
Therapeutic Strategies to Activate p53

Abstract
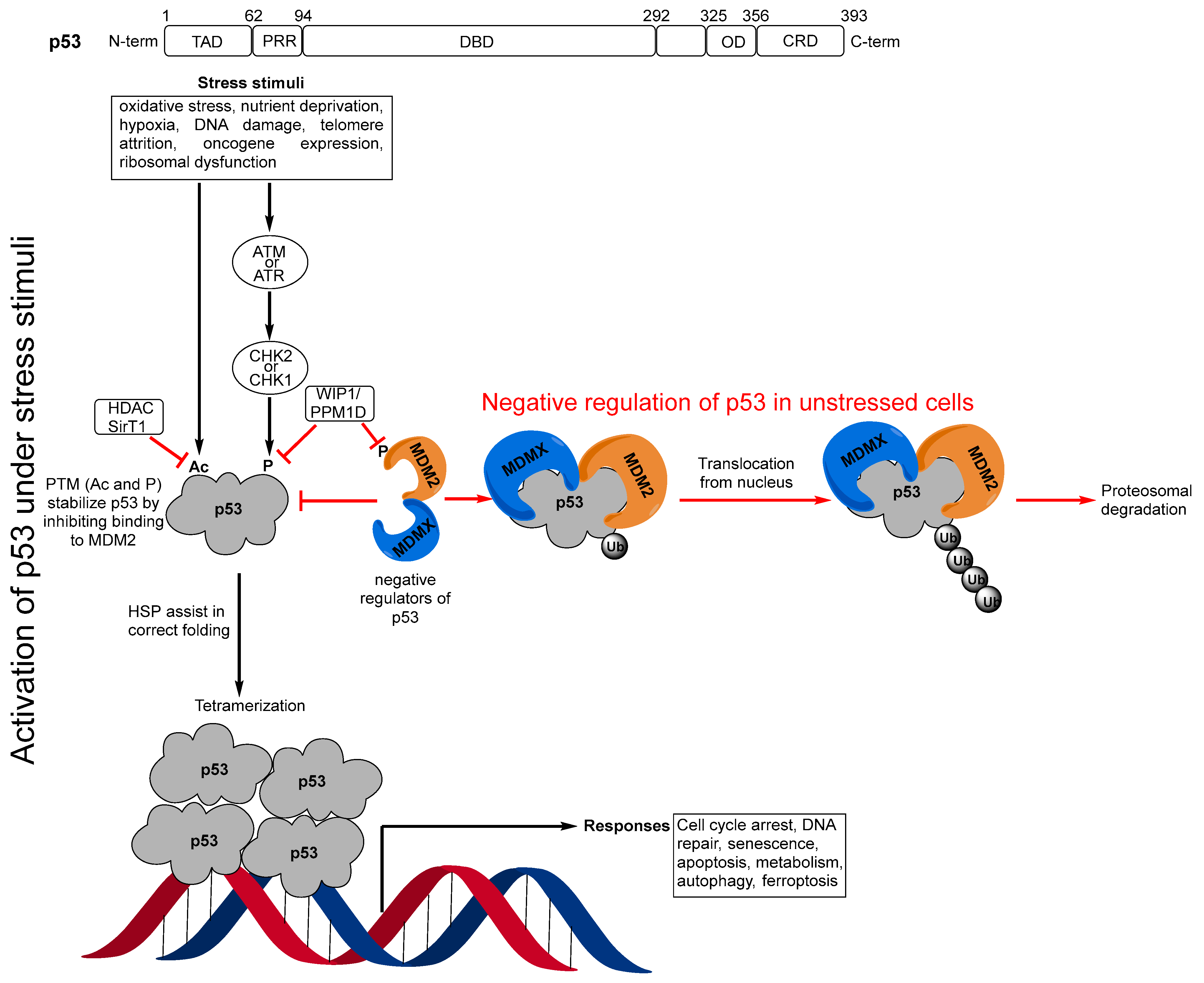
1. Introduction
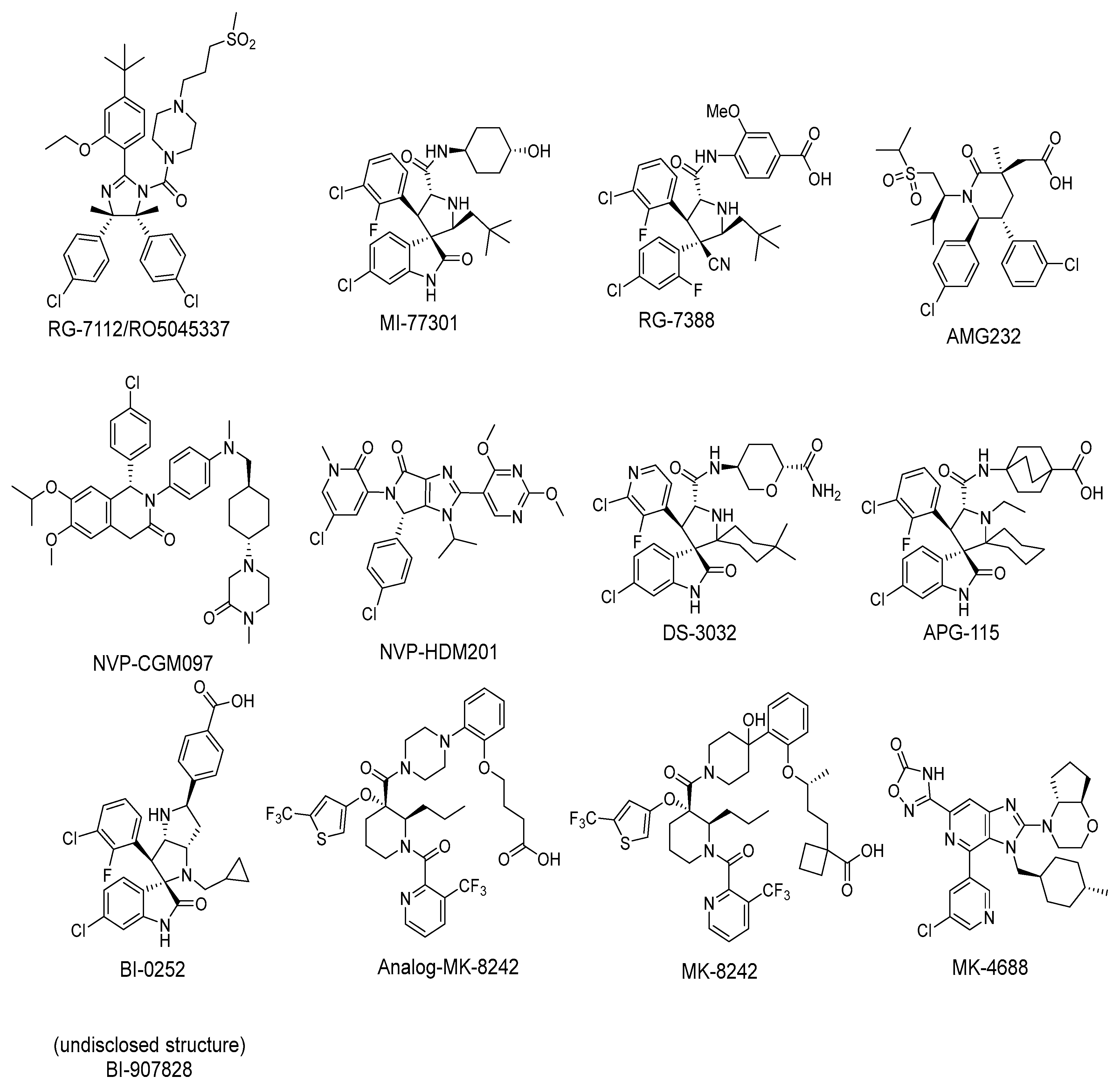
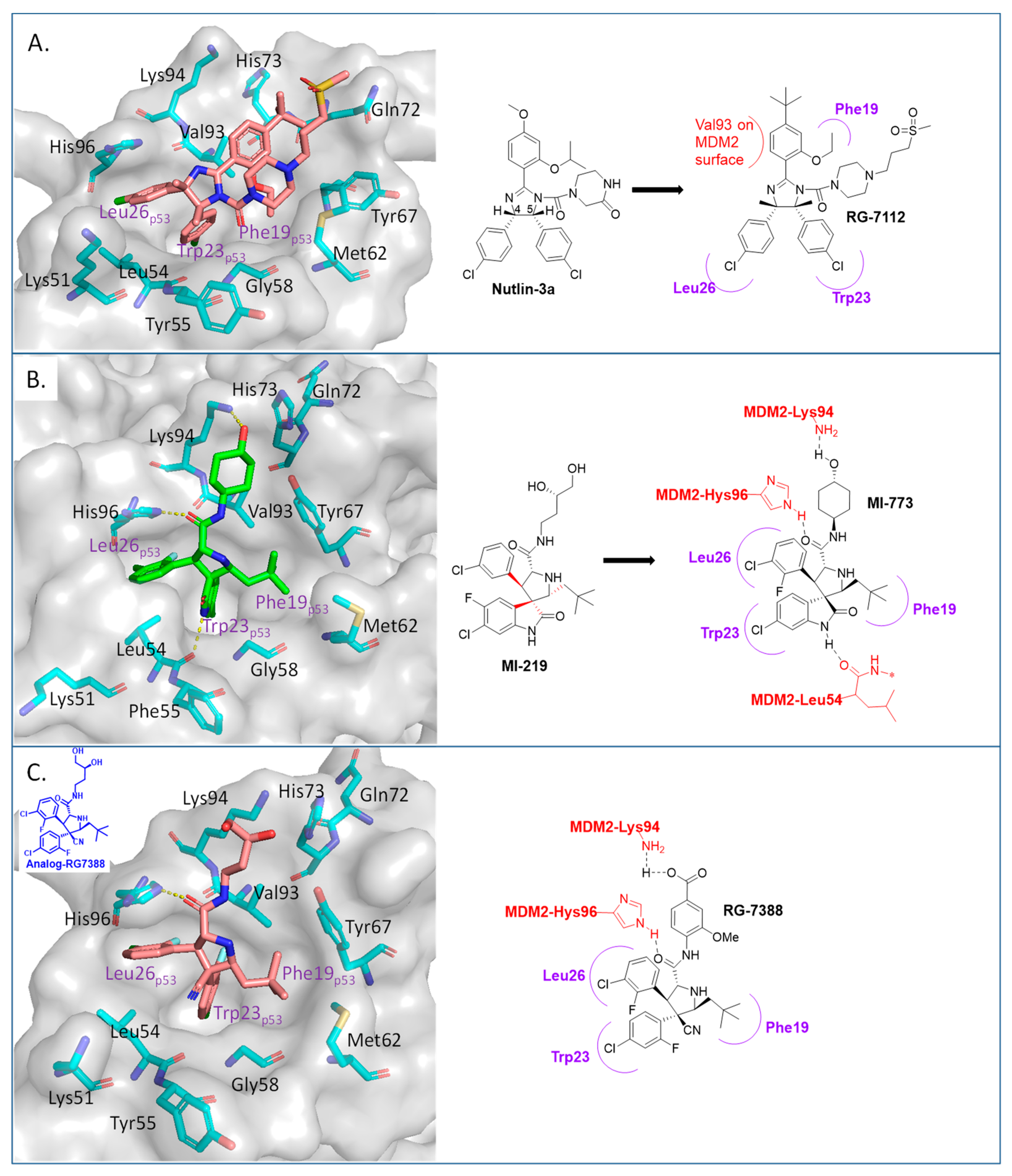
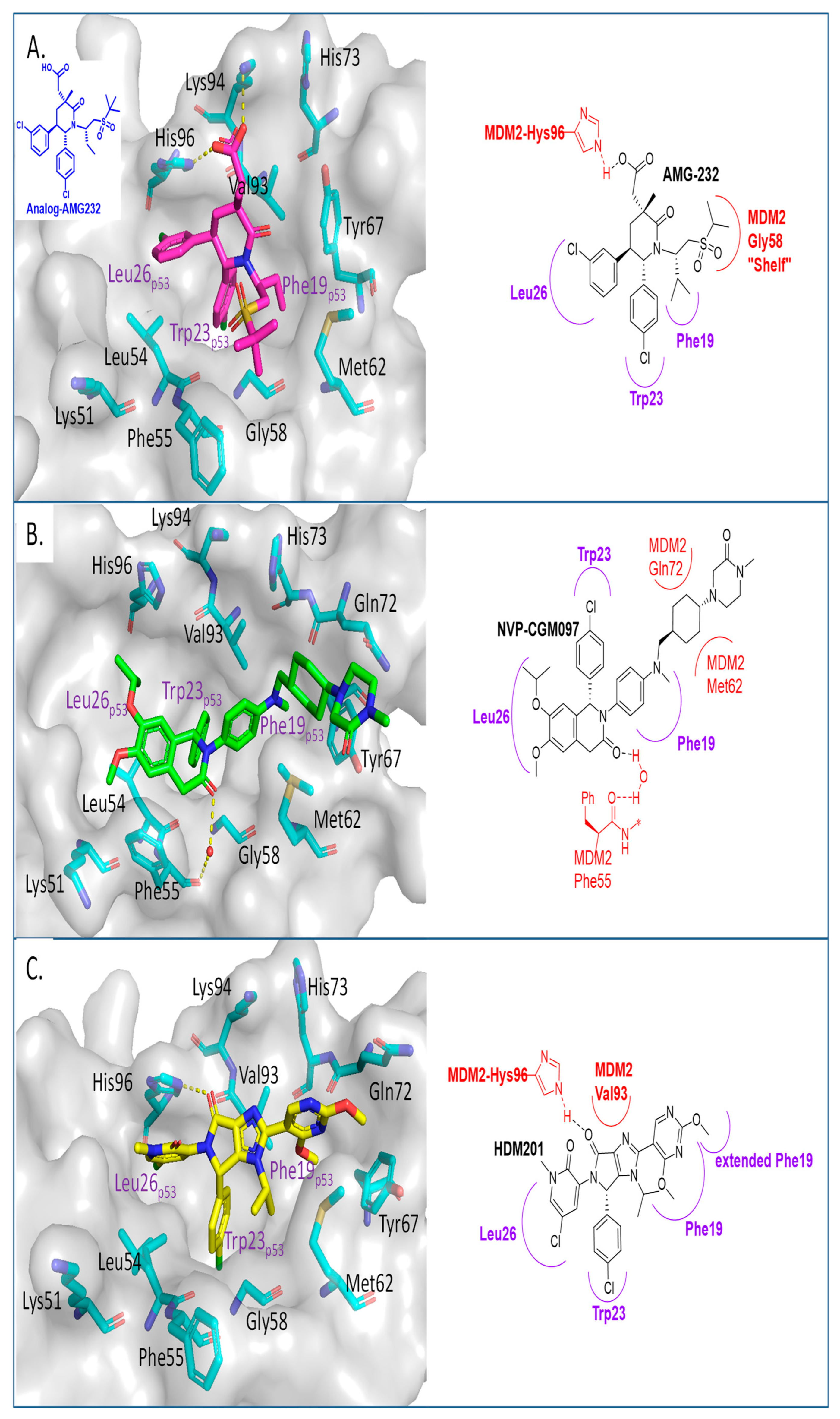
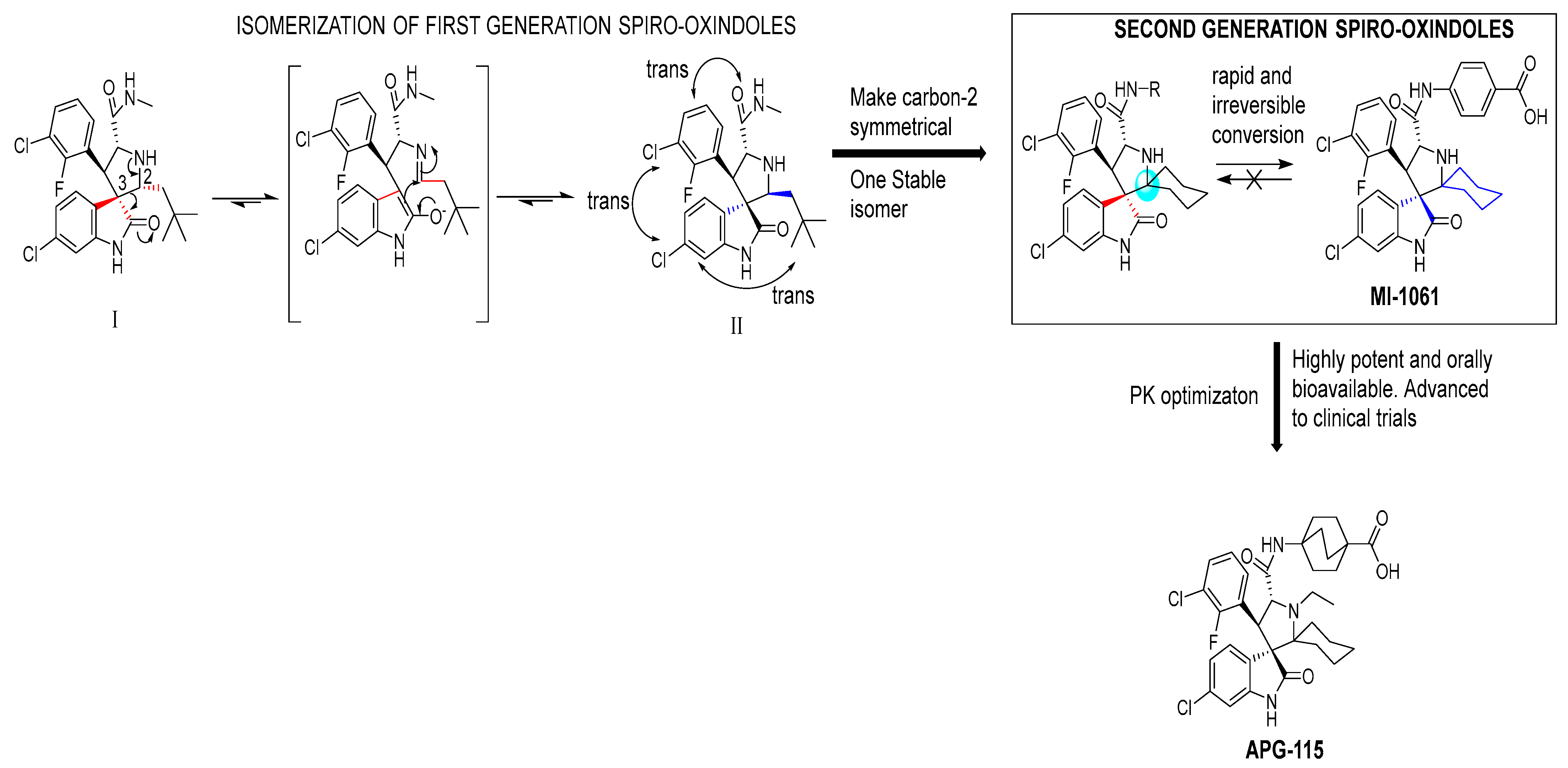
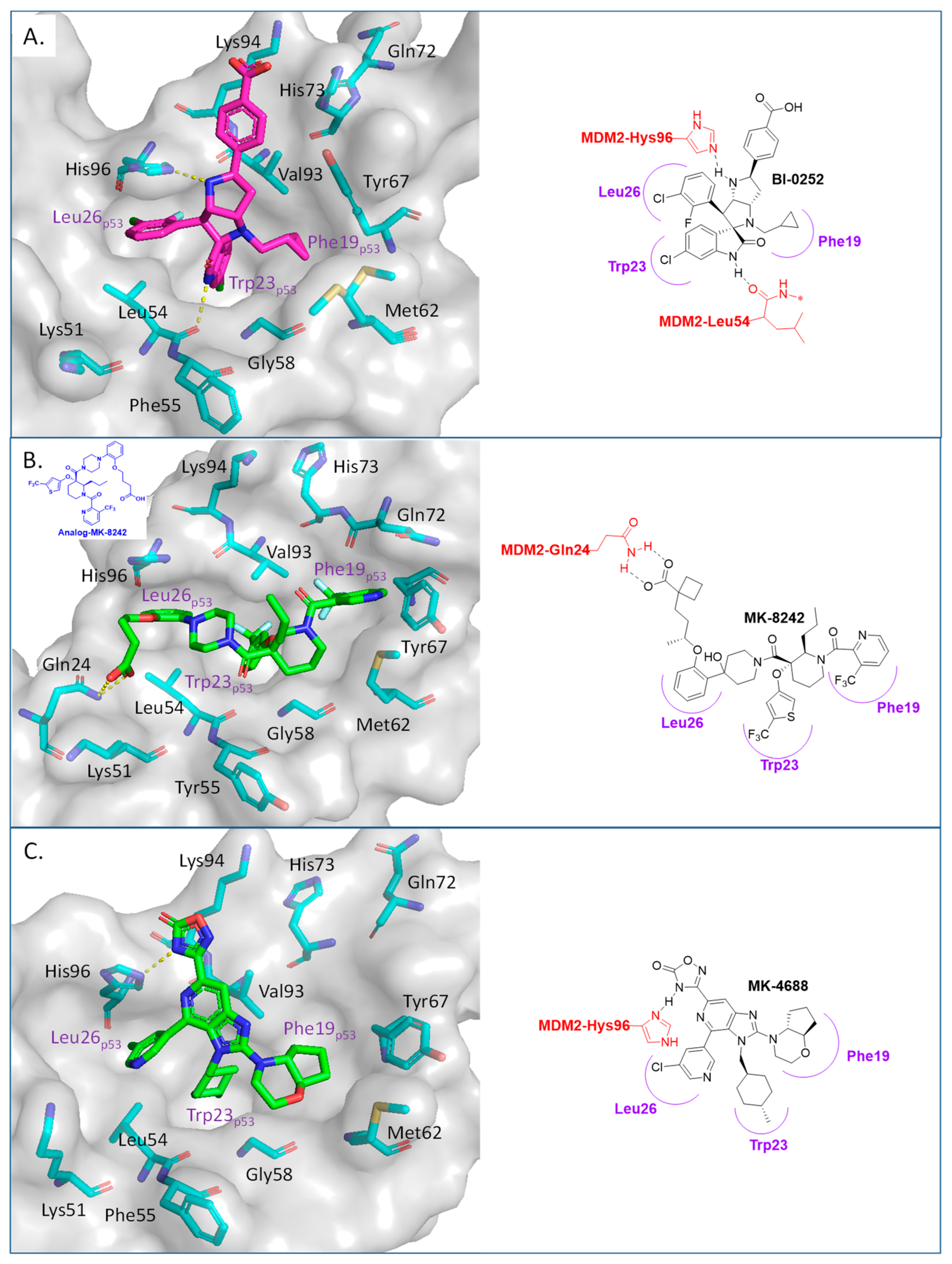
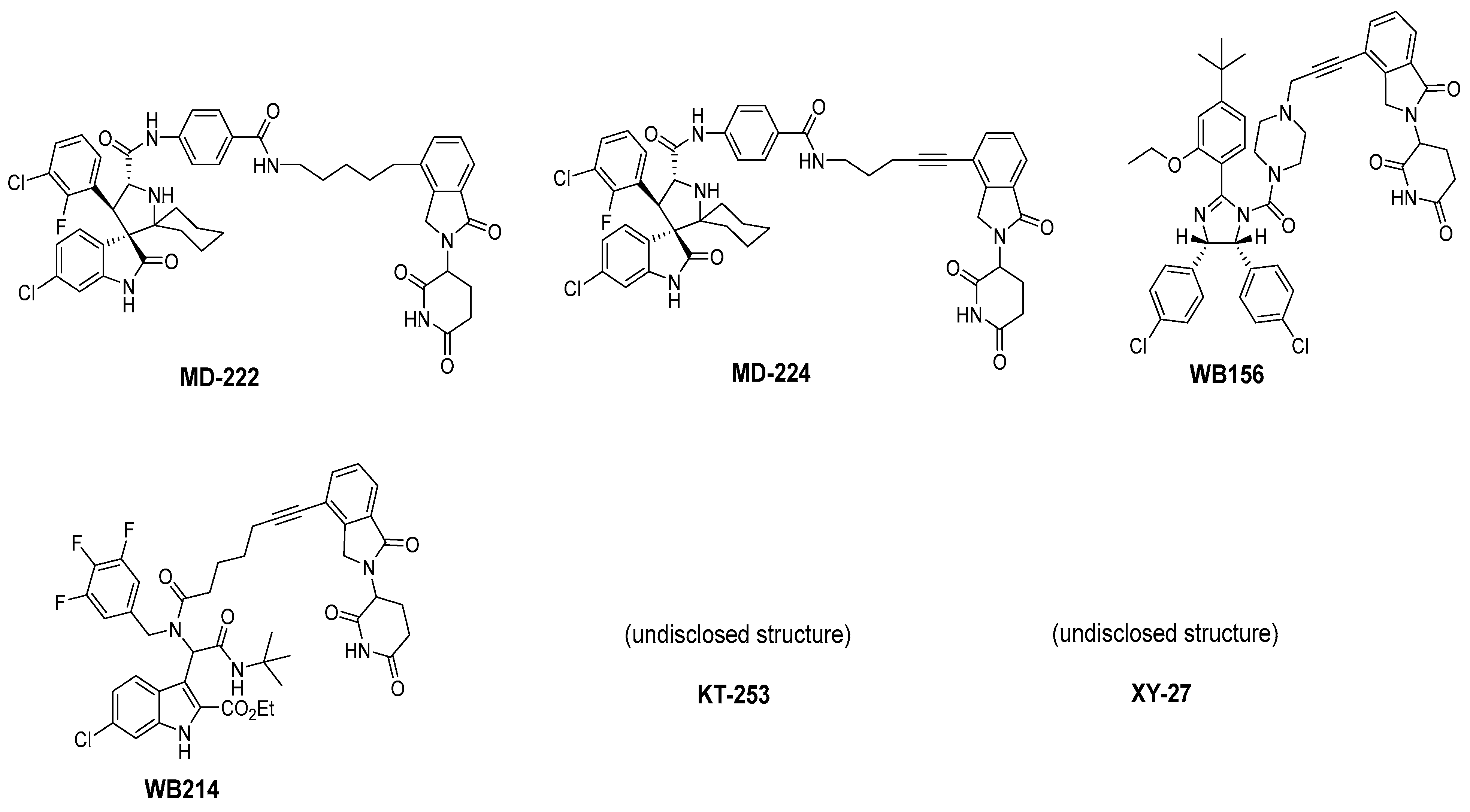
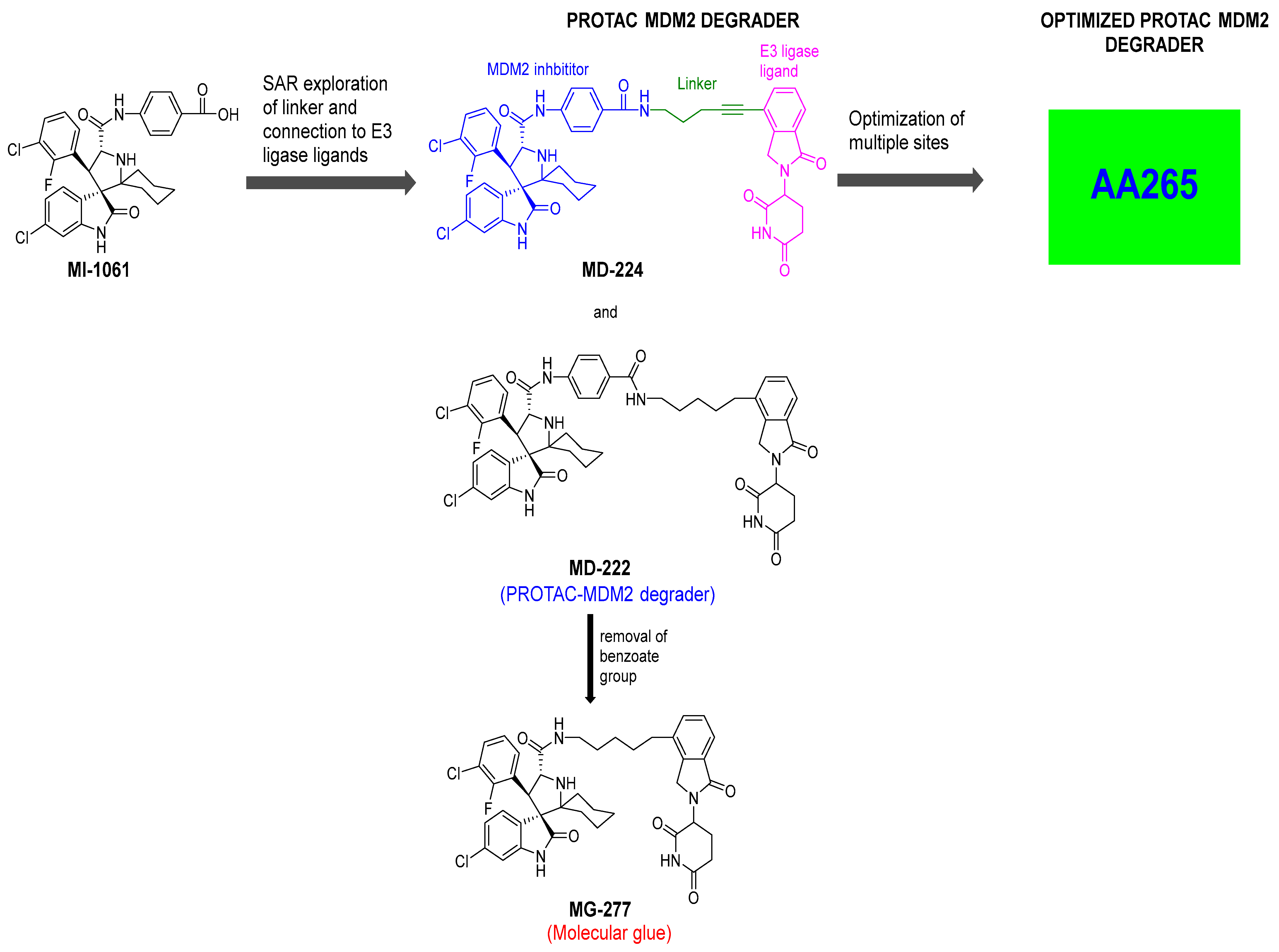
2. Targeting the Protein–Protein Interaction between MDM2 and p53
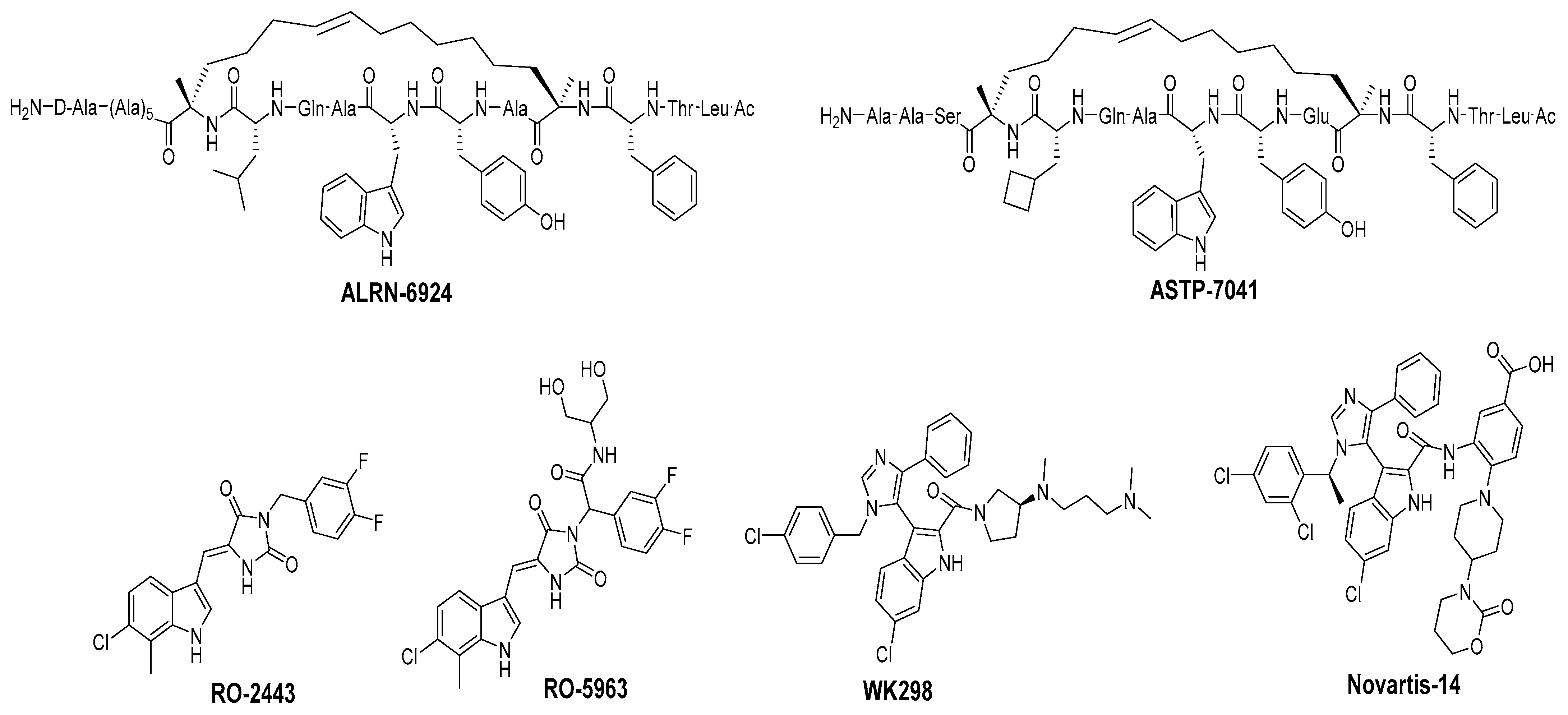
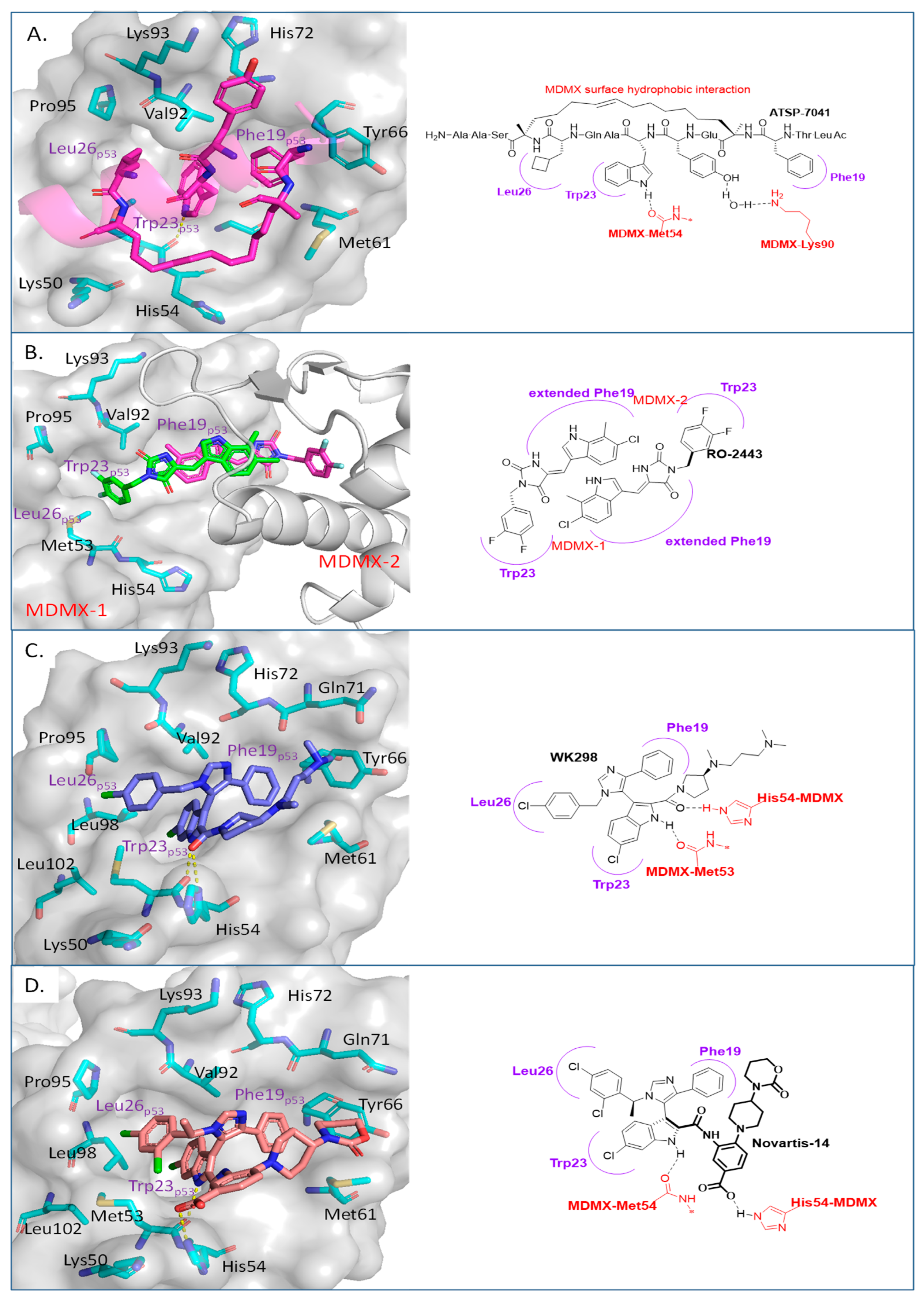
Disrupting MDMX–p53 Protein–Protein Interactions
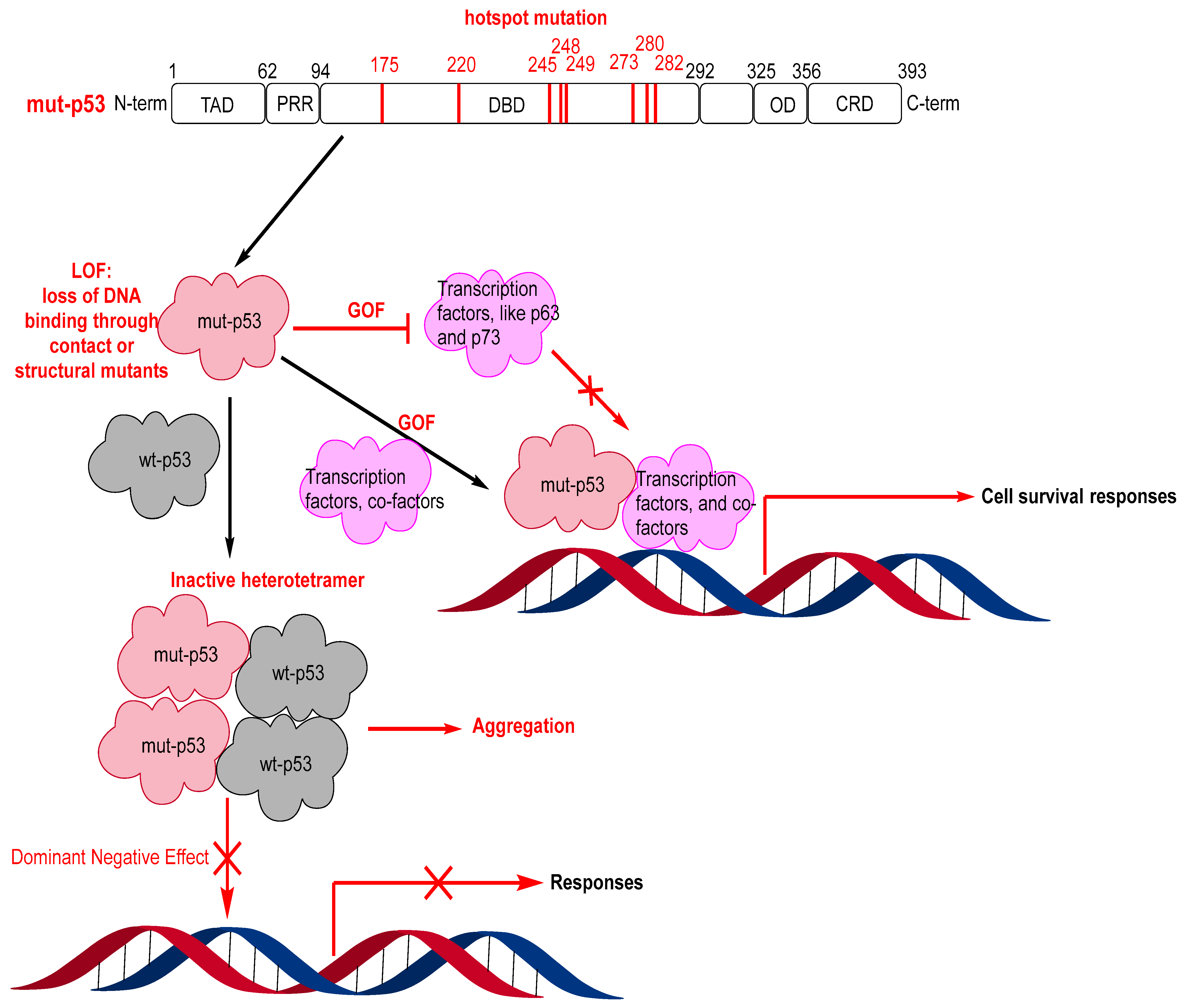
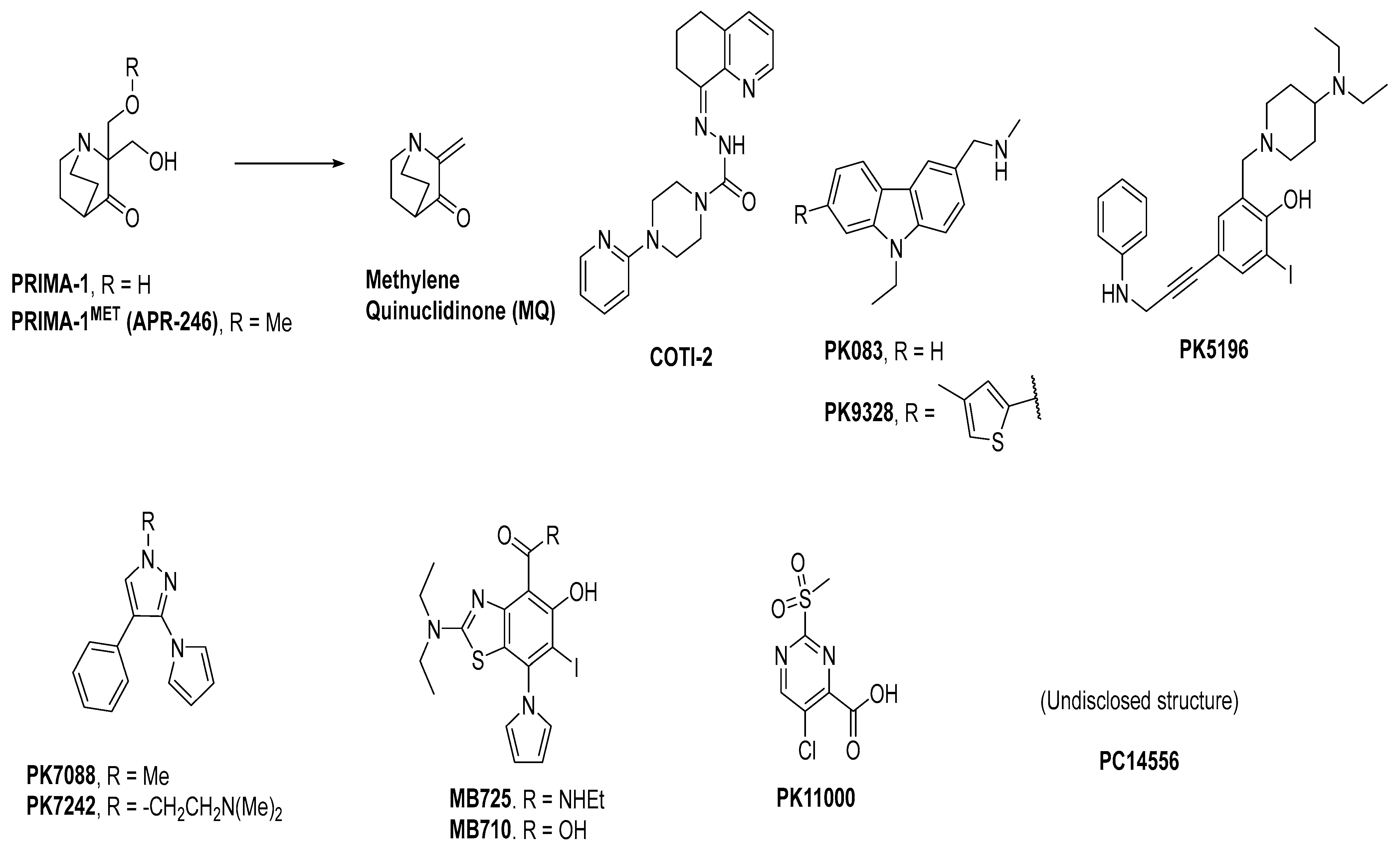
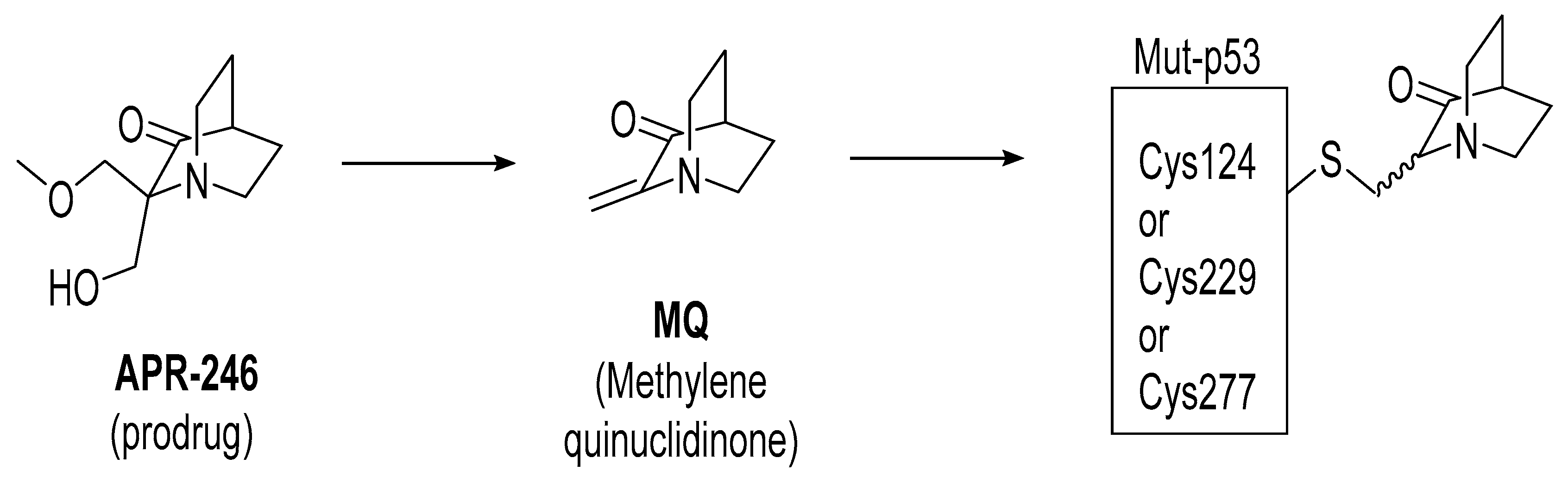
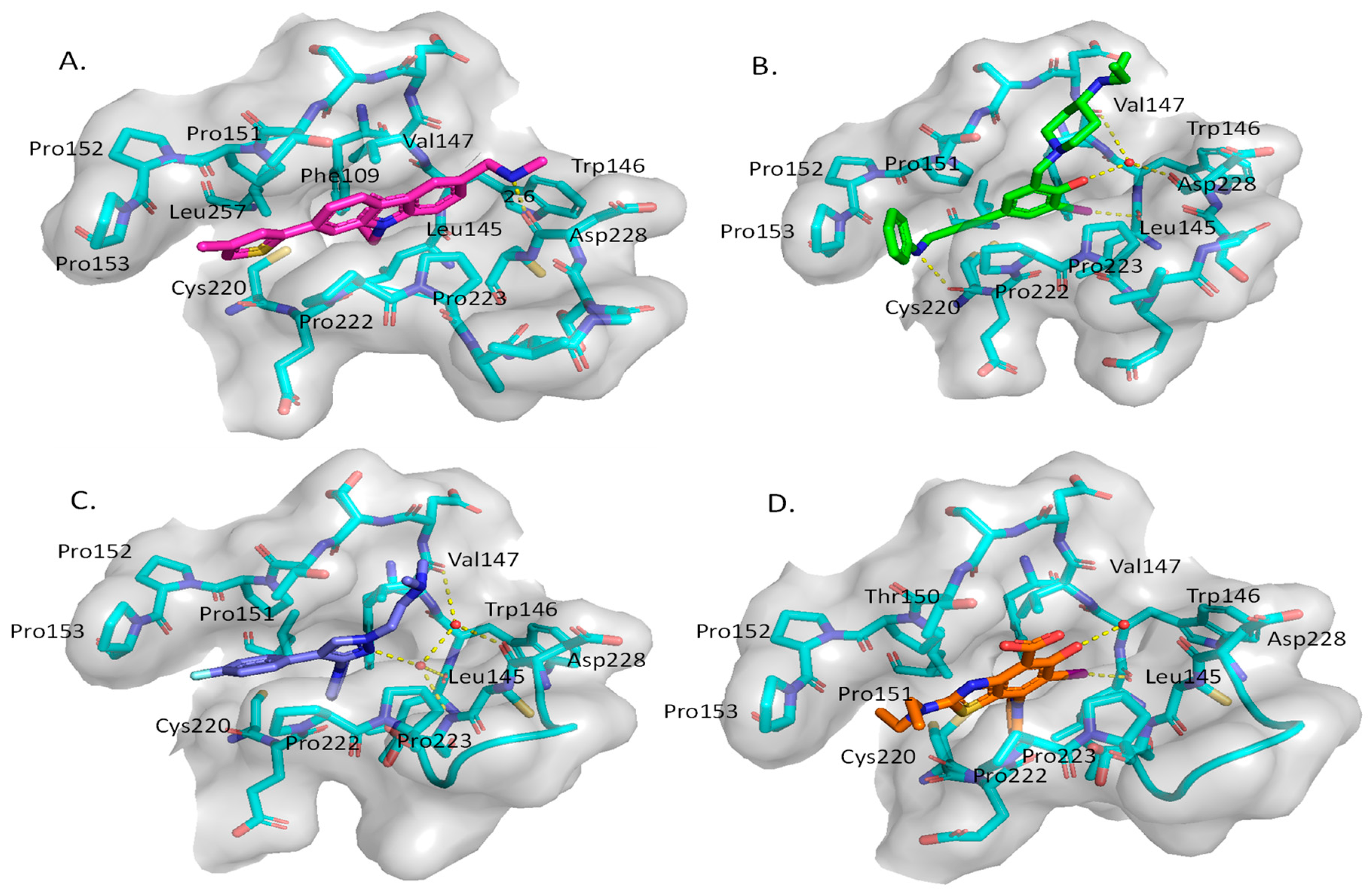
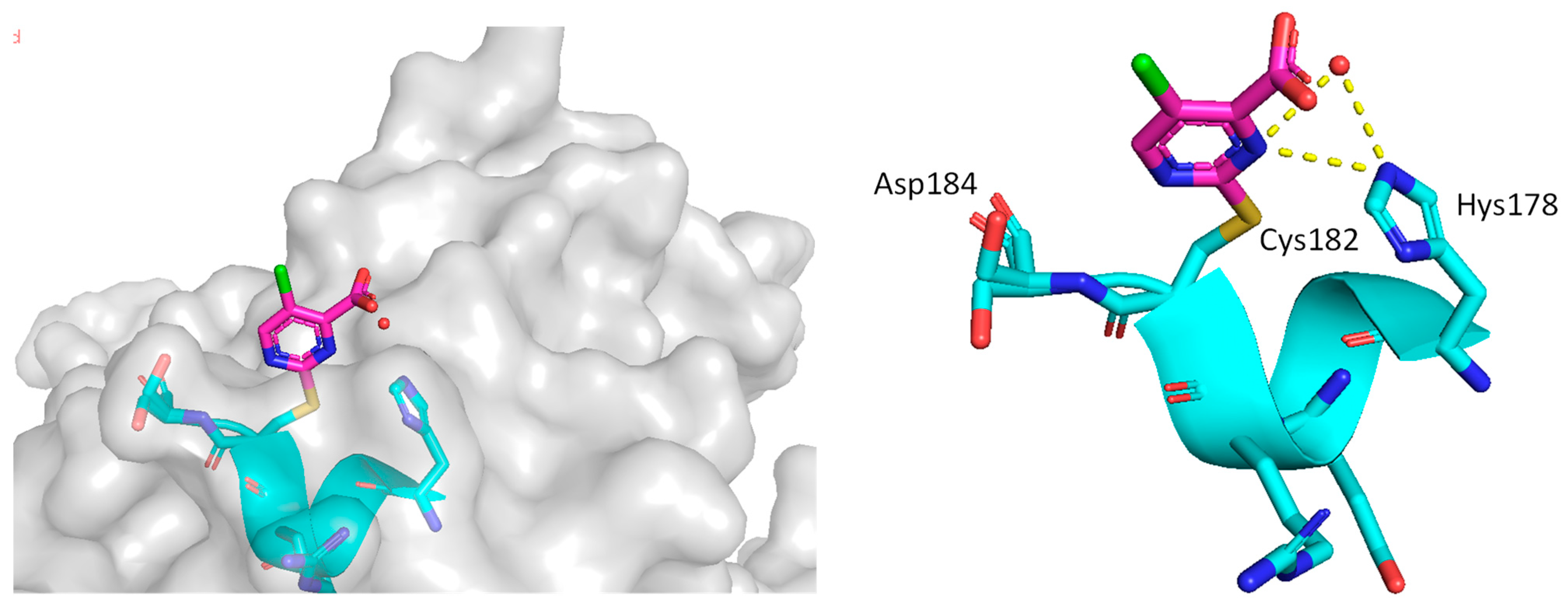
3. Mutant p53 Binders as Mutant p53 Activators
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lane, D.P. Cancer. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Nguyen, D.; Liao, W.; Zeng, S.X.; Lu, H. Reviving the Guardian of the Genome: Small Molecule Activators of P53. Pharmacol. Ther. 2017, 178, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Ladds, M.; Lain, S. Small Molecule Activators of the P53 Response. J. Mol. Cell Biol. 2019, 11, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Reed, M.; Wang, P.; Stenger, J.E.; Mayr, G.; Anderson, M.E.; Schwedes, J.F.; Tegtmeyer, P. P53 Domains: Identification and Characterization of Two Autonomous DNA-Binding Regions. Genes Dev. 1993, 7, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Klein, C.; Muller, L.; Hansen, S.; Buchner, J. P53 Contains Large Unstructured Regions in Its Native State. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Hock, A.; Vousden, K.H. Regulation of the P53 Pathway by Ubiquitin and Related Proteins. Int. J. Biochem. Cell Biol. 2010, 42, 1618–1621. [Google Scholar] [CrossRef]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The P53 Inhibitors Mdm2/Mdmx Complex Is Required for Control of P53 Activity in Vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.S.; et al. Activation of the P53 Pathway by Small-Molecule-Induced Mdm2 and Mdmx Dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The Human Homologs of Checkpoint Kinases Chk1 and Cds1 (Chk2) Phosphorylate P53 at Multiple DNA Damage-Inducible Sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [CrossRef]
- Shiloh, Y. Atm and Related Protein Kinases: Safeguarding Genome Integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of P53 Inhibits Its Ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of P53 Augments Its Site-Specific DNA Binding Both in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic Stability of Wild-Type and Mutant P53 Core Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed]
- Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution Structure of P53 Core Domain: Structural Basis for Its Instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Olszewski, M.B.; Gutkowska, M.; Helwak, A.; Zylicz, M.; Zylicz, A. Hsp70 Molecular Chaperones Are Required to Support P53 Tumor Suppressor Activity under Stress Conditions. Oncogene 2009, 28, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Gutkowska, M.; Klejman, M.P.; Wawrzynow, B.; Tracz, Z.; Wiech, M.; Zylicz, M.; Zylicz, A. Atp Binding to Hsp90 Is Sufficient for Effective Chaperoning of P53 Protein. J. Biol. Chem. 2010, 285, 32020–32028. [Google Scholar] [CrossRef]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the Guardian of the Genome. The Two-Faced Role of Molecular Chaperones in P53 Tumor Suppressor Action. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 161–174. [Google Scholar] [CrossRef]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary Structures of Tumor Suppressor P53 and a Specific P53 DNA Complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef]
- Melero, R.; Rajagopalan, S.; Lazaro, M.; Joerger, A.C.; Brandt, T.; Veprintsev, D.B.; Lasso, G.; Gil, D.; Scheres, S.H.; Carazo, J.M.; et al. Electron Microscopy Studies on the Quaternary Structure of P53 Reveal Different Binding Modes for P53 Tetramers in Complex with DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 557–562. [Google Scholar] [CrossRef]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W. Biogenesis of P53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. Implications on the Dominant Negative Effect. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative Binding of Tetrameric P53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by P53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant P53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of P53 Inhibitors: Progress, Challenges and Perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small Molecule Compounds Targeting the P53 Pathway: Are We Finally Making Progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef]
- Silva, J.L.; Lima, C.G.S.; Rangel, L.P.; Ferretti, G.D.S.; Pauli, F.P.; Ribeiro, R.C.B.; da Silva, T.B.; da Silva, F.C.; Ferreira, V.F. Recent Synthetic Approaches Towards Small Molecule Reactivators of P53. Biomolecules 2020, 10, 635. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The P53-Mdm-2 Autoregulatory Feedback Loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The Mdm-2 Oncogene Product Forms a Complex with the P53 Protein and Inhibits P53-Mediated Transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the Mdm2 Oncoprotein. Cell Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Oren, M. Mdm2: The Ups and Downs. Mol. Med. 1999, 5, 71–83. [Google Scholar] [CrossRef]
