
Current Treatment Options in Homozygous Familial Hypercholesterolemia


Abstract
1. Introduction
2. Treatment of Patients with HoFH
3. Conventional Lipid Lowering Therapy
4. PCSK9 Inhibitors
4.1. Alirocumab and Evolocumab
4.2. Inclisiran
4.3. Lerodalcibep
5. Pharmacologic Agents Acting Independent of LDL-Receptors
5.1. Anti-Apo-B Therapies
5.1.1. Lomitapide
5.1.2. Mipomersen
5.2. ANGPTL3 Inhibitors
5.2.1. Evinacumab
5.2.2. RNA Based Treatments Targeting ANGPTL3
6. Interventions to Lower LDL Independent of LDL-Receptor
6.1. Lipoprotein Apheresis
6.2. Liver Transplantation
7. Future or near Future Aspects
7.1. CRISPR-Based Genome Editing
7.2. Gene Transfer
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kayikcioglu, M.; Kısmalı, E.; Can, L.; Payzin, S. Long-term follow-up in patients with homozygous familial hypercholesterolemia; 13-year experience of a university hospital lipid clinic. Turk. Kardiyol. Dernegi Arsivi-Arch. Turk. Soc. Cardiol. 2014, 42, 599–611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bajaj, A.; Cuchel, M. Advancements in the Treatment of Homozygous Familial Hypercholesterolemia. J. Atheroscler. Thromb. 2022, 29, 1125–1135. [Google Scholar] [CrossRef]
- Tokgozoglu, L.; Kayikcioglu, M. Familial Hypercholesterolemia: Global Burden and Approaches. Curr. Cardiol. Rep. 2021, 23, 151. [Google Scholar] [CrossRef]
- Chemello, K.; García-Nafría, J.; Gallo, A.; Martín, C.; Lambert, G.; Blom, D. Lipoprotein metabolism in familial hypercholesterolemia. J. Lipid Res. 2021, 62, 100062. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R.; Blom, D.J.; Marais, A.D.; Seed, M.; Pilcher, G.J.; Raal, F.J. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur. Heart J. 2018, 39, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Evinacumab: A new option in the treatment of homozygous familial hypercholesterolemia. Expert Opin. Biol. Ther. 2022, 22, 813–820. [Google Scholar] [CrossRef]
- Kayikcioglu, M.; Tokgozoglu, L.; Yilmaz, M.; Kaynar, L.; Aktan, M.; Durmuş, R.B.; Gokce, C.; Temizhan, A.; Ozcebe, O.I.; Akyol, T.K.; et al. A nation-wide survey of patients with homozygous familial hypercholesterolemia phenotype undergoing LDL-apheresis in Turkey (A-HIT 1 registry). Atherosclerosis 2018, 270, 42–48. [Google Scholar] [CrossRef]
- Tunçel, Ö.K.; Kayıkçıoğlu, M.; Pırıldar, Ş.; Yılmaz, M.; Kaynar, L.; Aktan, M.; Durmuş, R.B.; Gökçe, C.; Temizhan, A.; Özcebe, O.İ.; et al. Mental status and physical activity in patients with homozygous familial hypercholesterolemia: A subgroup analysis of a nationwide survey (A-HIT1 registry). J. Clin. Lipidol. 2020, 14, 361–370. [Google Scholar] [CrossRef]
- Kayikcioglu, M.; Kuman-Tunçel, O.; Pirildar, S.; Yílmaz, M.; Kaynar, L.; Aktan, M.; Durmus, R.B.; Gökçe, C.; Temizhan, A.; Özcebe, O.I.; et al. Clinical management, psychosocial characteristics, and quality of life in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis in Turkey: Results of a nationwide survey (A-HIT1 registry). J. Clin. Lipidol. 2019, 13, 455–467. [Google Scholar] [CrossRef]
