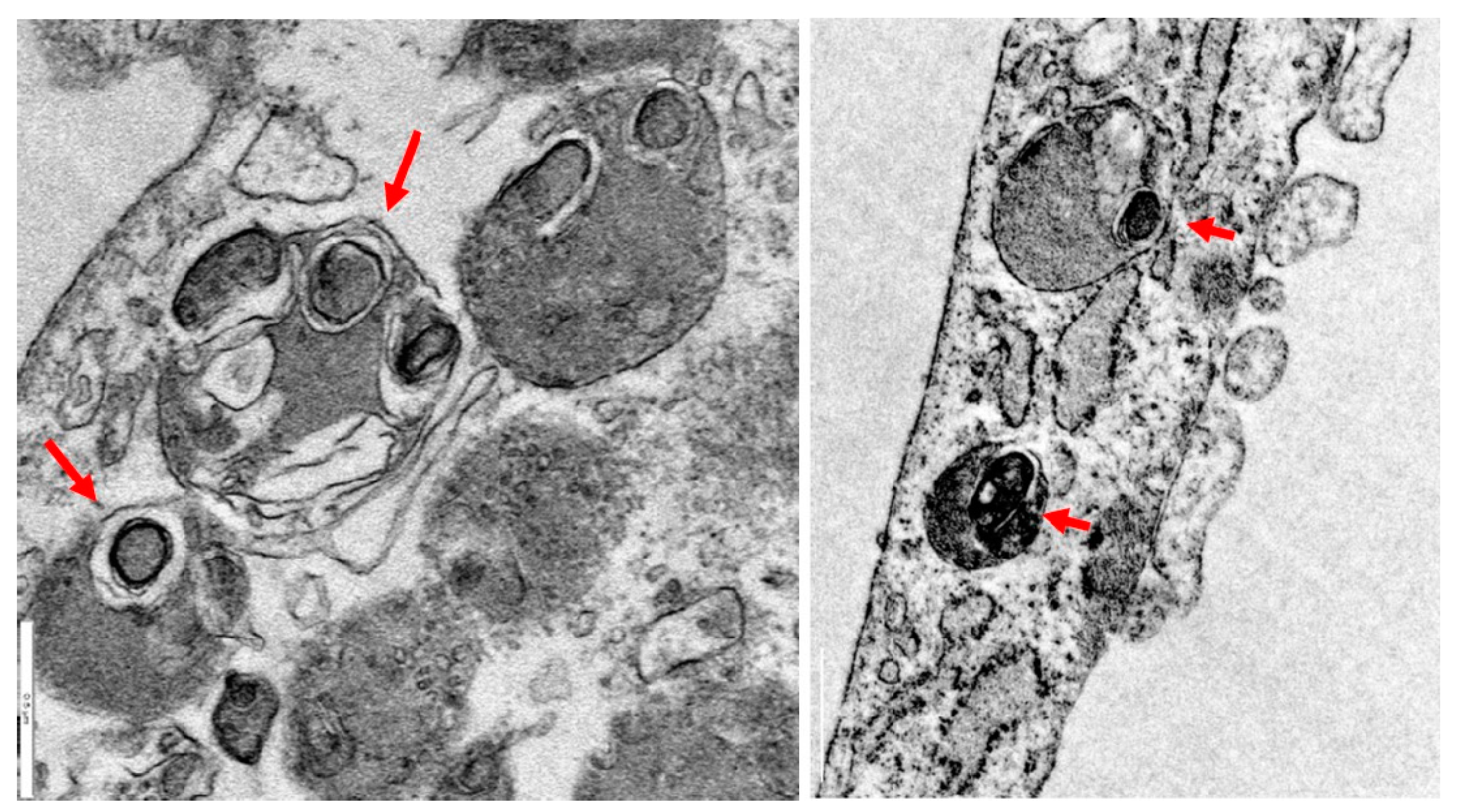
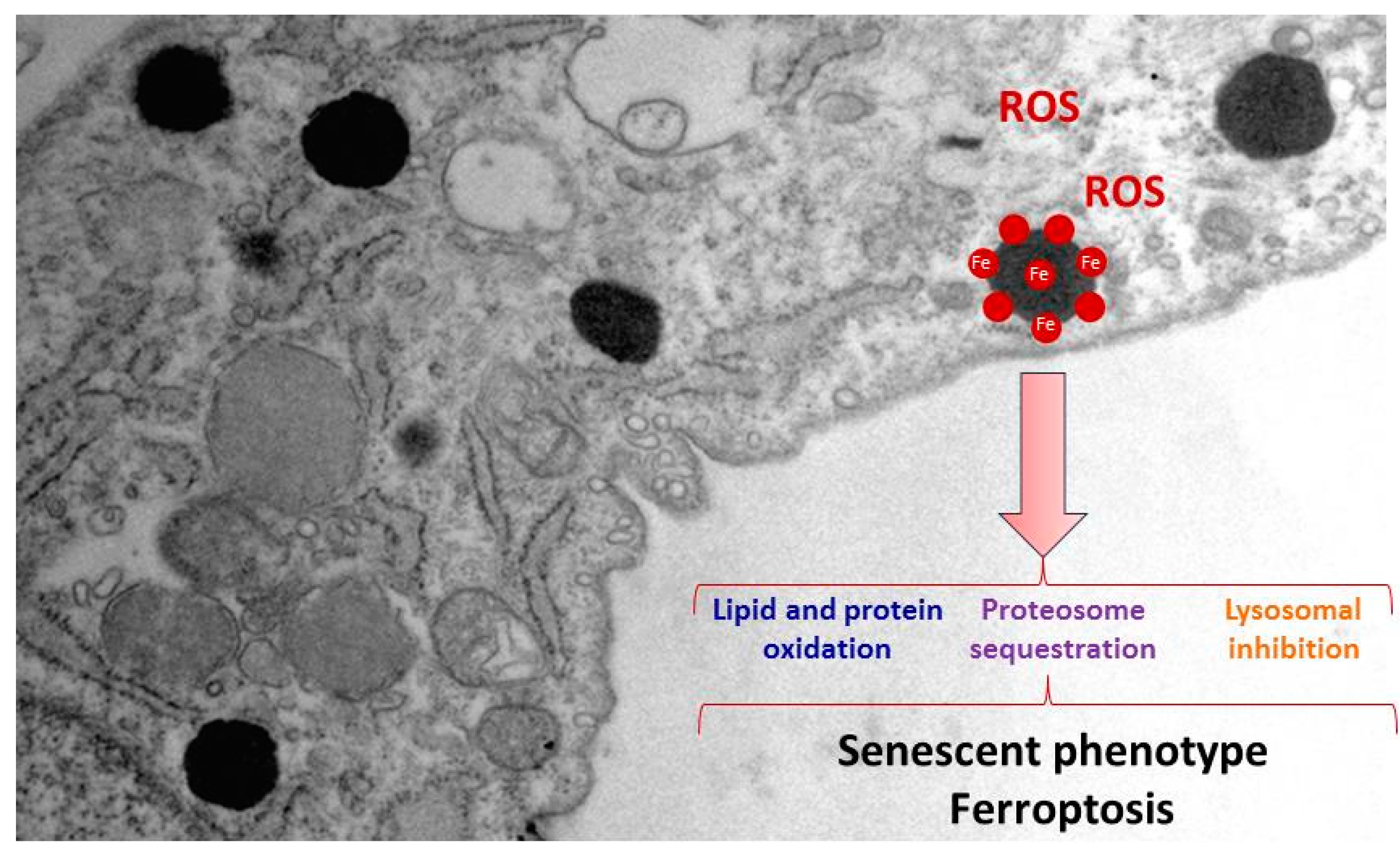
2.2. Iron/Lipofuscin Accumulation in PKAN
3. PKAN Disease Modeling
3.1. Modeling PKAN Disease in Biological Models
3.2. Patient-Derived Cellular Models
3.3. Induced Neurons
3.4. Alterations in Cellular Models of PKAN
4. Therapeutic Strategies for PKAN
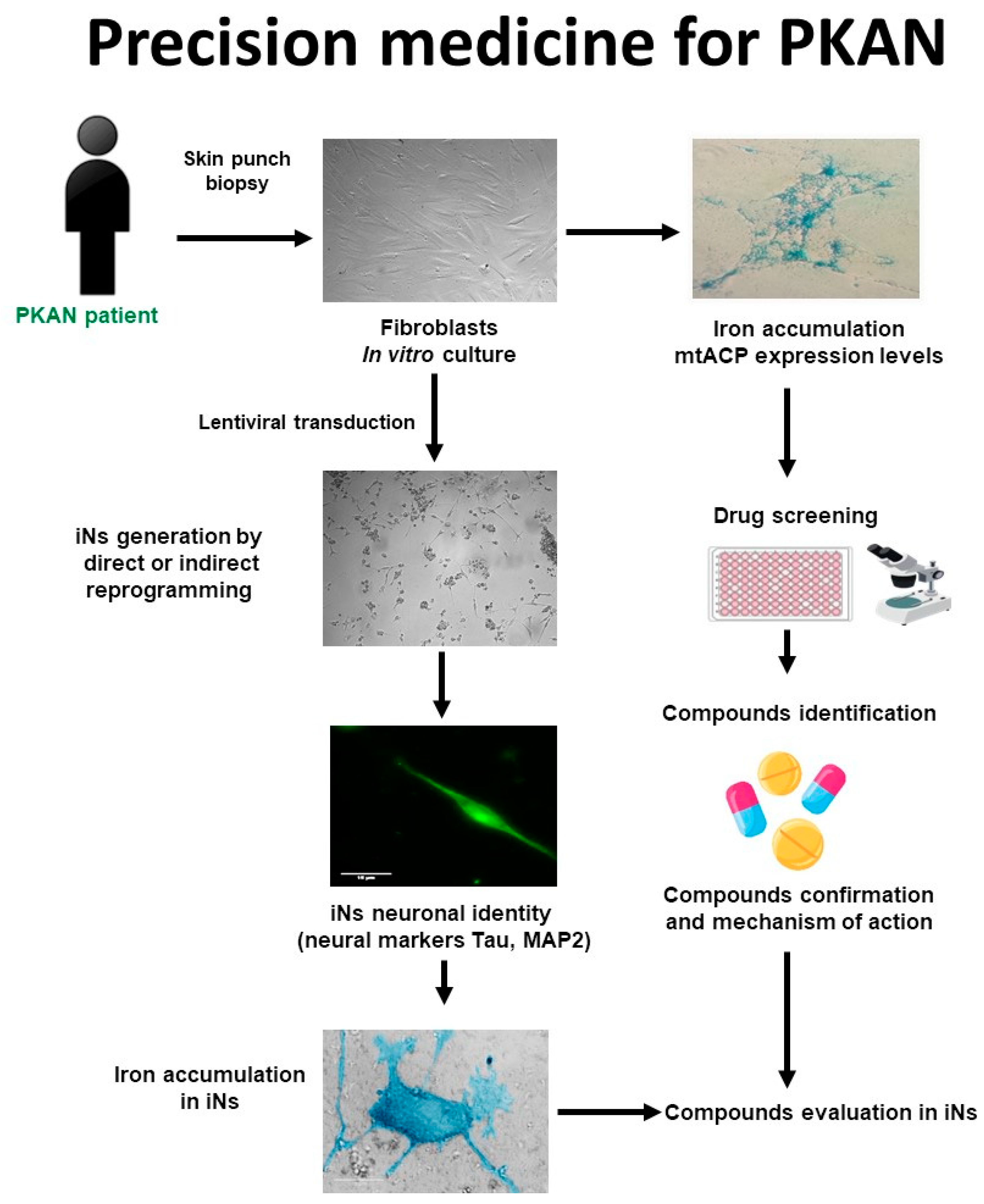
4.1. Strategy for Finding Alternative Treatments for PKAN Using Patient-Derived Cellular Models
4.2. Precision Medicine in PKAN
5. Polytarget Therapy in PKAN
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogarth, P.; Kurian, M.A.; Gregory, A.; Csanyi, B.; Zagustin, T.; Kmiec, T.; Wood, P.; Klucken, A.; Scalise, N.; Sofia, F.; et al. Consensus clinical management guideline for pantothenate kinase-associated neurodegeneration (PKAN). Mol. Genet. Metab. 2017, 120, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Polster, B.J.; Hayflick, S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009, 46, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J. Neurodegeneration with brain iron accumulation: From genes to pathogenesis. Semin. Pediatr. Neurol. 2006, 13, 182–185. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.; Gitschier, J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N. Engl. J. Med. 2003, 348, 33–40. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.M.; Lykidis, A.; Rock, C.O.; Jackowski, S. Localization and regulation of mouse pantothenate kinase 2. FEBS Lett. 2007, 581, 4639–4644. [Google Scholar] [CrossRef]
- Jackowski, S.; Rock, C.O. CoA regulation and metabolic control. Biochem 2015, 37, 4–8. [Google Scholar] [CrossRef]
- Huang, L.; Khusnutdinova, A.; Nocek, B.; Brown, G.; Xu, X.; Cui, H.; Petit, P.; Flick, R.; Zallot, R.; Balmant, K.; et al. A family of metal-dependent phosphatases implicated in metabolite damage-control. Nat. Chem. Biol. 2016, 12, 621–627. [Google Scholar] [CrossRef]
- Yao, J.; Subramanian, C.; Rock, C.O.; Jackowski, S. Human pantothenate kinase 4 is a pseudo-pantothenate kinase. Protein Sci. 2019, 28, 1031–1047. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Cavestro, C.; Diodato, D.; Tiranti, V.; Di Meo, I. Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments. Int. J. Mol. Sci. 2023, 24, 5951. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Hayflick, S.J. Neurodegeneration with brain iron accumulation. Folia Neuropathol. 2005, 43, 286–296. [Google Scholar] [PubMed]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar] [CrossRef]
- Yu, Y.; Moretti, I.F.; Grzeschik, N.A.; Sibon, O.C.M.; Schepers, H. Coenzyme A levels influence protein acetylation, CoAlation and 4’-phosphopantetheinylation: Expanding the impact of a metabolic nexus molecule. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118965. [Google Scholar] [CrossRef]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef]
- Garcia, M.; Leonardi, R.; Zhang, Y.M.; Rehg, J.E.; Jackowski, S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS ONE 2012, 7, e40871. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef]
- Beld, J.; Sonnenschein, E.C.; Vickery, C.R.; Noel, J.P.; Burkart, M.D. The phosphopantetheinyl transferases: Catalysis of a post-translational modification crucial for life. Nat. Prod. Rep. 2014, 31, 61–108. [Google Scholar] [CrossRef]
- Joshi, A.K.; Zhang, L.; Rangan, V.S.; Smith, S. Cloning, expression, and characterization of a human 4′-phosphopantetheinyl transferase with broad substrate specificity. J. Biol. Chem. 2003, 278, 33142–33149. [Google Scholar] [CrossRef] [PubMed]
- Bunkoczi, G.; Pasta, S.; Joshi, A.; Wu, X.; Kavanagh, K.L.; Smith, S.; Oppermann, U. Mechanism and substrate recognition of human holo ACP synthase. Chem. Biol. 2007, 14, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Witkowski, A.; Joshi, A.K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003, 42, 289–317. [Google Scholar] [CrossRef]
- Smith, S. The animal fatty acid synthase: One gene, one polypeptide, seven enzymes. FASEB J. 1994, 8, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, U.; Zensen, R.; Rohlen, D.; Jahnke, U.; Weiss, H. The acyl-carrier protein in Neurospora crassa mitochondria is a subunit of NADH:ubiquinone reductase (complex I). Eur. J. Biochem. 1991, 200, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Joshi, A.K.; Smith, S. Cloning, expression, characterization, and interaction of two components of a human mitochondrial fatty acid synthase. Malonyltransferase and acyl carrier protein. J. Biol. Chem. 2003, 278, 40067–40074. [Google Scholar] [CrossRef]