- Momand, J.; Wu, H.H.; Dasgupta, G. Mdm2—Master Regulator of the P53 Tumor Suppressor Protein. Gene 2000, 242, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.L.; Hu, W.W.; Levine, A.J. Mdm2 Is a Central Node in the P53 Pathway: 12 Years and Counting. Curr. Cancer Drug Targets 2005, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the Mdm2 Oncoprotein Bound to the P53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of Mdm2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of Rg7112: A Small-Molecule Mdm2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-Based Design of Potent Non-Peptide Mdm2 Inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-Based Design of Spiro-Oxindoles as Potent, Specific Small-Molecule Inhibitors of the Mdm2-P53 Interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal Activation of P53 by a Specific Mdm2 Inhibitor Is Selectively Toxic to Tumors and Leads to Complete Tumor Growth Inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomeric Spirooxindoles as Highly Potent and Efficacious Mdm2 Inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, S.; Sun, W.; Liu, L.; Lu, J.; McEachern, D.; Shargary, S.; Bernard, D.; Li, X.; Zhao, T.; et al. A Potent Small-Molecule Inhibitor of the Mdm2-P53 Interaction (Mi-888) Achieved Complete and Durable Tumor Regression in Mice. J. Med. Chem. 2013, 56, 5553–5561. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. Sar405838: An Optimized Inhibitor of Mdm2-P53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chu, X.J.; Liu, J.J.; Ding, Q.; Zhang, J.; Bartkovitz, D.; Jiang, N.; Karnachi, P.; So, S.S.; Tovar, C.; et al. Discovery of Potent and Orally Active P53-Mdm2 Inhibitors Ro5353 and Ro2468 for Potential Clinical Development. ACS Med. Chem. Lett. 2014, 5, 124–127. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of Rg7388, a Potent and Selective P53-Mdm2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of Amg 232, a Potent, Selective, and Orally Bioavailable Mdm2-P53 Inhibitor in Clinical Development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D. Discovery of a Small Molecule Mdm2 Inhibitor (Amg 232) for Treating Cancer. J. Med. Chem. 2014, 57, 6332–6341. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Chene, P.; De Pover, A.; Valat, T.S.; Lisztwan, J.H.; Kallen, J.; Masuya, K. The Central Valine Concept Provides an Entry in a New Class of Non Peptide Inhibitors of the P53-Mdm2 Interaction. Bioorg. Med. Chem. Lett. 2012, 22, 3498–3502. [Google Scholar] [CrossRef] [PubMed]
- Gessier, F.; Kallen, J.; Jacoby, E.; Chene, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Discovery of Dihydroisoquinolinone Derivatives as Novel Inhibitors of the P53-Mdm2 Interaction with a Distinct Binding Mode. Bioorg. Med. Chem. Lett. 2015, 25, 3621–3625. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (Nvp-Cgm097): A Highly Potent and Selective Mdm2 Inhibitor Undergoing Phase 1 Clinical Trials in P53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a Novel Class of Highly Potent Inhibitors of the P53-Mdm2 Interaction by Structure-Based Design Starting from a Conformational Argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef]