- Tromp, T.R.; Cuchel, M. New algorithms for treating homozygous familial hypercholesterolemia. Curr. Opin. Infect. Dis. 2022, 33, 326–335. [Google Scholar] [CrossRef]
- İncazli, S.P.B.; Özer, S.P.; Kayikçioğlu, M.M. Evaluation of the Effectiveness of Individually Tailored Lifestyle Intervention in Patients With Familial Hypercholesterolemia. J. Cardiovasc. Nurs. 2022, 37, 465–474. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Stefanutti, C. Lomitapide–a Microsomal Triglyceride Transfer Protein Inhibitor for Homozygous Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2020, 22, 38. [Google Scholar] [CrossRef]
- Raal, F.J.; Pilcher, G.J.; Panz, V.R.; van Deventer, H.E.; Brice, B.C.; Blom, D.J.; Marais, A.D. Reduction in Mortality in Subjects With Homozygous Familial Hypercholesterolemia Associated With Advances in Lipid-Lowering Therapy. Circulation 2011, 124, 2202–2207. [Google Scholar] [CrossRef]
- Wiegman, A.; Gidding, S.S.; Watts, G.F.; Chapman, M.J.; Ginsberg, H.N.; Cuchel, M.; Ose, L.; Averna, M.; Boileau, C.; Borén, J.; et al. Familial hypercholesterolaemia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Eur. Heart J. 2015, 36, 2425–2437. [Google Scholar] [CrossRef]
- Othman, R.A.; Myrie, S.B.; Mymin, D.; Roullet, J.-B.; Steiner, R.D.; Jones, P.J. Effect of ezetimibe on low- and high-density lipoprotein subclasses in sitosterolemia. Atherosclerosis 2017, 260, 27–33. [Google Scholar] [CrossRef][Green Version]
- I Kramer, A.; E Akioyamen, L.; Lee, S.; Bélanger, A.; Ruel, I.; Hales, L.; Genest, J.; Brunham, L.R. Major adverse cardiovascular events in homozygous familial hypercholesterolaemia: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2021, 29, 817–828. [Google Scholar] [CrossRef]
- Ray, K.K.; Dhalwani, N.; Sibartie, M.; Bridges, I.; Ebenbichler, C.; Perrone-Filardi, P.; Villa, G.; Vogt, A.; Bruckert, E. Low-density lipoprotein cholesterol levels exceed the recommended European threshold for PCSK9i initiation: Lessons from the HEYMANS study. Eur. Heart J. Qual. Care Clin. Outcomes 2022, 8, 447–460. [Google Scholar] [CrossRef]
- Stein, E.A.; Honarpour, N.; Wasserman, S.M.; Xu, F.; Scott, R.; Raal, F.J. Effect of the Proprotein Convertase Subtilisin/Kexin 9 Monoclonal Antibody, AMG 145, in Homozygous Familial Hypercholesterolemia. Circulation 2013, 128, 2113–2120. [Google Scholar] [CrossRef]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; A Stein, E. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef]
- Santos, R.D.; Stein, E.A.; Hovingh, G.K.; Blom, D.J.; Soran, H.; Watts, G.F.; López, J.A.G.; Bray, S.; Kurtz, C.E.; Hamer, A.W.; et al. Long-Term Evolocumab in Patients With Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2020, 75, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Blom, D.J.; Harada-Shiba, M.; Rubba, P.; Gaudet, D.; Kastelein, J.J.; Charng, M.-J.; Pordy, R.; Donahue, S.; Ali, S.; Dong, Y.; et al. Efficacy and Safety of Alirocumab in Adults With Homozygous Familial Hypercholesterolemia: The ODYSSEY HoFH Trial. J. Am. Coll. Cardiol. 2020, 76, 131–142. [Google Scholar] [CrossRef] [PubMed]