- Schneider, R.; Brors, B.; Massow, M.; Weiss, H. Mitochondrial fatty acid synthesis: A relic of endosymbiontic origin and a specialized means for respiration. FEBS Lett. 1997, 407, 249–252. [Google Scholar] [CrossRef]
- Schneider, R.; Massow, M.; Lisowsky, T.; Weiss, H. Different respiratory-defective phenotypes of Neurospora crassa and Saccharomyces cerevisiae after inactivation of the gene encoding the mitochondrial acyl carrier protein. Curr. Genet. 1995, 29, 10–17. [Google Scholar] [CrossRef]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial fatty acid synthesis type II: More than just fatty acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J. Neurosci. 2005, 25, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cordoba, M.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; Sanchez-Alcazar, J.A. Down regulation of the expression of mitochondrial phosphopantetheinyl-proteins in pantothenate kinase-associated neurodegeneration: Pathophysiological consequences and therapeutic perspectives. Orphanet J. Rare Dis. 2021, 16, 201. [Google Scholar] [CrossRef] [PubMed]
- Cronan, J.E. Assembly of Lipoic Acid on Its Cognate Enzymes: An Extraordinary and Essential Biosynthetic Pathway. Microbiol. Mol. Biol. Rev. 2016, 80, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Feichtinger, R.G.; Tort, F.; Ribes, A.; Sperl, W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 2014, 37, 553–563. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Nowinski, S.M.; Clowers, K.J.; Jeong, M.Y.; Ouyang, Y.; Berg, J.A.; Gygi, J.P.; Gygi, S.P.; Winge, D.R.; Rutter, J. ACP Acylation Is an Acetyl-CoA-Dependent Modification Required for Electron Transport Chain Assembly. Mol. Cell 2018, 71, 567–580.e564. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef]
- Cory, S.A.; Van Vranken, J.G.; Brignole, E.J.; Patra, S.; Winge, D.R.; Drennan, C.L.; Rutter, J.; Barondeau, D.P. Structure of human Fe-S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP-ISD11 interactions. Proc. Natl. Acad. Sci. USA 2017, 114, E5325–E5334. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; van der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4′-Phosphopantetheine corrects CoA, iron, and dopamine metabolic defects in mammalian models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Jeong, M.Y.; Wei, P.; Chen, Y.C.; Gygi, S.P.; Winge, D.R.; Rutter, J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 2016, 5, e17828. [Google Scholar] [CrossRef]
- Chen, O.S.; Hemenway, S.; Kaplan, J. Inhibition of Fe-S cluster biosynthesis decreases mitochondrial iron export: Evidence that Yfh1p affects Fe-S cluster synthesis. Proc. Natl. Acad. Sci. USA 2002, 99, 12321–12326. [Google Scholar] [CrossRef]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef]
- Nunez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. BioMetals 2012, 25, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Chen, J.; Chai, Z.F.; Hu, Y. The neurotoxicity of iron, copper and cobalt in Parkinson’s disease through ROS-mediated mechanisms. BioMetals 2016, 29, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Salvador, G.A.; Uranga, R.M.; Giusto, N.M. Iron and mechanisms of neurotoxicity. Int. J. Alzheimer’s Dis. 2010, 2011, 720658. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Kruer, M.C. The neuropathology of neurodegeneration with brain iron accumulation. Int. Rev. Neurobiol. 2013, 110, 165–194. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Villanueva-Paz, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Abril-Jaramillo, J.; Vintimilla-Tosi, A.B.; Sanchez-Alcazar, J.A. Precision medicine in pantothenate kinase-associated neurodegeneration. Neural Regen. Res. 2019, 14, 1177–1185. [Google Scholar] [CrossRef]
- Matsunaga, T.; Kotamraju, S.; Kalivendi, S.V.; Dhanasekaran, A.; Joseph, J.; Kalyanaraman, B. Ceramide-induced intracellular oxidant formation, iron signaling, and apoptosis in endothelial cells: Protective role of endogenous nitric oxide. J. Biol. Chem. 2004, 279, 28614–28624. [Google Scholar] [CrossRef]