- Holzer, P. Discovery of Potent and Selective P53-Mdm2 Protein-Protein Interaction Inhibitors as Anticancer Drugs. Chimia 2017, 71, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the P53-Mdm2 Inhibitor Hdm201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wunschel, J.; Toedling, J.; Deubzer, H.E.; Kunkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating Tp53 Signaling by the Novel Mdm2 Inhibitor Ds-3032b as a Therapeutic Option for High-Risk Neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Sun, W.; Liu, L.; Lu, J.; McEachern, D.; Bernard, D.; Deschamps, J.R.; Wang, S. Design of Chemically Stable, Potent, and Efficacious Mdm2 Inhibitors That Exploit the Retro-Mannich Ring-Opening-Cyclization Reaction Mechanism in Spiro-Oxindoles. J. Med. Chem. 2014, 57, 10486–10498. [Google Scholar] [CrossRef]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B.; et al. Discovery of 4-((3′r,4′s,5′r)-6″-Chloro-4′-(3-Chloro-2-Fluorophenyl)-1′-Ethyl-2″-Oxodispiro[ Cyclohexane-1,2′-Pyrrolidine-3′,3″-Indoline]-5′-Carboxamido)Bicyclo[2.2.2]Octane -1-Carboxylic Acid (Aa-115/Apg-115): A Potent and Orally Active Murine Double Minute 2 (Mdm2) Inhibitor in Clinical Development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar]
- Rudolph, D.; Reschke, M.; Blake, S.; Rinnenthal, J.; Wernitznig, A.; Weyer-Czernilofsky, U.; Gollner, A.; Haslinger, C.; Garin-Chesa, P.; Quant, J.; et al. Bi 907828: A Novel, Potent Mdm2 Inhibitor That Induces Antitumor Immunologic Memory and Acts Synergistically with an Anti-Pd-1 Antibody in Syngeneic Mouse Models of Cancer. Cancer Res. 2018, 78, 4866. [Google Scholar] [CrossRef]
- Cornillie, J.; Wozniak, A.; Li, H.; Gebreyohannes, Y.K.; Wellens, J.; Hompes, D.; Debiec-Rychter, M.; Sciot, R.; Schoffski, P. Anti-Tumor Activity of the Mdm2-Tp53 Inhibitor Bi-907828 in Dedifferentiated Liposarcoma Patient-Derived Xenograft Models Harboring Mdm2 Amplification. Clin. Transl. Oncol. 2020, 22, 546–554. [Google Scholar] [CrossRef]
- Rinnenthal, J.; Rudolph, D.; Blake, S.; Gollner, A.; Wernitznig, A.; Weyer-Czernilofsky, U.; Haslinger, C.; Garin-Chesa, P.; Moll, J.; Kraut, N.; et al. Bi 907828: A Highly Potent Mdm2 Inhibitor with Low Human Dose Estimation, Designed for High-Dose Intermittent Schedules in the Clinic. Cancer Res. 2018, 78, 4865. [Google Scholar] [CrossRef]
- Gollner, A.; Rudolph, D.; Arnhof, H.; Bauer, M.; Blake, S.M.; Boehmelt, G.; Cockroft, X.L.; Dahmann, G.; Ettmayer, P.; Gerstberger, T.; et al. Discovery of Novel Spiro[3h-Indole-3,2′-Pyrrolidin]-2(1h)-One Compounds as Chemically Stable and Orally Active Inhibitors of the Mdm2-P53 Interaction. J. Med. Chem. 2016, 59, 10147–10162. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Wu, J.; Lyalin, D.; et al. Initial Testing (Stage 1) of Mk-8242-a Novel Mdm2 Inhibitor-by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2016, 63, 1744–1752. [Google Scholar] [CrossRef]