- France, M.; Rees, A.; Datta, D.; Thompson, G.; Capps, N.; Ferns, G.; Ramaswami, U.; Seed, M.; Neely, D.; Cramb, R.; et al. HEART UK medical scientific and research committee. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis. 2016, 255, 128–139. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Giugliano, R.P.; Wiviott, S.D.; Atar, D.; Keech, A.C.; Kuder, J.F.; Im, K.; Murphy, S.A.; Flores-Arredondo, J.H.; López, J.A.G.; et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation 2022, 146, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Kayikcioglu, M.; Tokgozoglu, L.; Tuncel, O.K.; Pirildar, S.; Can, L. Negative impact of COVID-19 pandemic on the lifestyle and management of patients with homozygous familial hypercholesterolemia. J. Clin. Lipidol. 2020, 14, 751–755. [Google Scholar] [CrossRef]
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. Pooled Patient-Level Analysis of Inclisiran Trials in Patients With Familial Hypercholesterolemia or Atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Lepor, N.E.; Kallend, D.; Stoekenbroek, R.M.; Wijngaard, P.L.; Raal, F.J. Inclisiran Durably Lowers Low-Density Lipoprotein Cholesterol and Proprotein Convertase Subtilisin/Kexin Type 9 Expression in Homozygous Familial Hypercholesterolemia: The ORION-2 Pilot Study. Circulation 2020, 141, 1829–1831. [Google Scholar] [CrossRef]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. 2012, 9, 14. [Google Scholar] [CrossRef]
- Cuchel, M.; A Meagher, E.; Theron, H.D.T.; Blom, D.J.; Marais, A.D.; A Hegele, R.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2012, 381, 40–46. [Google Scholar] [CrossRef]
- Nohara, A.; Otsubo, Y.; Yanagi, K.; Yoshida, M.; Ikewaki, K.; Harada-Shiba, M.; Jurecka, A. Safety and Efficacy of Lomitapide in Japanese Patients with Homozygous Familial Hypercholesterolemia (HoFH): Results from the AEGR-733-301 Long-Term Extension Study. J. Atheroscler. Thromb. 2019, 26, 368–377. [Google Scholar] [CrossRef]
- Blom, D.J.; Averna, M.; Meagher, E.A.; Theron, H.D.T.; Sirtori, C.R.; Hegele, R.A.; Shah, P.K.; Gaudet, D.; Stefanutti, C.; Vigna, G.; et al. Long-Term Efficacy and Safety of the Microsomal Triglyceride Transfer Protein Inhibitor Lomitapide in Patients With Homozygous Familial Hypercholesterolemia. Circulation 2017, 136, 332–335. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Gallo, A.; Cefalù, A.B.; Di Costanzo, A.; Saheb, S.; Giammanco, A.; Averna, M.; Buonaiuto, A.; Iannuzzo, G.; Fortunato, G.; et al. Long-term efficacy of lipoprotein apheresis and lomitapide in the treatment of homozygous familial hypercholesterolemia (HoFH): A cross-national retrospective survey. Orphanet J. Rare Dis. 2021, 16, 381. [Google Scholar] [CrossRef]
- Kayikcioglu, M. LDL Apheresis and Lp (a) Apheresis: A Clinician’s Perspective. Curr. Atheroscler. Rep. 2021, 23, 15. [Google Scholar] [CrossRef]