- Perry, T.L.; Norman, M.G.; Yong, V.W.; Whiting, S.; Crichton, J.U.; Hansen, S.; Kish, S.J. Hallervorden-Spatz disease: Cysteine accumulation and cysteine dioxygenase deficiency in the globus pallidus. Ann. Neurol. 1985, 18, 482–489. [Google Scholar] [CrossRef]
- Biosa, A.; Arduini, I.; Soriano, M.E.; Giorgio, V.; Bernardi, P.; Bisaglia, M.; Bubacco, L. Dopamine Oxidation Products as Mitochondrial Endotoxins, a Potential Molecular Mechanism for Preferential Neurodegeneration in Parkinson’s Disease. ACS Chem. Neurosci. 2018, 9, 2849–2858. [Google Scholar] [CrossRef]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef]
- Napolitano, A.; Crescenzi, O.; Pezzella, A.; Prota, G. Generation of the neurotoxin 6-hydroxydopamine by peroxidase/H2O2 oxidation of dopamine. J. Med. Chem. 1995, 38, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yagnik, G.; Jiang, D.; Shi, S.; Chang, P.; Zhou, F. Separation of intermediates of iron-catalyzed dopamine oxidation reactions using reversed-phase ion-pairing chromatography coupled in tandem with UV-visible and ESI-MS detections. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 911, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, A.; d’Ischia, M.; Napolitano, A.; Misuraca, G.; Prota, G. Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: A possible contributory mechanism for neuronal degeneration in Parkinson’s disease. J. Med. Chem. 1997, 40, 2211–2216. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dryhurst, G. Irreversible inhibition of mitochondrial complex I by 7-(2-aminoethyl)-3,4-dihydro-5-hydroxy-2H-1,4-benzothiazine-3-carboxyli c acid (DHBT-1): A putative nigral endotoxin of relevance to Parkinson’s disease. J. Neurochem. 1997, 69, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for Parkinson’s disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Zhang, F.; Dryhurst, G. Effects of L-cysteine on the oxidation chemistry of dopamine: New reaction pathways of potential relevance to idiopathic Parkinson’s disease. J. Med. Chem. 1994, 37, 1084–1098. [Google Scholar] [CrossRef]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar] [CrossRef]
- Jolly, R.D.; Douglas, B.V.; Davey, P.M.; Roiri, J.E. Lipofuscin in bovine muscle and brain: A model for studying age pigment. Gerontology 1995, 41 (Suppl. S2), 283–295. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, distribution, and metabolic consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef]
- Konig, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.L.; Grune, T.; Hohn, A. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017, 11, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Frolova, M.S.; Surin, A.M.; Braslavski, A.V.; Vekshin, N.L. Degradation of Mitochondria to Lipofuscin upon Heating and Illumination. Biofizika 2015, 60, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Salmonowicz, H.; Passos, J.F. Detecting senescence: A new method for an old pigment. Aging Cell 2017, 16, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Powell, S.R.; Wang, P.; Divald, A.; Teichberg, S.; Haridas, V.; McCloskey, T.W.; Davies, K.J.; Katzeff, H. Aggregates of oxidized proteins (lipofuscin) induce apoptosis through proteasome inhibition and dysregulation of proapoptotic proteins. Free Radic. Biol. Med. 2005, 38, 1093–1101. [Google Scholar] [CrossRef]
- Hohn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Pan, C.; Banerjee, K.; Lehmann, G.L.; Almeida, D.; Hajjar, K.A.; Benedicto, I.; Jiang, Z.; Radu, R.A.; Thompson, D.H.; Rodriguez-Boulan, E.; et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc. Natl. Acad. Sci. USA 2021, 118, e2100122118. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Privitera, D.; Guaraldo, M.; Rovelli, E.; Barzaghi, C.; Garavaglia, B.; Santambrogio, P.; Cozzi, A.; Levi, S. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum. Mol. Genet. 2012, 21, 4049–4059. [Google Scholar] [CrossRef]
- Luckenbach, M.W.; Green, W.R.; Miller, N.R.; Moser, H.W.; Clark, A.W.; Tennekoon, G. Ocular clinicopathologic correlation of Hallervorden-Spatz syndrome with acanthocytosis and pigmentary retinopathy. Am. J. Ophthalmol. 1983, 95, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Srinivasan, V.; Muhlenhoff, U. The role of mitochondria in cytosolic-nuclear iron-sulfur protein biogenesis and in cellular iron regulation. Curr. Opin. Microbiol. 2014, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef]