- Ravandi, F.; Gojo, I.; Patnaik, M.M.; Minden, M.D.; Kantarjian, H.; Johnson-Levonas, A.O.; Fancourt, C.; Lam, R.; Jones, M.B.; Knox, C.D.; et al. A Phase I Trial of the Human Double Minute 2 Inhibitor (Mk-8242) in Patients with Refractory/Recurrent Acute Myelogenous Leukemia (Aml). Leuk. Res. 2016, 48, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lahue, B.R.; Shipps, G.W., Jr.; Brookes, J.; Wang, Y. Substituted Piperidines as Hdm2 Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Lahue, B.R.; Ma, Y.; Nair, L.G.; Shipps, G.W., Jr.; Wang, Y.; Doll, R.; Bogen, S.L. Core Modification of Substituted Piperidines as Novel Inhibitors of Hdm2-P53 Protein-Protein Interaction. Bioorg. Med. Chem. Lett. 2014, 24, 1983–1986. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lahue, B.R.; Gibeau, C.R.; Shipps, G.W., Jr.; Bogen, S.L.; Wang, Y.; Guo, Z.; Guzi, T.J. Pivotal Role of an Aliphatic Side Chain in the Development of an Hdm2 Inhibitor. ACS Med. Chem. Lett. 2014, 5, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Bogen, S.L.; Pan, W.; Gibeau, C.R.; Lahue, B.R.; Ma, Y.; Nair, L.G.; Seigel, E.; Shipps, G.W., Jr.; Tian, Y.; Wang, Y.; et al. Discovery of Novel 3,3-Disubstituted Piperidines as Orally Bioavailable, Potent, and Efficacious Hdm2-P53 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 324–329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reutershan, M.H.; Machacek, M.R.; Altman, M.D.; Bogen, S.; Cai, M.; Cammarano, C.; Chen, D.; Christopher, M.; Cryan, J.; Daublain, P.; et al. Discovery of Mk-4688: An Efficient Inhibitor of the Hdm2-P53 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 16213–16241. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the Mdm2 Antagonist Rg7112 on the P53 Pathway in Patients with Mdm2-Amplified, Well-Differentiated or Dedifferentiated Liposarcoma: An Exploratory Proof-of-Mechanism Study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Iancu-Rubin, C.; Mosoyan, G.; Glenn, K.; Gordon, R.E.; Nichols, G.L.; Hoffman, R. Activation of P53 by the Mdm2 Inhibitor Rg7112 Impairs Thrombopoiesis. Exp. Hematol. 2014, 42, 137–145.e5. [Google Scholar] [CrossRef]
- Siu, L.L.; Italiano, A.; Miller, W.H.; Blay, J.Y.; Gietema, J.A.; Bang, Y.J.; Mileshkin, L.R.; Hirte, H.W.; Reckner, M.; Higgins, B.; et al. Phase 1 Dose Escalation, Food Effect, and Biomarker Study of Rg7388, a More Potent Second-Generation Mdm2 Antagonist, in Patients (Pts) with Solid Tumors. J. Clin. Oncol. 2014, 32, 2535. [Google Scholar] [CrossRef]
- De Weger, V.; Lolkema, M.P.; Dickson, M.; Le Cesne, A.; Wagner, A.; Merqui-Roelvink, M.; Varga, A.; Tap, W.; Schwartz, G.; Demetri, G.; et al. A First-in-Human (Fih) Safety and Pharmacological Study of Sar405838, a Novel Hdm2 Antagonist, in Patients with Solid Malignancies. Eur. J. Cancer 2014, 50, 121–122. [Google Scholar] [CrossRef]
