
Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors


 and
and
Abstract
1. Introduction
2. Results and Discussion
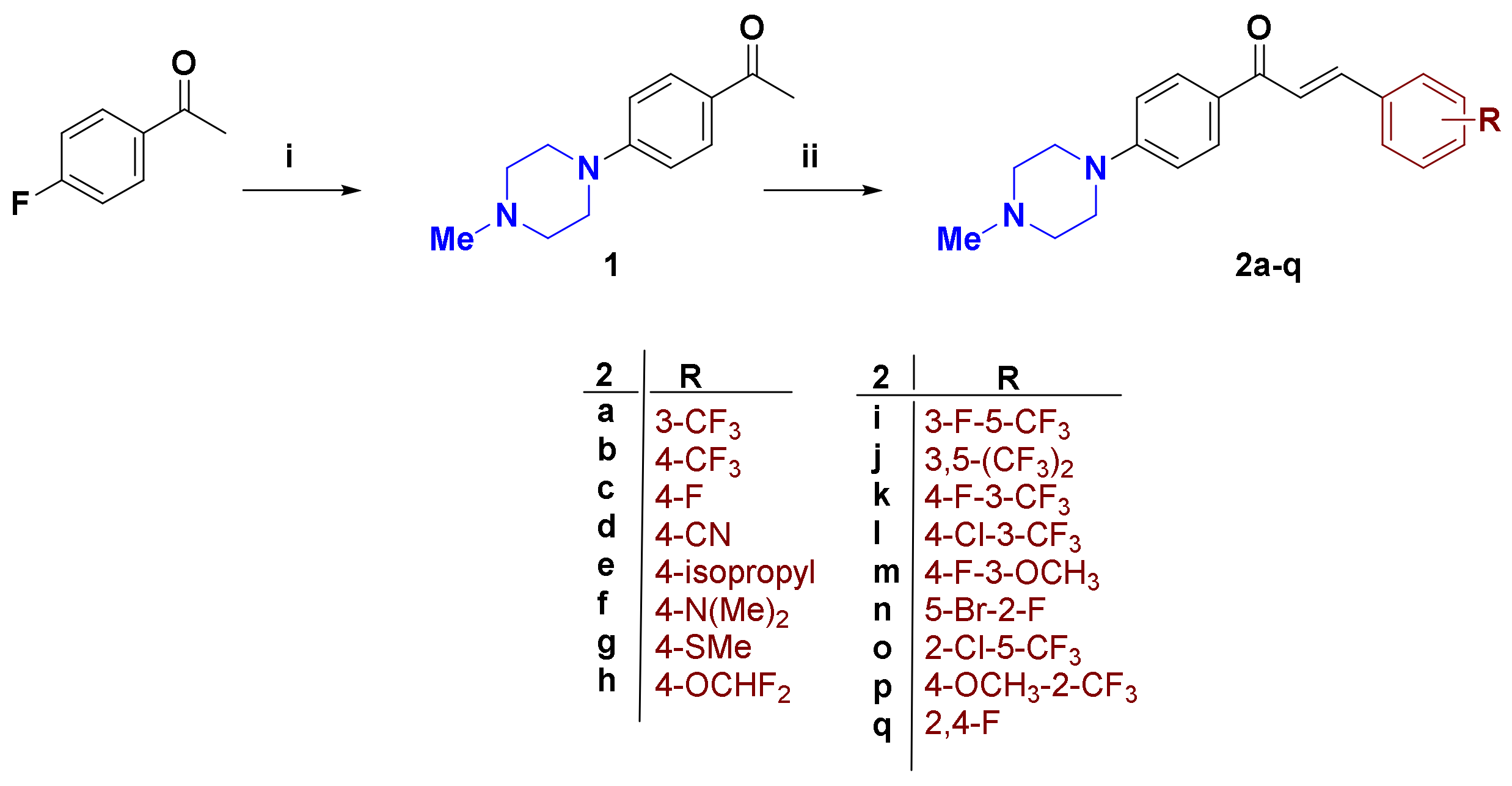
2.1. Synthesis of Compounds
2.2. Inhibitory Activities against MAO-A/B, AchE, and BChE
2.2.1. Overview of the Activity
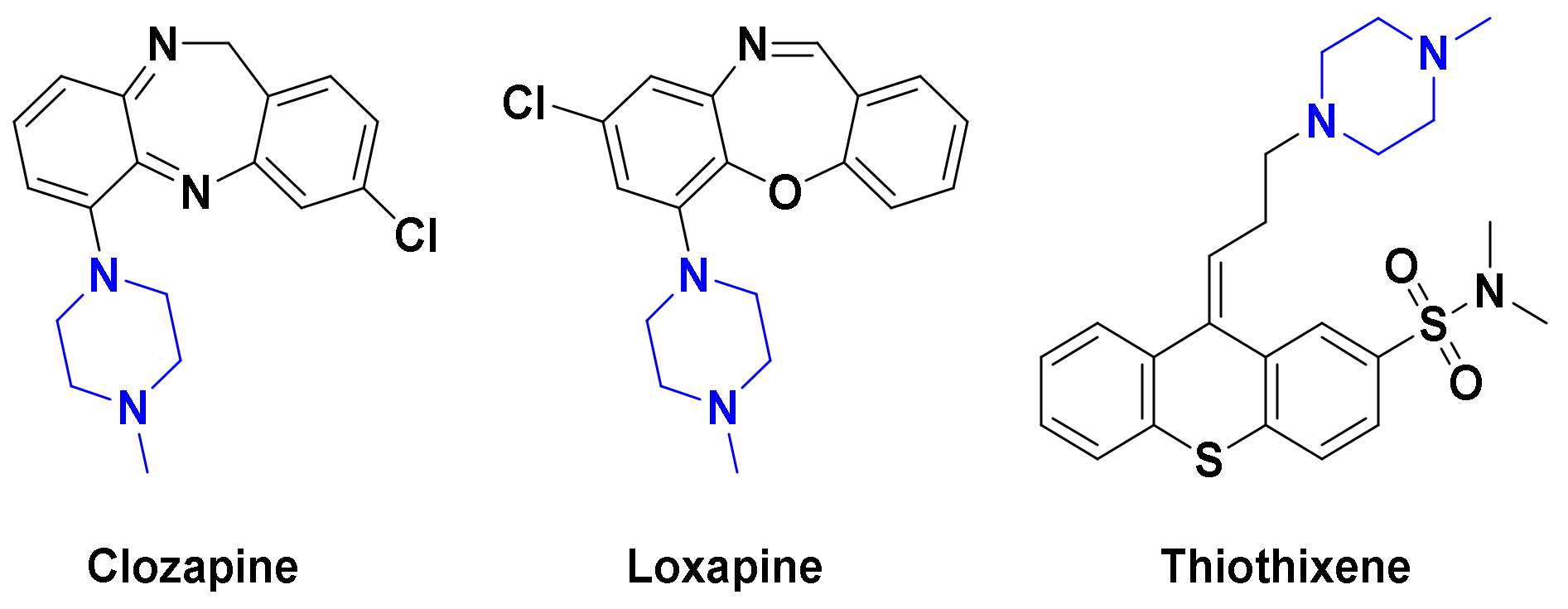
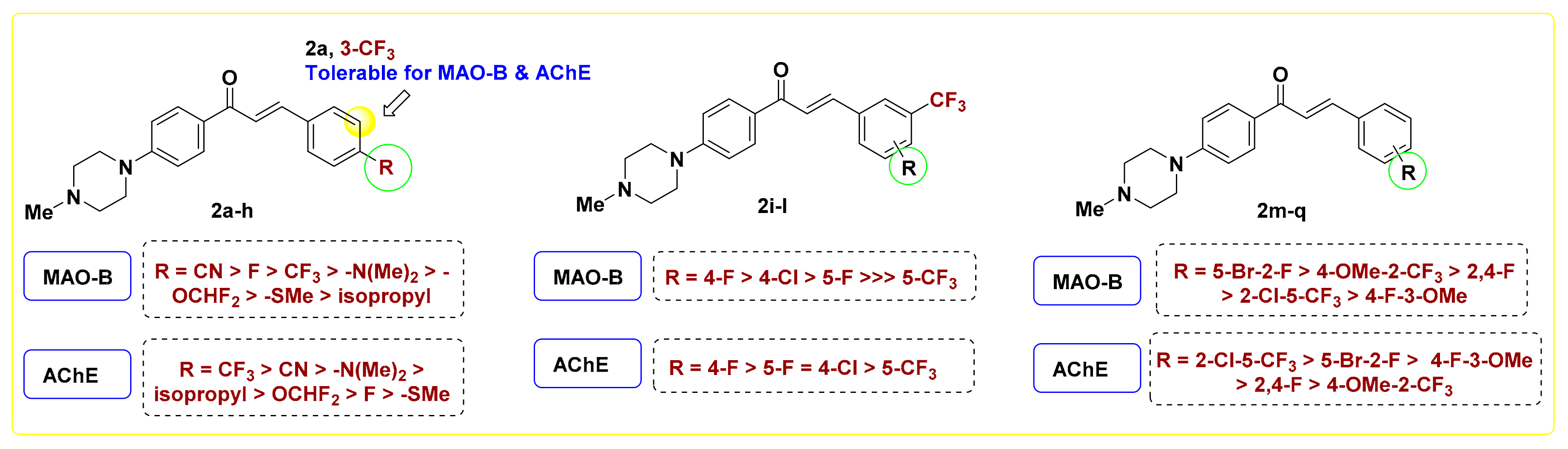
2.2.2. SAR