- Poli, M.; Derosas, M.; Luscieti, S.; Cavadini, P.; Campanella, A.; Verardi, R.; Finazzi, D.; Arosio, P. Pantothenate kinase-2 (Pank2) silencing causes cell growth reduction, cell-specific ferroportin upregulation and iron deregulation. Neurobiol. Dis. 2010, 39, 204–210. [Google Scholar] [CrossRef]
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef]
- Bosveld, F.; Rana, A.; van der Wouden, P.E.; Lemstra, W.; Ritsema, M.; Kampinga, H.H.; Sibon, O.C. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008, 17, 2058–2069. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef]
- Chiu, J.E.; Thekkiniath, J.; Mehta, S.; Muller, C.; Bracher, F.; Ben Mamoun, C. The yeast pantothenate kinase Cab1 is a master regulator of sterol metabolism and of susceptibility to ergosterol biosynthesis inhibitors. J. Biol. Chem. 2019, 294, 14757–14767. [Google Scholar] [CrossRef]
- Khatri, D.; Mignani, L.; Zizioli, D.; Ritelli, M.; Monti, E.; Finazzi, D. Abnormal Vasculature Development in Zebrafish Embryos with Reduced Expression of Pantothenate Kinase 2 Gene. Bull. Exp. Biol. Med. 2020, 170, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.J.; Kayser, O.; Sibon, O.C. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Yao, J.; Frank, M.W.; Rock, C.O.; Jackowski, S. A pantothenate kinase-deficient mouse model reveals a gene expression program associated with brain coenzyme a reduction. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165663. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, C.; Lv, S.; Zhou, B. Pantothenate kinase-associated neurodegeneration: Insights from a Drosophila model. Hum. Mol. Genet. 2009, 18, 3659–3672. [Google Scholar] [CrossRef]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef]
- Johnson, M.A.; Kuo, Y.M.; Westaway, S.K.; Parker, S.M.; Ching, K.H.; Gitschier, J.; Hayflick, S.J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Hayflick, S.J.; Gitschier, J. Deprivation of pantothenic acid elicits a movement disorder and azoospermia in a mouse model of pantothenate kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2007, 30, 310–317. [Google Scholar] [CrossRef]
- Munshi, M.I.; Yao, S.J.; Ben Mamoun, C. Redesigning therapies for pantothenate kinase-associated neurodegeneration. J. Biol. Chem. 2022, 298, 101577. [Google Scholar] [CrossRef] [PubMed]
- Afshar, K.; Gonczy, P.; DiNardo, S.; Wasserman, S.A. fumble encodes a pantothenate kinase homolog required for proper mitosis and meiosis in Drosophila melanogaster. Genetics 2001, 157, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Olzhausen, J.; Schubbe, S.; Schuller, H.J. Genetic analysis of coenzyme A biosynthesis in the yeast Saccharomyces cerevisiae: Identification of a conditional mutation in the pantothenate kinase gene CAB1. Curr. Genet. 2009, 55, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli Berti, C.; Gilea, A.I.; De Gregorio, M.A.; Goffrini, P. Exploring Yeast as a Study Model of Pantothenate Kinase-Associated Neurodegeneration and for the Identification of Therapeutic Compounds. Int. J. Mol. Sci. 2020, 22, 293. [Google Scholar] [CrossRef]
- Connolly, G.P. Fibroblast models of neurological disorders: Fluorescence measurement studies. Trends Pharmacol. Sci. 1998, 19, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Colman, A.; Dreesen, O. Pluripotent stem cells and disease modeling. Cell Stem Cell 2009, 5, 244–247. [Google Scholar] [CrossRef]
- Lee, C.T.; Bendriem, R.M.; Wu, W.W.; Shen, R.F. 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef]
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef]
- Takeda, Y.; Harada, Y.; Yoshikawa, T.; Dai, P. Chemical compound-based direct reprogramming for future clinical applications. Biosci. Rep. 2018, 38, BSR20171650. [Google Scholar] [CrossRef]
- Ladewig, J.; Koch, P.; Brustle, O. Leveling Waddington: The emergence of direct programming and the loss of cell fate hierarchies. Nat. Rev. 2013, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Sudhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Lau, S.; Brattas, P.L.; Rylander Ottosson, D.; Pircs, K.; Grassi, D.A.; Collins, L.M.; Vuono, R.; Andersson Sjoland, A.; Westergren-Thorsson, G.; et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017, 9, 1117–1131. [Google Scholar] [CrossRef]