- de Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Mace, S.; Tuffal, G.; et al. A Phase I Study of Sar405838, a Novel Human Double Minute 2 (Hdm2) Antagonist, in Patients with Solid Tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.A.; Fancourt, C.; Levonas, A.; Lam, R.; Meister, A.; Kemp, R.K.; et al. A Phase I Trial of the Human Double Minute 2 (Hdm2) Inhibitor Mk-8242 in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 10564. [Google Scholar] [CrossRef]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I Trial of the Human Double Minute 2 Inhibitor Mk-8242 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S., Jr.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Crews, C.M.; Georg, G.; Wang, S.M. Inducing Protein Degradation as a Therapeutic Strategy. J. Med. Chem. 2016, 59, 5129–5130. [Google Scholar] [CrossRef][Green Version]
- Bekes, M.; Langley, D.R.; Crews, C.M. Protac Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Chan, K.H.; Zengerle, M.; Testa, A.; Ciulli, A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (Bet) Degraders Derived from Triazolodiazepine (Jq1) and Tetrahydroquinoline (I-Bet726) Bet Inhibitor Scaffolds. J. Med. Chem. 2018, 61, 504–513. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of Protac Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule Protacs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.Y.; Wang, M.; Han, X.; Wang, S. Discovery of Md-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Aguilar, A.; Liu, Z.; Yang, C.Y.; Wang, S. Simple Structural Modifications Converting a Bona Fide Mdm2 Protac Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of Protac Degraders. J. Med. Chem. 2019, 62, 9471–9487. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, S.; Liu, J.; Yang, K.; Xie, H.; Tang, W. Development of Selective Small Molecule Mdm2 Degraders Based on Nutlin. Eur. J. Med. Chem. 2019, 176, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, J.; Tandon, I.; Wu, S.; Teng, P.; Liao, J.; Tang, W. Development of Mdm2 Degraders Based on Ligands Derived from Ugi Reactions: Lessons and Discoveries. Eur. J. Med. Chem. 2021, 219, 113425. [Google Scholar] [CrossRef]
- Marcellino, B.; Yang, X.B.; Chen, H.; Chen, K.; Brady, C.; Clementelli, C.; Li, Z.J.; Hoffman, R.; Liu, J.; Kaniskan, H.U.; et al. Development of an Mdm2 Degrader for Treatment of Acute Leukemias. Blood 2021, 138, 1866. [Google Scholar] [CrossRef]
- Chutake, Y.; Mayo, M.; Chen, D.; Enerson, B.; Cho, P.; Filiatrault, J.; Brown, C.; Placke, M.; Adams, M.; Karnik, R.; et al. Abstract 3934: Kt-253, a Highly Potent and Selective Heterobifunctional Mdm2 Degrader for the Treatment of Wildtype P53 Tumors with Superior Potency and Differentiated Biological Activity Compared to Small Molecule Inhibitors (Smi). Cancer Res. 2022, 82, 3934. [Google Scholar] [CrossRef]