- Underberg, J.A.; Cannon, C.P.; Larrey, D.; Makris, L.; Blom, D.; Phillips, H. Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: Five-year data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER). J. Clin. Lipidol. 2020, 14, 807–817. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Cefalù, A.B.; Noto, D.; Giammanco, A.; Averna, M.; Pintus, P.; Medde, P.; Vigna, G.B.; Sirtori, C.; Calabresi, L.; et al. Efficacy of Lomitapide in the Treatment of Familial Homozygous Hypercholesterolemia: Results of a Real-World Clinical Experience in Italy. Adv. Ther. 2017, 34, 1200–1210. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Steward, K.; Cefalù, A.B.; Di Costanzo, A.; Boersma, E.; Bini, S.; Arca, M.; van Lennep, J.R.; Giammanco, A.; Averna, M.; et al. Efficacy and safety of lomitapide in homozygous familial hypercholesterolaemia: The pan-European retrospective observational study. Eur. J. Prev. Cardiol. 2021, 29, 832–841. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Masana, L.; Kolovou, G.; Ariceta, G.; Nóvoa, F.J.; Lund, A.M.; Bogsrud, M.P.; Araujo, M.; Hussein, O.; Ibarretxe, D.; et al. Real-World Outcomes with Lomitapide Use in Paediatric Patients with Homozygous Familial Hypercholesterolaemia. Adv. Ther. 2019, 36, 1786–1811. [Google Scholar] [CrossRef]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome Sequencing, ANGPTL3 Mutations, and Familial Combined Hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Watts, G.; Schwabe, C.; Scott, R.; Gladding, P.; Sullivan, D.; Baker, J.; Clifton, P.; Hamilton, J.; Given, B.; Martin, J.S.; et al. RNAi inhibition of angiopoietin-like protein 3 (ANGPTL3) with ARO-ANG3 mimics the lipid and lipoprotein profile of familial combined hypolipidemia. Eur. Heart J. 2020, 41 (Suppl. S2), ehaa946.3331. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, C.; Thompson, G.R. Lipoprotein Apheresis in the Management of Familial Hypercholesterolaemia: Historical Perspective and Recent Advances. Curr. Atheroscler. Rep. 2014, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tromp, T.R.; Hartgers, M.L.; Hovingh, G.K.; Vallejo-Vaz, A.J.; Ray, K.K.; Soran, H.; Freiberger, T.; Bertolini, S.; Harada-Shiba, M.; Blom, D.J.; et al. Worldwide experience of homozygous familial hypercholesterolaemia: Retrospective cohort study. Lancet 2022, 399, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Kroon, A.A.; Aengevaeren, W.R.; Van Der Werf, T.; Uijen, G.J.; Reiber, J.H.; Bruschke, A.V.; Stalenhoef, A.F. LDL-Apheresis Atherosclerosis Regression Study (LAARS). Circulation 1996, 93, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Kawagishi, N.; Hasegawa, Y.; Sawada, S.; Katagiri, H.; Satomi, S.; Oikawa, S. Liver Transplantation for Homozygous Familial Hypercholesterolemia. J. Atheroscler. Thromb. 2019, 26, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Brodlie, S.; Griesemer, A.; Kato, T.; Harren, P.; Gordon, B.; Parker, T.; Levine, D.; Tyberg, T.; Starc, T.; et al. Effects of Liver Transplantation on Lipids and Cardiovascular Disease in Children With Homozygous Familial Hypercholesterolemia. Am. J. Cardiol. 