- Ladewig, J.; Mertens, J.; Kesavan, J.; Doerr, J.; Poppe, D.; Glaue, F.; Herms, S.; Wernet, P.; Kogler, G.; Muller, F.J.; et al. Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 2012, 9, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Ek, F.; Lang, S.; Soneji, S.; Olsson, R.; Parmar, M. Small molecules increase direct neural conversion of human fibroblasts. Sci. Rep. 2016, 6, 38290. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5, e18648. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Torper, O.; Pfisterer, U.; Wolf, D.A.; Pereira, M.; Lau, S.; Jakobsson, J.; Björklund, A.; Grealish, S.; Parmar, M. Generation of induced neurons via direct conversion in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 7038–7043. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Masserdotti, G.; Gascon, S.; Gotz, M. Direct neuronal reprogramming: Learning from and for development. Development 2016, 143, 2494–2510. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, M.; Santambrogio, P.; Racchetti, G.; Cozzi, A.; Di Meo, I.; Tiranti, V.; Levi, S. PKAN hiPS-Derived Astrocytes Show Impairment of Endosomal Trafficking: A Potential Mechanism Underlying Iron Accumulation. Front. Cell. Neurosci. 2022, 16, 878103. [Google Scholar] [CrossRef]
- Santambrogio, P.; Ripamonti, M.; Cozzi, A.; Raimondi, M.; Cavestro, C.; Di Meo, I.; Rubio, A.; Taverna, S.; Tiranti, V.; Levi, S. Massive iron accumulation in PKAN-derived neurons and astrocytes: Light on the human pathological phenotype. Cell Death Dis. 2022, 13, 185. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Povea-Cabello, S.; Talaveron-Rey, M.; Villanueva-Paz, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Legault, E.M.; Nilsson, F.; Pircs, K.; Bouquety, J.; Petit, F.; Shrigley, S.; Birtele, M.; Pereira, M.; Storm, P.; et al. Age-related pathological impairments in directly reprogrammed dopaminergic neurons derived from patients with idiopathic Parkinson’s disease. Stem Cell Rep. 2022, 17, 2203–2219. [Google Scholar] [CrossRef]
- Pircs, K.; Drouin-Ouellet, J.; Horvath, V.; Gil, J.; Rezeli, M.; Garza, R.; Grassi, D.A.; Sharma, Y.; St-Amour, I.; Harris, K.; et al. Distinct subcellular autophagy impairments in induced neurons from patients with Huntington’s disease. Brain 2022, 145, 3035–3057. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Alvarez-Cordoba, M.; de la Mata, M.; Talaveron-Rey, M.; Jackson, S.; Sanchez-Alcazar, J.A. Pathophysiological characterization of MERRF patient-specific induced neurons generated by direct reprogramming. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 861–881. [Google Scholar] [CrossRef]
- Lin, D.S.; Huang, Y.W.; Ho, C.S.; Huang, T.S.; Lee, T.H.; Wu, T.Y.; Huang, Z.D.; Wang, T.J. Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons. Cells 2022, 12, 15. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Pinero-Perez, R.; et al. Therapeutic approach with commercial supplements for pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2022, 17, 311. [Google Scholar] [CrossRef]
- Talaveron-Rey, M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Gomez-Fernandez, D.; Romero-Gonzalez, A.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Cilleros-Holgado, P.; et al. Alpha-lipoic acid supplementation corrects pathological alterations in cellular models of pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2023, 18, 80. [Google Scholar] [CrossRef]
- Thakur, N.; Klopstock, T.; Jackowski, S.; Kuscer, E.; Tricta, F.; Videnovic, A.; Jinnah, H.A. Rational Design of Novel Therapies for Pantothenate Kinase-Associated Neurodegeneration. Mov. Disord. 2021, 36, 2005–2016. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Jeong, S.Y.; Sibon, O.C.M. PKAN pathogenesis and treatment. Mol. Genet. Metab. 2022, 137, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wan, Z.; Tang, Y.; Xu, J.; Laboret, B.; Nallamothu, S.; Yang, C.; Liu, B.; Lu, R.O.; Lu, B.; et al. Pantothenate kinase 2 interacts with PINK1 to regulate mitochondrial quality control via acetyl-CoA metabolism. Nat. Commun. 2022, 13, 2412. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef] [PubMed]
- Goldin, E.; Zheng, W.; Motabar, O.; Southall, N.; Choi, J.H.; Marugan, J.; Austin, C.P.; Sidransky, E. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS ONE 2012, 7, e29861. [Google Scholar] [CrossRef]
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–7176. [Google Scholar] [CrossRef]