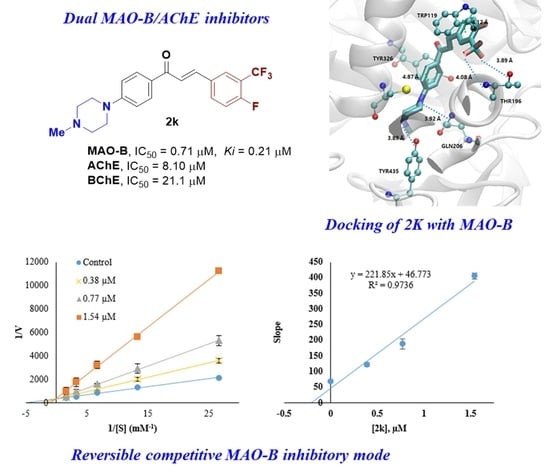
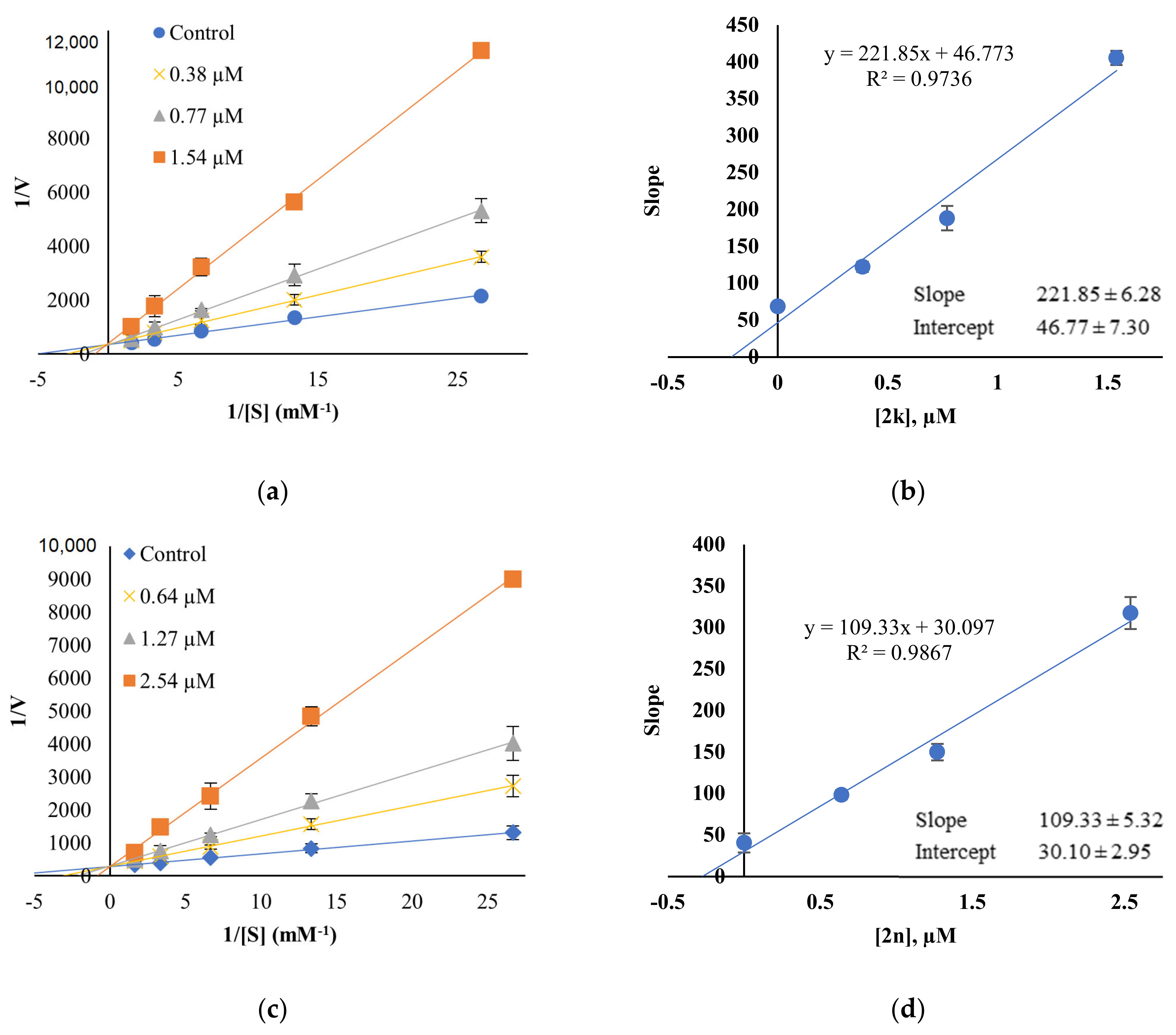
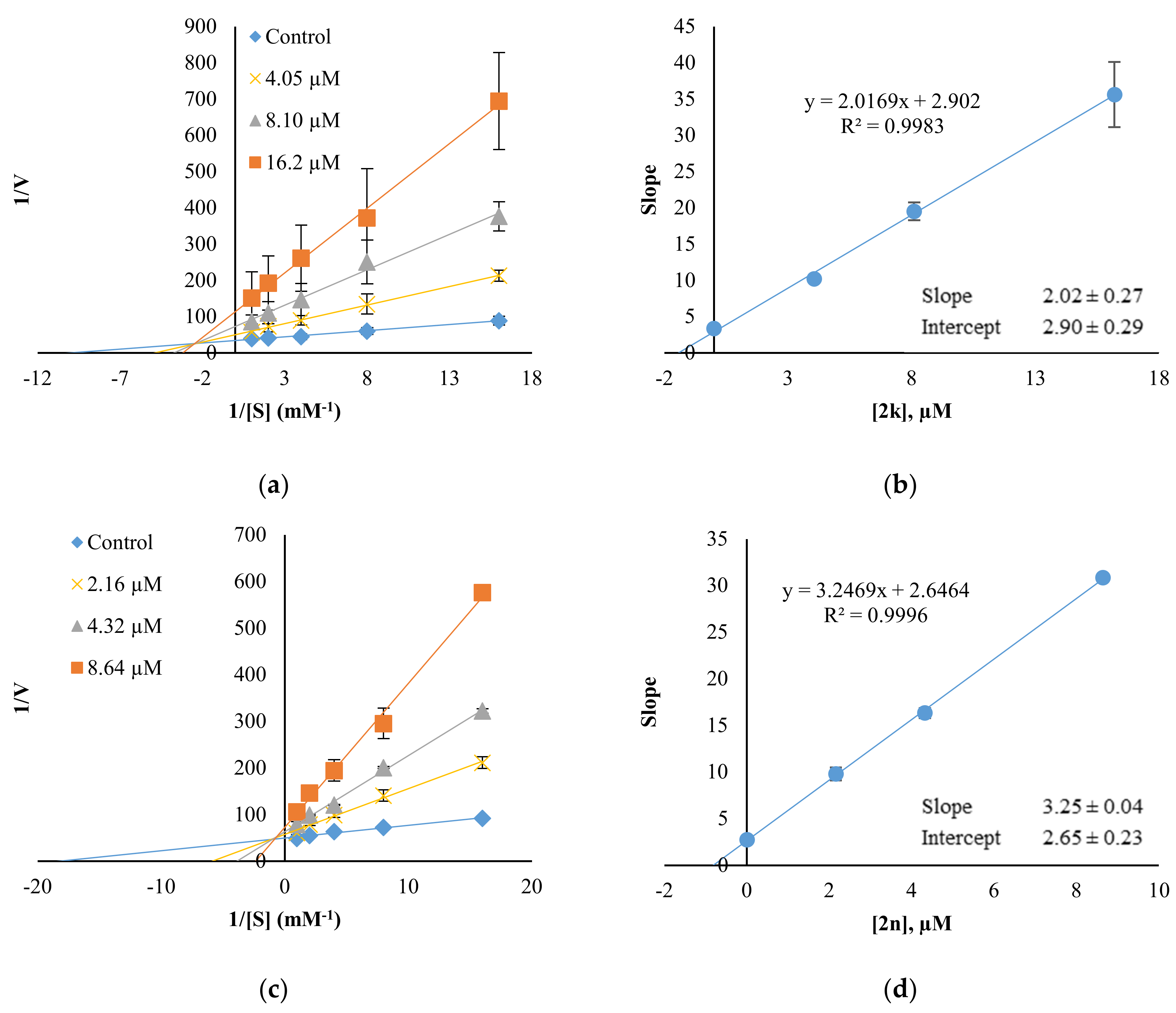
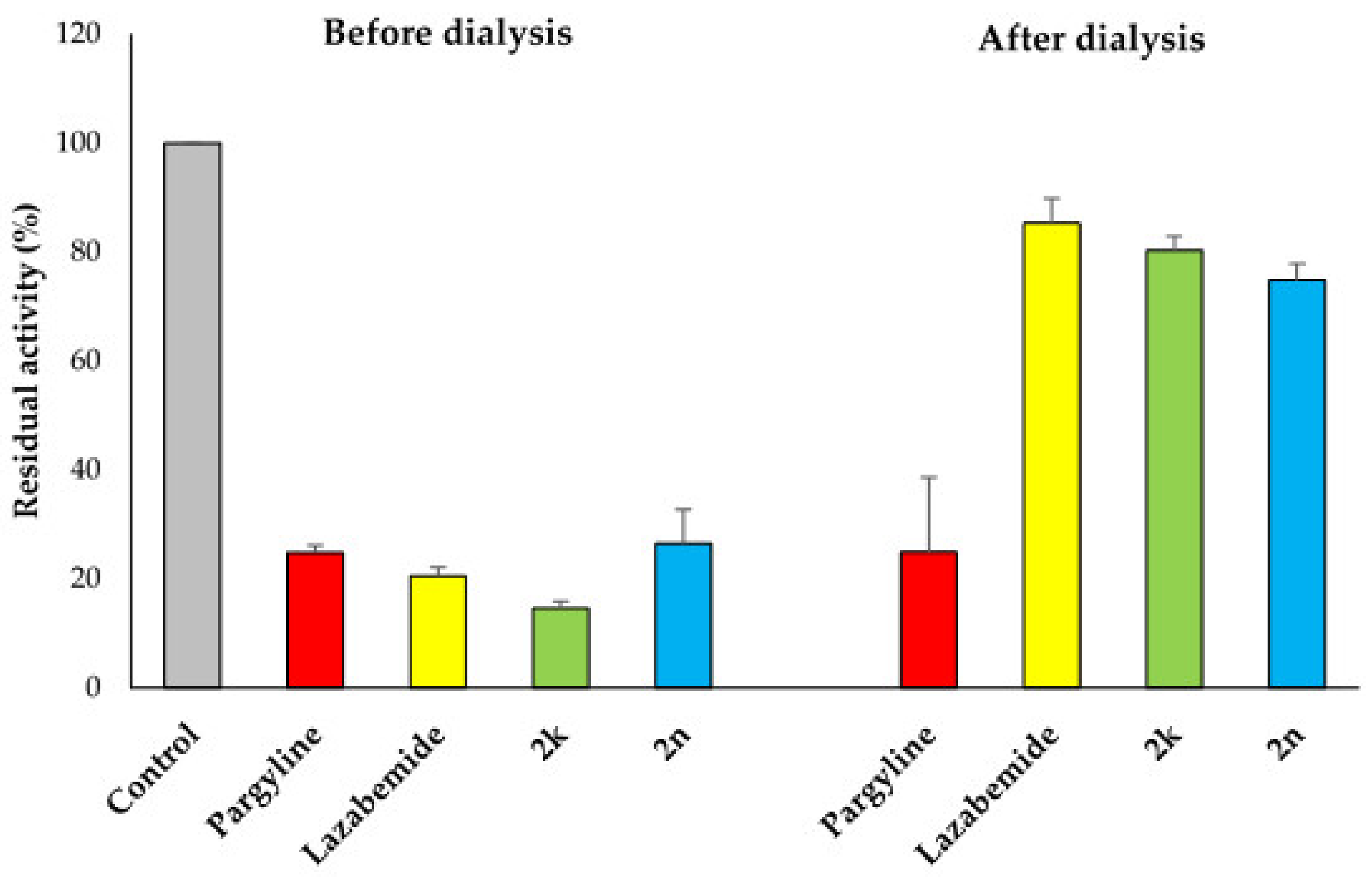
2.3. Inhibition Pattern Analysis of 2k and 2n
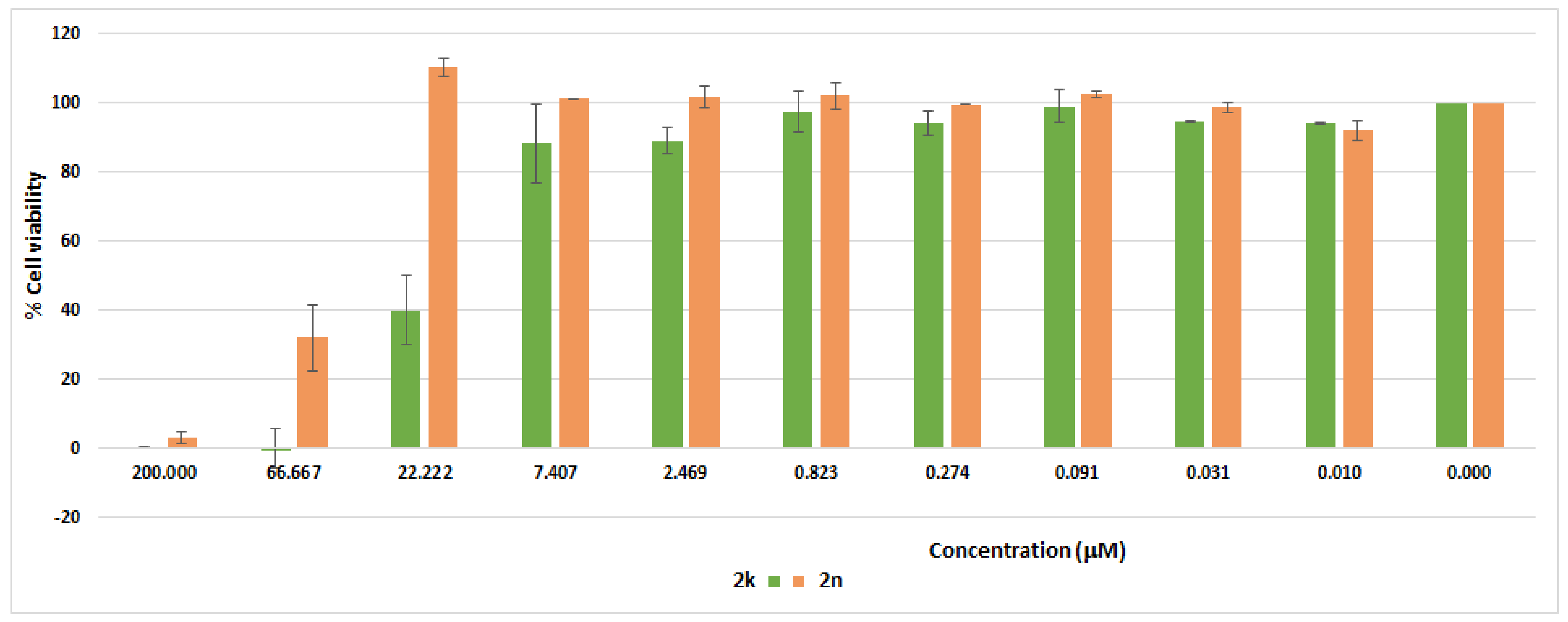
2.4. Cytotoxicity Study of 2k and 2n
2.5. Computational Studies
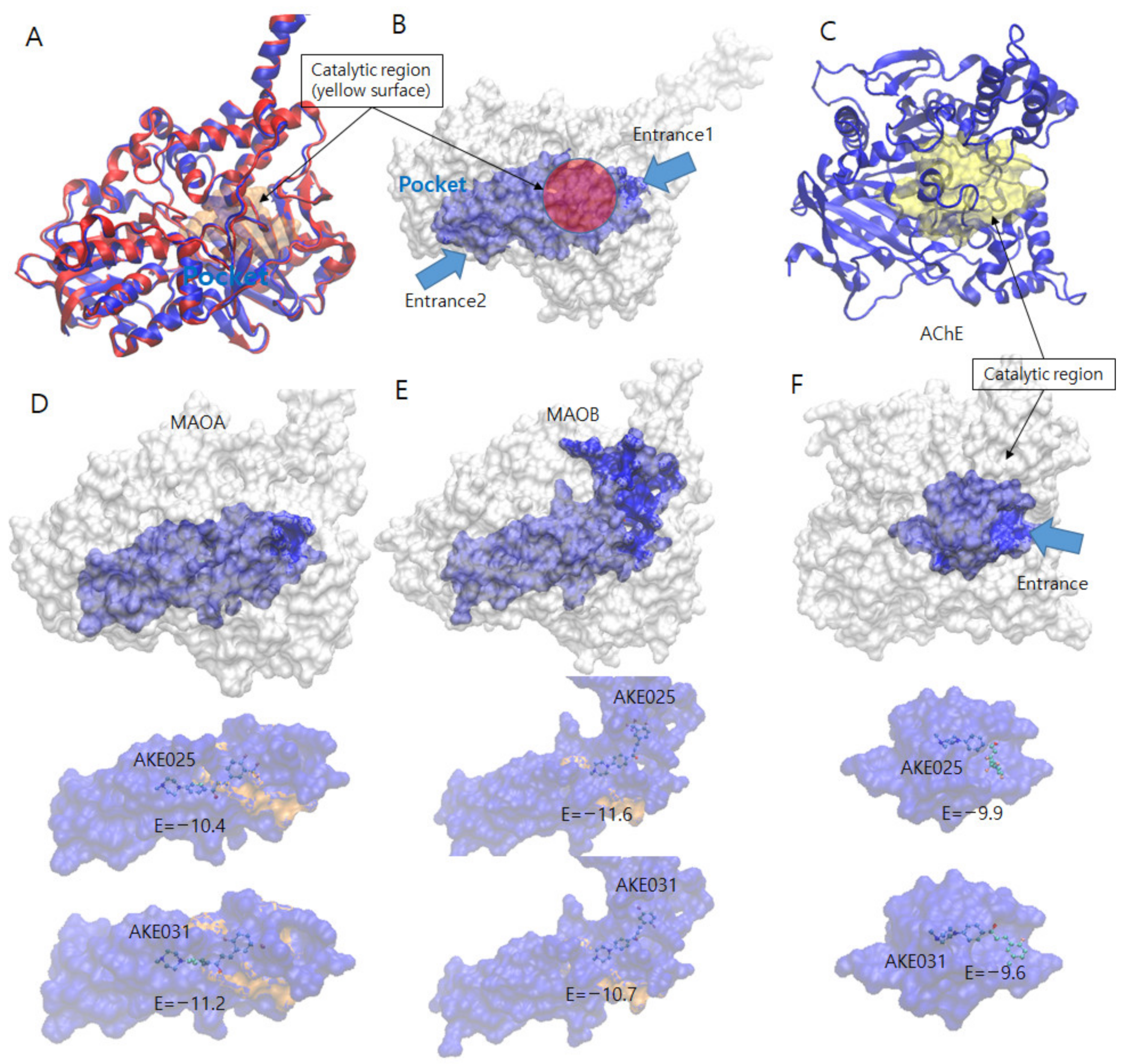
2.5.1. Docking Simulations of 2k and 2n
2.5.2. Pharmacokinetic Prediction of 2k and 2n
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis
3.2.1. 1-(4-(4-Methylpiperazin-1-yl)phenyl)ethan-1-one (1) [34]
3.2.2. General Procedure for Synthesis of Compounds 2a–q
- 1.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(3-(trifluoromethyl)phenyl)prop-2-en-1-one (2a)
- 2.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (2b)
- 3.
- (E)-3-(4-fluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2c)
- 4.
- (E)-4-(3-(4-(4-methylpiperazin-1-yl)phenyl)-3-oxoprop-1-en-1-yl)benzonitrile (2d)
- 5.
- (E)-3-(4-isopropylphenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2e)
- 6.
- (E)-3-(4-(dimethylamino)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2f)
- 7.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(4-(methylthio)phenyl)prop-2-en-1-one (2g)
- 8.
- (E)-3-(4-(difluoromethoxy)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2h)
- 9.
- (E)-3-(3-fluoro-5-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2i)
- 10.
- (E/Z)-3-(3,5-bis(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2j)
- 11.