2016, 118, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Al Dubayee, M.; Kayikcioglu, M.; van Lennep, J.R.; Hergli, N.; Mata, P. Is Liver Transplant Curative in Homozygous Familial Hypercholesterolemia? A Review of Nine Global Cases. Adv. Ther. 2022, 39, 3042–3057. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Evitt, N.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- Cuchel, M.; Bajaj, A.; Carr, R.; Sikora, T.; Duell, P.B.; Tardif, J.-C.; Roeters van Lennep, J.E.; Linton, M.F.; Averna, M.; Cho, Y.; et al. Use of prophylactic steroids to mitigate potential T-cell response in AAV8-mediated hLDLR gene transfer in subjects with homozygous familial hypercholesterolemia. Presented at the ASGCT 23rd Annual Meeting, Virtual, 12–15 May 2020. [Google Scholar]
- Kassim, S.H.; Li, H.; Vandenberghe, L.H.; Hinderer, C.; Bell, P.; Marchadier, D.; Wilson, A.; Cromley, D.; Redon, V.; Yu, H.; et al. Gene Therapy in a Humanized Mouse Model of Familial Hypercholesterolemia Leads to Marked Regression of Atherosclerosis. PLoS ONE 2010, 5, e13424. [Google Scholar] [CrossRef]
- Greig, J.A.; Limberis, M.P.; Bell, P.; Chen, S.J.; Calcedo, R.; Rader, D.J.; Wilson, J.M. Non-Clinical Study Examining AAV8.TBG.hLDLR Vector-Associated Toxicity in Chow-Fed Wild-Type and LDLR+/- Rhesus Macaques. Hum. Gene Ther. Clin. Dev. 2017, 28, 39–50. [Google Scholar] [CrossRef]
- Greig, J.A.; Limberis, M.P.; Bell, P.; Chen, S.J.; Calcedo, R.; Rader, D.J.; Wilson, J.M. Nonclinical Pharmacology/Toxicology Study of AAV8.TBG.mLDLR and AAV8.TBG.hLDLR in a Mouse Model of Homozygous Familial Hypercholesterolemia. Hum. Gene Ther. Clin. Dev. 2017, 28, 28–38. [Google Scholar] [CrossRef]




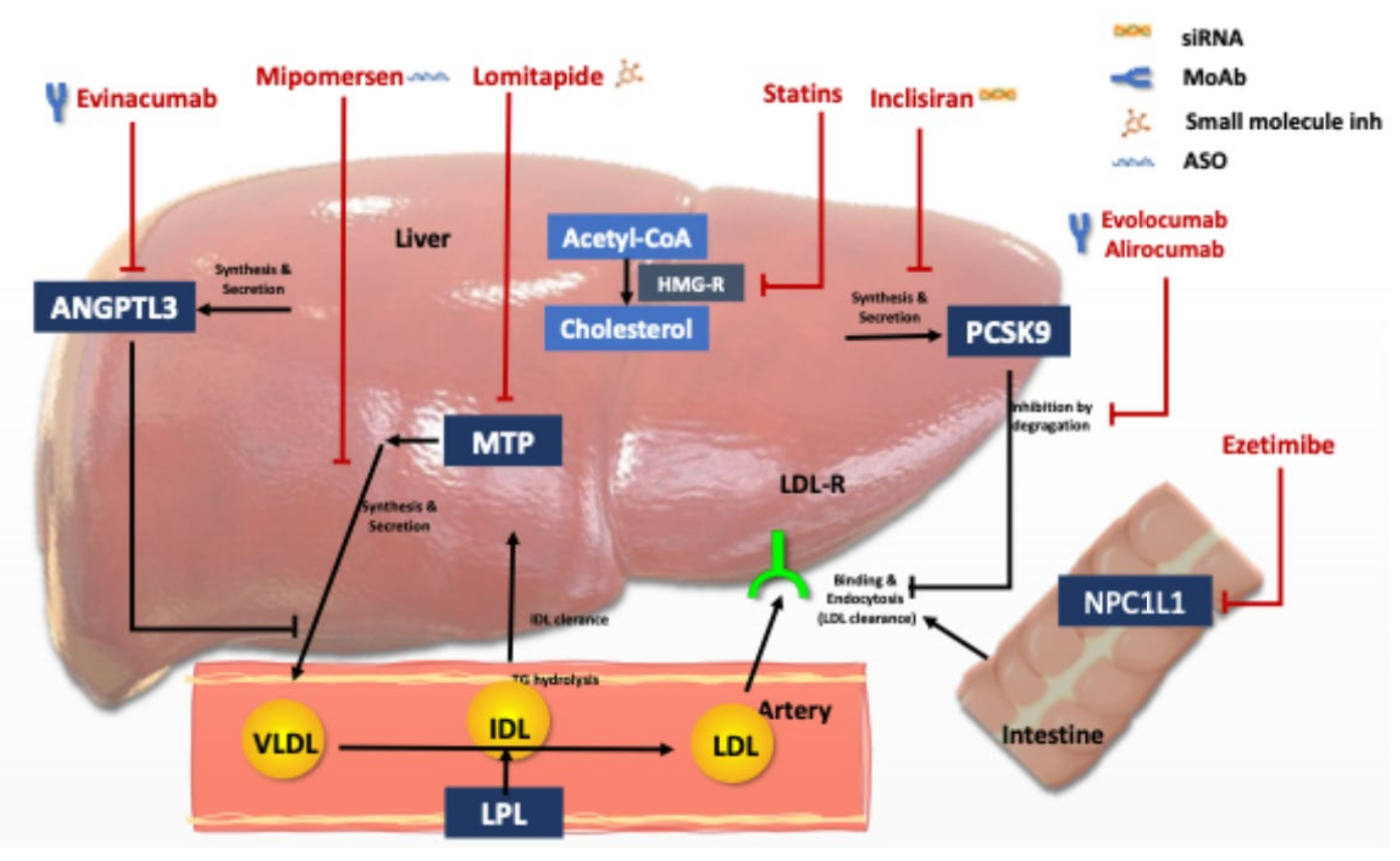{kind=link}
{kind=link}
{kind=link}
| Agent | Mechanism of Action | Route & Dose | Effect on Lipoprotein Levels in HoFH | Clinical Study Results | Guideline Recommendations | Comment |
|---|---|---|---|---|---|---|
| LDL-R dependent | ||||||
| Statins | -HMG-CoA reductase inhibition, -Reduce cholesterol synthesis, -Upregulate LDL-R -Increase LDL-C clearance | PO, Dose depends on the LDL-C target & type of statin | LDL-C 14–31%↓ | Lower both LDL-C & ASCVD risk in all statin trials in both primary & secondary prevention. | First line in HoFH with limited efficacy | - Cornerstone of LDL-C-lowering therapy in FH -More effective in ARH patients |
| Ezetimibe | -Inhibits NPCILI protein -Inhibits cholesterol absorption -Upregulate LDL-R -Increase LDL-C clearance | PO, 10 mg/day | LDL-C 5–14%↓ | Lowers both LDL-C & ASCVD outcomes on top of statins | Second line add on therapy for HoFH | -Approved for treatment of HeFH & HoFH patients either alone or in combination with statins -More effective in sitosterolemia |