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the responsiveness to pharmacological chaperones: Lysosomal human alpha-galactosidase, a case of study. Orphanet J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Mahuran, D. Diltiazem, a L-type Ca(2+) channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol. Genet. Metab. 2009, 96, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.; Hamilton, J.W. Altered biogenesis of deltaF508-CFTR following treatment with doxorubicin. Cell Physiol. Biochem. 2007, 20, 465–472. [Google Scholar] [CrossRef]
- Porto, C.; Ferrara, M.C.; Meli, M.; Acampora, E.; Avolio, V.; Rosa, M.; Cobucci-Ponzano, B.; Colombo, G.; Moracci, M.; Andria, G.; et al. Pharmacological enhancement of alpha-glucosidase by the allosteric chaperone N-acetylcysteine. Mol. Ther. 2012, 20, 2201–2211. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef]
- Martin, G.M.; Chen, P.C.; Devaraneni, P.; Shyng, S.L. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front. Physiol. 2013, 4, 386. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef]
- Ishihara, K.; Okuyama, S.; Kumano, S.; Iida, K.; Hamana, H.; Murakoshi, M.; Kobayashi, T.; Usami, S.; Ikeda, K.; Haga, Y.; et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear. Res. 2010, 270, 110–118. [Google Scholar] [CrossRef]
- Strafella, C.; Caputo, V.; Galota, M.R.; Zampatti, S.; Marella, G.; Mauriello, S.; Cascella, R.; Giardina, E. Application of Precision Medicine in Neurodegenerative Diseases. Front. Neurol. 2018, 9, 701. [Google Scholar] [CrossRef]
- Tan, L.; Jiang, T.; Tan, L.; Yu, J.T. Toward precision medicine in neurological diseases. Ann. Transl. Med. 2016, 4, 104. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Banning, A.; Tikkanen, R. Development of precision therapies for rare inborn errors of metabolism: Functional investigations in cell culture models. J. Inherit. Metab. Dis. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Schee Genannt Halfmann, S.; Mahlmann, L.; Leyens, L.; Reumann, M.; Brand, A. Personalized Medicine: What’s in it for Rare Diseases? Adv. Exp. Med. Biol. 2017, 1031, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Frasier, M.; Fiske, B.K.; Sherer, T.B. Precision medicine for Parkinson’s disease: The subtyping challenge. Front. Aging Neurosci. 2022, 14, 1064057. [Google Scholar] [CrossRef]
- Subramanian, C.; Yun, M.K.; Yao, J.; Sharma, L.K.; Lee, R.E.; White, S.W.; Jackowski, S.; Rock, C.O. Allosteric Regulation of Mammalian Pantothenate Kinase. J. Biol. Chem. 2016, 291, 22302–22314. [Google Scholar] [CrossRef]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme A metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef]
- Zano, S.P.; Pate, C.; Frank, M.; Rock, C.O.; Jackowski, S. Correction of a genetic deficiency in pantothenate kinase 1 using phosphopantothenate replacement therapy. Mol. Genet. Metab. 2015, 116, 281–288. [Google Scholar] [CrossRef]
- Balibar, C.J.; Hollis-Symynkywicz, M.F.; Tao, J. Pantethine rescues phosphopantothenoylcysteine synthetase and phosphopantothenoylcysteine decarboxylase deficiency in Escherichia coli but not in Pseudomonas aeruginosa. J. Bacteriol. 2011, 193, 3304–3312. [Google Scholar] [CrossRef]
- Evans, M.; Rumberger, J.A.; Azumano, I.; Napolitano, J.J.; Citrolo, D.; Kamiya, T. Pantethine, a derivative of vitamin B5, favorably alters total, LDL and non-HDL cholesterol in low to moderate cardiovascular risk subjects eligible for statin therapy: A triple-blinded placebo and diet-controlled investigation. Vasc. Health Risk Manag. 2014, 10, 89–100. [Google Scholar] [CrossRef]
- Chang, X.; Zhang, J.; Jiang, Y.; Yao, B.; Wang, J.; Wu, Y. Pilot trial on the efficacy and safety of pantethine in children with pantothenate kinase-associated neurodegeneration: A single-arm, open-label study. Orphanet J. Rare Dis. 2020, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998, 39, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016, 7, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Talaveron-Rey, M.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Pinero-Perez, R.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196–1202. [Google Scholar] [CrossRef]
- Burton, G.W.; Joyce, A.; Ingold, K.U. First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 1982, 2, 327. [Google Scholar] [CrossRef]