- (E)-3-(4-fluoro-3-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2k)
- 12.
- (E)-3-(4-chloro-3-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2l)
- 13.
- (E)-3-(4-fluoro-3-methoxyphenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2m)
- 14.
- (E)-3-(5-bromo-2-fluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2n)
- 15.
- (E)-3-(2-chloro-5-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2o)
- 16.
- (E)-3-(4-methoxy-2-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2p)
- 17.
- (E)-3-(2,4-difluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2q)
3.3. Monoamine Oxidase (MAO) and Cholinesterase (ChE) Biological Studies
3.3.1. Enzyme Inhibition Assays
3.3.2. Kinetics and Reversibility Analysis
3.4. Cell Cytotoxicity Study
3.4.1. Materials
3.4.2. Method
3.5. Molecular Docking
3.6. Pharmacokinetic Prediction of the 2k and 2n Using the SwissADME Web Tool
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Xu, S.T.; Zhu, Z.Y.; Xu, J.Y. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Soacha, D.A.; Scheiner, M.; Decker, M. Multi-target-directed-ligands acting as enzyme inhibitors and receptor ligands. Eur. J. Med. Chem. 2019, 180, 690–706. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.D.; Dias, K.S.T.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C. Multi-Target Directed Drugs as a Modern Approach for Drug Design Towards Alzheimer’s Disease: An Update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 2012, 7057–7065. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 1996, 47, 161S–170S. [Google Scholar] [CrossRef]
- Robottom, B.J. Efficacy, safety, and patient preference of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease. Patient Prefer. Adher. 2011, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Secci, D.; Bolasco, A.; Chimenti, P.; D’Ascenzio, M. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2012, 22, 759–801. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Mathew, B.; Jat, R.K.; Nicolotti, O.; Kim, H. A Comprehensive Review of Monoamine Oxidase-A Inhibitors in their Syntheses and Poteneies. Comb. Chem. High Throughput Scr. 2020, 23, 898–914. [Google Scholar] [CrossRef]
- Park, J.-H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lu, C.J.; Sun, Y.; Mao, F.; Luo, Z.H.; Su, T.; Jiang, H.L.; Shan, W.J.; Li, X.S. Multitarget-Directed Benzylideneindanone Derivatives: Anti-beta-Amyloid (A beta) Aggregation, Antioxidant, Metal Chelation, and Monoamine Oxidase B (MAO-B) Inhibition Properties against Alzheimer’s Disease. J. Med. Chem. 2012, 55, 8483–8492. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Cisternas, P.; Oliva, C.A.; Sánchez-López, E.; Ettcheto, M.; Bartolini, M.; De Simone, A.; Ricchini, M. Discovery of a potent dual inhibitor of acetylcholinesterase and butyrylcholinesterase with antioxidant activity that alleviates Alzheimer-like pathology in old APP/PS1 mice. J. Med. Chem. 2020, 64, 812–839. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A. Quinolizidinyl derivatives of bi-and tricyclic systems as potent inhibitors of acetyl-and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef]
- Sang, Z.P.; Bai, P.; Ban, Y.J.; Wang, K.R.; Wu, A.G.; Mi, J.; Hu, J.Q.; Xu, R.; Zhu, G.F.; Wang, J.T.; et al. Novel donepezil-chalcone-rivastigmine hybrids as potential multifunctional anti-Alzheimer’s agents: Design, synthesis, In Vitro biological evaluation, In Vivo and In Silico studies. Bioorg. Chem. 2022, 127, 106007. [Google Scholar] [CrossRef]
- Sang, Z.P.; Song, Q.; Cao, Z.C.; Deng, Y.; Tan, Z.H.; Zhang, L. Design, synthesis and evaluation of novel dimethylamino chalcone-O-alkylamines derivatives as potential multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 216, 113310. [Google Scholar] [CrossRef]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Perez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-target-directed ligands for Alzheimer’s disease: Discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef]
- Yamali, C.; Engin, F.S.; Bilginer, S.; Tugrak, M.; Ozgun, D.O.; Ozli, G.; Levent, S.; Saglik, B.N.; Ozkay, Y.; Gul, H.I. Phenothiazine-based chalcones as potential dual-target inhibitors toward cholinesterases (AChE, BuChE) and monoamine oxidases (MAO-A, MAO-B). J. Heterocycl. Chem. 2021, 58, 161–171. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Shin, H.S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef]
- Brito, A.F.; Moreira, L.K.S.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fund Clin. Pharmacol. 2019, 33, 13–24. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macias, A.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2022, 65, 1047–1131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Mantha, A.K.; Kumar, V. Synthesis, biological evaluation and molecular modeling studies of phenyl-/benzhydrylpiperazine derivatives as potential MAO inhibitors. Bioorg. Chem. 2018, 77, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, I.I.; Lai, T.H.; Penjisevic, J.Z.; Dukic-Stefanovic, S.; Andric, D.B.; Brust, P.; Kostic-Rajacic, S.V.; Teodoro, R. Newly Synthesized Fluorinated Cinnamylpiperazines Possessing Low In Vitro MAO-B Binding. Molecules 2020, 25, 4941. [Google Scholar] [CrossRef] [PubMed]
- Modh, R.P.; Kumar, S.P.; Jasrai, Y.T.; Chikhalia, K.H. Design, Synthesis, Biological Evaluation, and Molecular Modeling of Coumarin-Piperazine Derivatives as Acetylcholinesterase Inhibitors. Arch. Pharm. 2013, 346, 793–804. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef]
- Zhuang, C.L.; Zhang, W.; Sheng, C.Q.; Zhang, W.N.; Xing, C.G.; Miao, Z.Y. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yanez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A Valid Scaffold for Monoamine Oxidases Inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef]
- Mathew, B.; Ucar, G.; Mathew, G.E.; Mathew, S.; Purapurath, P.K.; Moolayil, F.; Mohan, S.; Gupta, S.V. Monoamine Oxidase Inhibitory Activity: Methyl- versus Chlorochalcone Derivatives. Chemmedchem 2016, 11, 2649–2655. [Google Scholar] [CrossRef]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef]
- Lakshminarayanan, B.; Baek, S.C.; Lee, J.P.; Kannappan, N.; Mangiatordi, G.F.; Nicolotti, O.; Subburaju, T.; Kim, H.; Mathew, B. Ethoxylated Head of Chalcones as a New Class of Multi-Targeted MAO Inhibitors. Chemistryselect 2019, 4, 6614–6619. [Google Scholar] [CrossRef]
- Xiao, G.Y.; Li, Y.; Qiang, X.M.; Xu, R.; Zheng, Y.X.Z.; Cao, Z.C.; Luo, L.; Yang, X.; Sang, Z.P.; Su, F.; et al. Design, synthesis and biological evaluation of 4’-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.Q.; Qiang, X.M.; Song, Q.; Cao, Z.C.; Ye, C.Y.; He, Y.X.; Deng, Y.; Zhang, L. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2020, 94, 103477. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.S.; Parambi, D.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-substituted chalcones: A new class of MAO-B, AChE, and BACE-1 inhibitors for the treatment of neurological disorders. Environ. Sci. Pollut. Res. 2021, 28, 38855–38866. [Google Scholar] [CrossRef] [PubMed]
- Heiser, U.; Niestroj, A.; Zeitlmann, L. N-Pyridinyl Carboxamides as Cyclin-Dependent Kinase Inhibitors Useful in the Treatment of Diseases. 2011, WO2011110612.