| Bile acid sequestrants(Cholestyramine, Colestipol & Colesevelam) | -Decrease reabsorption of bile acids -Reduce cholesterol content in hepatocytes -Upregulate LDL-R -Increase LDL-C clearance | PO, Daily | LDL-C 0–10%↓ | No RCT on FH | None for FH | -Mostly preferred in children and pregnant women -Effect is weak. -Not absorbed |
| PCSK9 inhibitors (Alirocumab, Evolocumab) | -Monoclonal antibodies to PCSK9 -Inhibits PCSK9, -Upregulate LDL-R -Increase LDL-C clearance | Alirocumab SC, Biweekly 75–150 mg Evalocumab SC, Biweekly 140 mg, Monthly 420 mg | Alirocumab HoFH LDL-C 26%↓ Evalocumab HoFH LDL-C 15–32%↓ | -Reduced LDL-C & ASCVD outcomes in phase 3 studies. Alirocumab -ODYSSEY-HoFH Evalocumab -TESLA -HoFH short term -TAUSSING-HoFH&HeFH 4years sustained efficacy & safety | Treatment with a PCSK9 inhibitor is recommended in very-high-risk FH patients if the treatment goal is not achieved on maximal tolerated statin plus ezetimibe. | - Both are beneficial in HoFH patients with at least 2% of functional LDL-Rs. |
| Inclisirian | -SiRNA inhibiting the translation of PCSK9 -Upregulate LDL-R -Increase LDL-C clearance | SC, 300 mg on Days 1, 90 then every 6 months | HoFH LDL-C 12–37%↓ | -Orion Trial Program -Orion-5 Phase 3 RCT on HoFH-ongoing | -Not in the GLs yet -Approved for adults with primary hypercholesterolemia or mixed dyslipidemia by EC December 2020 | -Enables infrequent dosing & sustained effect -Promising use in young FH individuals with an improved compliance |
| LDL-R independent action | ||||||
| Lomitapide | -Inhibits MTP, thereby interfering with the assembly of lipoproteins -Decrease ApoB | PO, 10–60 mg/day | LDL-C 24–52%↓ | -Effective in phase 3 -LOWER registry (5-year real life data) showed efficacy even in low doses (10–40 mg/d) | -Approved by EMA &FDA as an adjunct to LLT in patients with HoFH ≥ 18 years with/without apheresis | -Shown to either reduce the frequency of apheresis or replace apheresis |
| Evinacumab | -Monoclonal antibody to ANGPTL3 | IV inf, monthly | LDL-C 47.1%↓ | -ECLIPSE study showed efficacy & safety in HoFH -ASCVD study not done yet. | -Not in the GLs yet - Approved by FDA for HoFH adults & children aged ≥ 12 years in December 2020 | -Effective also in Null variants -Advantage of monthly injections -Effective in lowering TGs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayikcioglu, M.; Tokgozoglu, L. Current Treatment Options in Homozygous Familial Hypercholesterolemia. Pharmaceuticals 2023, 16, 64. https://doi.org/10.3390/ph16010064
Kayikcioglu M, Tokgozoglu L. Current Treatment Options in Homozygous Familial Hypercholesterolemia. Pharmaceuticals. 2023; 16(1):64. https://doi.org/10.3390/ph16010064
Chicago/Turabian StyleKayikcioglu, Meral, and Lale Tokgozoglu. 2023. "Current Treatment Options in Homozygous Familial Hypercholesterolemia" Pharmaceuticals 16, no. 1: 64. https://doi.org/10.3390/ph16010064
APA StyleKayikcioglu, M., & Tokgozoglu, L. (2023). Current Treatment Options in Homozygous Familial Hypercholesterolemia. Pharmaceuticals, 16(1), 64. https://doi.org/10.3390/ph16010064