- Traber, M.G. Vitamin E: Necessary nutrient for neural development and cognitive function. Proc. Nutr. Soc. 2021, 80, 319–326. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Argellati, F.; Pronzato, M.A.; Domenicotti, C. Vitamin E and neurodegenerative diseases. Mol. Asp. Med. 2007, 28, 591–606. [Google Scholar] [CrossRef]
- Ulatowski, L.M.; Manor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Vento, M.; Baquero, M.; Chafer-Pericas, C. Lipid peroxidation in neurodegeneration. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 497, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Espinos, C.; Galindo, M.I.; Garcia-Gimeno, M.A.; Ibanez-Cabellos, J.S.; Martinez-Rubio, D.; Millan, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Heshmati, J.; Morvaridzadeh, M.; Maroufizadeh, S.; Akbari, A.; Yavari, M.; Amirinejad, A.; Maleki-Hajiagha, A.; Sepidarkish, M. Omega-3 fatty acids supplementation and oxidative stress parameters: A systematic review and meta-analysis of clinical trials. Pharmacol. Res. 2019, 149, 104462. [Google Scholar] [CrossRef]
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Cole, G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 fatty acids in neurodegenerative diseases: Focus on mitochondria. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 105–114. [Google Scholar] [CrossRef]
- Da Silva, E.P., Jr.; Nachbar, R.T.; Levada-Pires, A.C.; Hirabara, S.M.; Lambertucci, R.H. Omega-3 fatty acids differentially modulate enzymatic anti-oxidant systems in skeletal muscle cells. Cell Stress. Chaperones 2016, 21, 87–95. [Google Scholar] [CrossRef]
- Calviello, G.; Su, H.M.; Weylandt, K.H.; Fasano, E.; Serini, S.; Cittadini, A. Experimental evidence of omega-3 polyunsaturated fatty acid modulation of inflammatory cytokines and bioactive lipid mediators: Their potential role in inflammatory, neurodegenerative, and neoplastic diseases. Biomed. Res. Int. 2013, 2013, 743171. [Google Scholar] [CrossRef]
- Cardoso, C.; Afonso, C.; Bandarra, N.M. Dietary DHA and health: Cognitive function ageing. Nutr. Res. Rev. 2016, 29, 281–294. [Google Scholar] [CrossRef]
- Moore, K.; Hughes, C.F.; Ward, M.; Hoey, L.; McNulty, H. Diet, nutrition and the ageing brain: Current evidence and new directions. Proc. Nutr. Soc. 2018, 77, 152–163. [Google Scholar] [CrossRef]
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Syndr. 2014, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Berkay Yilmaz, Y.; Antika, G.; Boyunegmez Tumer, T.; Fawzi Mahomoodally, M.; Lobine, D.; Akram, M.; Riaz, M.; Capanoglu, E.; Sharopov, F.; et al. Insights on the Use of alpha-Lipoic Acid for Therapeutic Purposes. Biomolecules 2019, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef]
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.O. Lipoic Acid: Its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 2015, 15, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959. [Google Scholar] [CrossRef]
- Molz, P.; Schroder, N. Potential Therapeutic Effects of Lipoic Acid on Memory Deficits Related to Aging and Neurodegeneration. Front. Pharmacol. 2017, 8, 849. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef]
- Perham, R.N. Swinging arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annu. Rev. Biochem. 2000, 69, 961–1004. [Google Scholar] [CrossRef]
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef]
- Rezaei Zonooz, S.; Hasani, M.; Morvaridzadeh, M.; Beatriz Pizarro, A.; Heydari, H.; Yosaee, S.; Rezamand, G.; Heshmati, J. Effect of alpha-lipoic acid on oxidative stress parameters: A systematic review and meta-analysis. J. Funct. Foods 2021, 87, 104774. [Google Scholar] [CrossRef]
- Suh, J.H.; Moreau, R.; Heath, S.H.; Hagen, T.M. Dietary supplementation with (R)-alpha-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 2005, 10, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; Di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. alpha-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20, 609. [Google Scholar] [CrossRef] [PubMed]
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2021, 32, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Modanloo, M.; Shokrzadeh, M. Analyzing Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis: Potential Role of L-carnitine. Iran. J. Kidney Dis. 2019, 13, 74–86. [Google Scholar]