- Rangarajan, T.; Mathew, B. Recent Updates on Pyrazoline Derivatives as Promising Candidates for Neuropsychiatric and Neurodegenerative Disorders. Curr. Trends Med. Chem. 2021, 21, 2695–2714. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Baek, S.C.; Lee, J.P.; Tondo, A.R.; Nicolotti, O.; Kim, H.; Mathew, B. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019, 93, 103335. [Google Scholar]
- Jeong, G.S.; Kaipakasseri, S.; Lee, S.R.; Marraiki, N.; Batiha, G.E.S.; Dev, S.; Palakkathondi, A.; Kavully, F.S.; Gambacorta, N.; Nicolotti, O.; et al. Selected 1, 3-benzodioxine-containing chalcones as multipotent oxidase and acetylcholinesterase inhibitors. ChemMedChem 2020, 15, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; Cipolloni, M.; Catto, M.; Colliva, C.; Miniero, D.V.; Latronico, T.; De Candia, M.; Benicchi, T.; Linusson, A.; Giacchè, N. Probing Fluorinated Motifs onto Dual AChE-MAO B Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Early-ADME Studies. J. Med. Chem. 2022, 65, 3962–3977. [Google Scholar] [CrossRef]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B.; et al. Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: Synthesis and biochemical investigations. J. Enzym. Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Son, S.Y.; Ma, A.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-angstrom resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Soto, C.S.; Honig, B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. USA 2002, 99, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Ryu, H.W.; Kang, M.-G.; Park, D.; Oh, S.-R.; Kim, H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016, 26, 4714–4719. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Oh, J.M.; Jang, H.-J.; Kim, W.J.; Kang, M.-G.; Baek, S.C.; Lee, J.P.; Park, D.; Oh, S.-R.; Kim, H. Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int. J. Biol. Macromol. 2020, 151, 441–448. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Residual Activities at 10 μM (%) | |||
|---|---|---|---|---|
| MAO-A | MAO-B | AChE | BChE | |
| 2a | 117.97 ± 7.73 | 12.78 ± 0.79 | 34.94 ± 5.19 | 58.65 ± 6.80 |
| 2b | 88.19 ± 6.87 | 21.02 ± 4.02 | 18.99 ± 0.55 | 58.50 ± 1.38 |
| 2c | 103.47 ± 8.84 | 22.73 ± 1.61 | 49.42 ± 4.66 | 49.67 ± 3.70 |
| 2d | 72.35 ± 2.50 | 16.67 ± 4.71 | 24.26 ± 0.95 | 71.53 ± 5.25 |
| 2e | 60.56 ± 3.98 | 44.09 ± 3.04 | 17.75 ± 1.85 | 49.32 ± 1.28 |
| 2f | 83.58 ± 4.22 | 21.33 ± 3.77 | 26.37 ± 1.85 | 76.51 ± 3.35 |
| 2g | 97.18 ± 1.99 | 40.00 ± 1.67 | 50.39 ± 4.43 | 74.66 ± 4.48 |
| 2h | 55.22 ± 4.12 | 17.33 ± 3.77 | 37.99 ± 0.92 | 73.92 ± 3.35 |
| 2i | 114.29 ± 6.06 | 20.35 ± 2.47 | 51.49 ± 4.92 | 63.72 ± 6.24 |
| 2j | 97.14 ± 4.04 | 67.78 ± 5.36 | 57.12 ± 5.39 | 86.03 ± 1.04 |
| 2k | 96.43 ± 3.03 | −8.72 ± 0.82 | 46.85 ± 2.58 | 61.76 ± 5.54 |
| 2l | 85.88 ± 1.66 | 0.00 ± 1.79 | 68.50 ± 1.32 | 73.02 ± 1.75 |
| 2m | 78.82 ± 1.66 | 39.87 ± 6.26 | 27.61 ± 1.90 | 66.83 ± 2.10 |
| 2n | 55.63 ± 1.00 | 6.45 ± 3.04 | 30.55 ± 1.11 | 43.21 ± 2.88 |
| 2o | 92.54 ± 4.23 | 22.87 ± 2.26 | 37.60 ± 0.00 | 6.46 ± 3.05 |
| 2p | 44.53 ± 3.31 | 15.00 ± 3.93 | 37.84 ± 7.64 | 28.85 ± 4.08 |
| 2q | 72.66 ± 3.31 | 13.11 ± 2.06 | 33.78 ± 0.27 | 55.29 ± 0.68 |
| Compound No. | IC50 (μM) | SI (MAO) | SI (ChE) | |||
|---|---|---|---|---|---|---|
| MAO-A | MAO-B | AChE | BChE | |||
| 2a | >40 | 2.58 ± 0.19 | 5.70 ± 0.22 | 13.89 ± 2.22 | 15.50 | 2.44 |
| 2b | >40 | 3.10 ± 0.04 | 2.26 ± 0.29 | 13.37 ± 0.27 | 12.90 | 5.92 |