- Laurie, N.A.; Donovan, S.L.; Shih, C.S.; Zhang, J.; Mills, N.; Fuller, C.; Teunisse, A.; Lam, S.; Ramos, Y.; Mohan, A.; et al. Inactivation of the P53 Pathway in Retinoblastoma. Nature 2006, 444, 61–66. [Google Scholar] [CrossRef]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of Mdmx (or Mdm4) Directly Contributes to Tumor Formation by Inhibiting P53 Tumor Suppressor Activity. Mol. Cell Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. Mdm4 Is a Key Therapeutic Target in Cutaneous Melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. Mdmx: A Novel P53-Binding Protein with Some Functional Properties of Mdm2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. Mdm2 Interacts with Mdmx through Their Ring Finger Domains. FEBS Lett. 1999, 447, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Leslie, P.L.; Ke, H.; Zhang, Y. The Mdm2 Ring Domain and Central Acidic Domain Play Distinct Roles in Mdm2 Protein Homodimerization and Mdm2-Mdmx Protein Heterodimerization. J. Biol. Chem. 2015, 290, 12941–12950. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Kawai, H.; Nie, L.; Kitao, H.; Wiederschain, D.; Jochemsen, A.G.; Parant, J.; Lozano, G.; Yuan, Z.M. Mutual Dependence of Mdm2 and Mdmx in Their Functional Inactivation of P53. J. Biol. Chem. 2002, 277, 19251–19254. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping P53 in Check: Essential and Synergistic Functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Czarna, A.; Holak, T.A. Structure of the Human Mdmx Protein Bound to the P53 Tumor Suppressor Transactivation Domain. Cell Cycle 2008, 7, 2441–2443. [Google Scholar] [CrossRef]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled Alpha-Helical Peptide Drug Development: A Potent Dual Inhibitor of Mdm2 and Mdmx for P53-Dependent Cancer Therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual Inhibition of Mdmx and Mdm2 as a Therapeutic Strategy in Leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef]
- Kallen, J.; Izaac, A.; Chau, S.; Wirth, E.; Schoepfer, J.; Mah, R.; Schlapbach, A.; Stutz, S.; Vaupel, A.; Guagnano, V.; et al. Structural States of Hdm2 and Hdmx: X-Ray Elucidation of Adaptations and Binding Interactions for Different Chemical Compound Classes. ChemMedChem 2019, 14, 1305–1314. [Google Scholar] [CrossRef]
- Zhang, S.; Lou, J.; Li, Y.; Zhou, F.; Yan, Z.; Lyu, X.; Zhao, Y. Recent Progress and Clinical Development of Inhibitors That Block Mdm4/P53 Protein-Protein Interactions. J. Med. Chem. 2021, 64, 10621–10640. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Domling, A.; Holak, T.A. Structures of Low Molecular Weight Inhibitors Bound to Mdmx and Mdm2 Reveal New Approaches for P53-Mdmx/Mdm2 Antagonist Drug Discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Joerg, K.; Julien, L.; Keiichi, M. 3-Imidazolylindoles as Mdm2 and Mdm4 Inhibitors and Their Preparation; Novartis: Basel, Switzerland, 2012; 333p. [Google Scholar]