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000, 468, 1–5. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I.P. Mitochondrial Dysfunction and Neurodegenerative Disorders: Role of Nutritional Supplementation. Int. J. Mol. Sci. 2022, 23, 2603. [Google Scholar] [CrossRef]
- Lonsdale, D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid. Based Complement. Altern. Med. 2006, 3, 49–59. [Google Scholar] [CrossRef]
- Marsac, C.; Benelli, C.; Desguerre, I.; Diry, M.; Fouque, F.; De Meirleir, L.; Ponsot, G.; Seneca, S.; Poggi, F.; Saudubray, J.M.; et al. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1 alpha deficiency. Hum. Genet. 1997, 99, 785–792. [Google Scholar] [CrossRef]
- Naito, E.; Ito, M.; Takeda, E.; Yokota, I.; Yoshijima, S.; Kuroda, Y. Molecular analysis of abnormal pyruvate dehydrogenase in a patient with thiamine-responsive congenital lactic acidemia. Pediatr. Res. 1994, 36, 340–346. [Google Scholar] [CrossRef][Green Version]
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Chen, S.; Maehara, M.; Kuroda, Y. Concomitant administration of sodium dichloroacetate and thiamine in west syndrome caused by thiamine-responsive pyruvate dehydrogenase complex deficiency. J. Neurol. Sci. 1999, 171, 56–59. [Google Scholar] [CrossRef]
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Matsuda, J.; Ogawa, Y.; Kitamura, S.; Takada, E.; Horii, Y.; Kuroda, Y. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim. Biophys. Acta 2002, 1588, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Matsuda, J.; Osaka, H.; Kimura, S.; Kuroda, Y. Biochemical and molecular analysis of an X-linked case of Leigh syndrome associated with thiamin-responsive pyruvate dehydrogenase deficiency. J. Inherit. Metab. Dis. 1997, 20, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.J.; Yoon, M.Y.; Hayflick, S.J. Characterization of the human PANK2 promoter. Gene 2010, 465, 53–60. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.J. Multi-target therapeutics and new drug discovery. Yao Xue Xue Bao 2009, 44, 226–230. [Google Scholar]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78. [Google Scholar] [CrossRef]
- Borisy, A.A.; Elliott, P.J.; Hurst, N.W.; Lee, M.S.; Lehar, J.; Price, E.R.; Serbedzija, G.; Zimmermann, G.R.; Foley, M.A.; Stockwell, B.R.; et al. Systematic discovery of multicomponent therapeutics. Proc. Natl. Acad. Sci. USA 2003, 100, 7977–7982. [Google Scholar] [CrossRef] [PubMed]
- Butcher, E.C. Can cell systems biology rescue drug discovery? Nat. Rev. Drug Discov. 2005, 4, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed. Res. Int. 2020, 2020, 5120230. [Google Scholar] [CrossRef]
- Bawa, P.; Pradeep, P.; Kumar, P.; Choonara, Y.E.; Modi, G.; Pillay, V. Multi-target therapeutics for neuropsychiatric and neurodegenerative disorders. Drug Discov. Today 2016, 21, 1886–1914. [Google Scholar] [CrossRef]
- Jackowski, S. Proposed Therapies for Pantothenate-Kinase-Associated Neurodegeneration. J. Exp. Neurosci. 2019, 13, 1179069519851118. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Córdoba, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Romero-González, A.; et al. Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration. Pharmaceuticals 2023, 16, 1359. https://doi.org/10.3390/ph16101359
Álvarez-Córdoba M, Talaverón-Rey M, Povea-Cabello S, Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, Reche-López D, Munuera-Cabeza M, Suárez-Carrillo A, Romero-González A, et al. Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration. Pharmaceuticals. 2023; 16(10):1359. https://doi.org/10.3390/ph16101359
Chicago/Turabian StyleÁlvarez-Córdoba, Mónica, Marta Talaverón-Rey, Suleva Povea-Cabello, Paula Cilleros-Holgado, David Gómez-Fernández, Rocío Piñero-Pérez, Diana Reche-López, Manuel Munuera-Cabeza, Alejandra Suárez-Carrillo, Ana Romero-González, and et al. 2023. "Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration" Pharmaceuticals 16, no. 10: 1359. https://doi.org/10.3390/ph16101359
APA StyleÁlvarez-Córdoba, M., Talaverón-Rey, M., Povea-Cabello, S., Cilleros-Holgado, P., Gómez-Fernández, D., Piñero-Pérez, R., Reche-López, D., Munuera-Cabeza, M., Suárez-Carrillo, A., Romero-González, A., Romero-Domínguez, J. M., López-Cabrera, A., Armengol, J. Á., & Sánchez-Alcázar, J. A. (2023). Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration. Pharmaceuticals, 16(10), 1359. https://doi.org/10.3390/ph16101359