| 2c | >40 | 2.98 ± 0.18 | 10.30 ± 1.03 | 18.41 ± 3.17 | 13.42 | 1.79 |
| 2d | >40 | 2.20 ± 0.06 | 2.38 ± 0.13 | 24.71 ± 2.00 | 18.18 | 10.38 |
| 2e | 16.21 ± 1.74 | 8.36 ± 0.39 | 3.75 ± 0.82 | 14.36 ± 0.40 | 1.94 | 3.83 |
| 2f | >40 | 3.29 ± 0.08 | 3.03 ± 0.31 | >40 | 12.16 | >13.20 |
| 2g | >40 | 6.74 ± 0.03 | 12.41 ± 1.96 | >40 | 5.93 | >3.22 |
| 2h | >40 | 6.14 ± 0.43 | 5.82 ± 0.51 | >40 | 6.51 | >6.87 |
| 2i | >40 | 2.33 ± 0.14 | 13.93 ± 0.08 | 25.30 ± 2.96 | 17.17 | 1.82 |
| 2j | >40 | 15.70 ± 1.17 | 15.36 ± 2.43 | >40 | 2.55 | >2.60 |
| 2k | >40 | 0.71 ± 0.03 | 8.10 ± 0.69 | 21.09 ± 0.23 | 56.34 | 2.60 |
| 2l | >40 | 1.41 ± 0.04 | 14.07 ± 0.54 | 26.09 ± 1.28 | 28.37 | 1.85 |
| 2m | >40 | 6.76 ± 0.82 | 4.45 ± 0.24 | 18.61 ± 1.35 | 5.92 | 4.18 |
| 2n | 17.80 ± 0.28 | 1.11 ± 0.06 | 4.32 ± 0.39 | 9.44 ± 0.05 | 16.04 | 2.19 |
| 2o | >40 | 3.20 ± 0.32 | 3.87 ± 0.18 | 1.19 ± 0.06 | 12.50 | 0.31 |
| 2p | 6.67 ± 0.63 | 2.18 ± 0.01 | 7.17 ± 0.60 | 7.58 ± 0.25 | 3.06 | 1.06 |
| 2q | 16.52 ± 1.75 | 3.03 ± 0.13 | 6.37 ± 0.17 | 10.89 ± 0.90 | 5.45 | 1.71 |
| Toloxatone | 1.08 ± 0.03 | - | - | - | - | - |
| Lazabemide | 0.11 ± 0.02 | - | - | - | - | |
| Clorgyline | 0.007 ± 0.001 | - | - | - | - | - |
| Pargyline | - | 0.14 ± 0.01 | - | - | - | - |
| Tacrine | - | - | 0.27 ± 0.02 | 0.060 ± 0.002 | - | - |
| Donepezil | - | - | 0.010 ± 0.002 | 0.180 ± 0.004 | - | - |
| PC5 * | 34.1 ± 2.51 | 2.31 ± 0.46 | - | - | 14.8 | |
| PC10 * | 31.4 ± 3.50 | 0.65 ± 0.023 | 28.0 ± 2.43 | 36.4 ± 2.36 | ||
| PC11 * | 34.9 ± 4.10 | 0.71 ± 0.0035 | 26.3 ± 1.29 | 36.2 ± 3.65 | ||
| Enzyme | Cpds | No. of Cpds | Ave. Energy | Lowest Energy (Rank) | Free Energy | No. of HB | No. of π-π/Aro. |
|---|---|---|---|---|---|---|---|
| MAO-A | 2k | 32 | −5.97 ± 2.56 | −10.40 (2) | −12.48 | 4 | 1/5 |
| 2n | 50 | −5.88 ± 2.42 | −11.20 (2) | −13.55 | 2 | 1/4 | |
| MAO-B | 2k | 10 | −5.47 ± 3.70 | −11.60 (1) | −12.98 | 4 | 2/7 |
| 2n | 16 | −5.33 ± 3.31 | −10.70 (1) | −12.36 | 2 | 2/7 | |
| AChE | 2k | 41 | −7.81 ± 1.15 | −9.90 (1) | −12.13 | 7 | 1/9 |
| 2n | 39 | −7.74 ± 1.25 | −9.60 (1) | −11.80 | 4 | 1/9 |
| Compound No. | GI Absorption | BBB Permeant | P-gp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Log Kp (Skin Permeation) |
|---|---|---|---|---|---|---|---|---|---|
| 2k | High | Yes | No | No | Yes | No | Yes | Yes | −5.42 cm/s |
| 2n | High | Yes | No | No | Yes | Yes | Yes | No | −5.62 cm/s |
| Compound No. | Mw | cLog Po/w | HBD | HBA | TPSA (Å2) | RB | Lipinski Violations * |
|---|---|---|---|---|---|---|---|
| 2k | 392.40 | 4.48 | 0 | 6 | 23.55 | 5 | 0 |
| 2n | 403.30 | 4.03 | 0 | 3 | 23.55 | 4 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Damasy, A.K.; Park, J.E.; Kim, H.J.; Lee, J.; Bang, E.-K.; Kim, H.; Keum, G. Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals 2023, 16, 83. https://doi.org/10.3390/ph16010083
El-Damasy AK, Park JE, Kim HJ, Lee J, Bang E-K, Kim H, Keum G. Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals. 2023; 16(1):83. https://doi.org/10.3390/ph16010083
Chicago/Turabian StyleEl-Damasy, Ashraf K., Jong Eun Park, Hyun Ji Kim, Jinhyuk Lee, Eun-Kyoung Bang, Hoon Kim, and Gyochang Keum. 2023. "Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors" Pharmaceuticals 16, no. 1: 83. https://doi.org/10.3390/ph16010083
APA StyleEl-Damasy, A. K., Park, J. E., Kim, H. J., Lee, J., Bang, E.-K., Kim, H., & Keum, G. (2023). Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals, 16(1), 83. https://doi.org/10.3390/ph16010083