- Jung, J.; Lee, J.S.; Dickson, M.A.; Schwartz, G.K.; Le Cesne, A.; Varga, A.; Bahleda, R.; Wagner, A.J.; Choy, E.; de Jonge, M.J.; et al. Tp53 Mutations Emerge with Hdm2 Inhibitor Sar405838 Treatment in De-Differentiated Liposarcoma. Nat. Commun. 2016, 7, 12609. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Momand, J.; Finlay, C.A. The P53 Tumour Suppressor Gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the P53 Tumor Suppressor Gene: Clues to Cancer Etiology and Molecular Pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The Tp53 Website: An Integrative Resource Centre for the Tp53 Mutation Database and Tp53 Mutant Analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Therapeutic Targeting of P53: All Mutants Are Equal, but Some Mutants Are More Equal Than Others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (Gof) Mutant P53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. P53 from Complexity to Simplicity: Mutant P53 Stabilization, Gain-of-Function, and Dominant-Negative Effect. FASEB J. 2000, 14, 1901–1907. [Google Scholar] [CrossRef]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive Full-Length P53 Mutants Lacking Dominant Wild-Type P53 Inhibition Highlight Loss of Heterozygosity as an Important Aspect of P53 Status in Human Cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of Function of Mutant P53 by Coaggregation with Multiple Tumor Suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant P53: An Oncogenic Transcription Factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of P53 Function Leads to Tumour Regression in Vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting Mutant P53 for Cancer Therapy: Direct and Indirect Strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of Mutant P53 Functional Properties on Tp53 Mutation Patterns and Tumor Phenotype: Lessons from Recent Developments in the Iarc Tp53 Database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. Tp53 Variations in Human Cancers: New Lessons from the Iarc Tp53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. Tp53 Mutations in Human Cancers: Functional Selection and Impact on Cancer Prognosis and Outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structure-Function-Rescue: The Diverse Nature of Common P53 Cancer Mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative Analysis of Residual Folding and DNA Binding in Mutant P53 Core Domain: Definition of Mutant States for Rescue in Cancer Therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a P53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Bullock, A.N.; Fersht, A.R. Rescuing the Function of Mutant P53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor P53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The P53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. Prima-1(Met) Synergizes with Cisplatin to Induce Tumor Cell Apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; Koropatnick, J. Coti-2, a New Anticancer Drug Currently under Clinical Investigation, Targets Mutant P53 and Negatively Modulates the Pi3k/Akt/Mtor Pathway. Eur. J. Cancer 2016, 69, S19. [Google Scholar] [CrossRef]
- Westin, S.N.; Nieves-Neira, W.; Lynam, C.; Salim, K.Y.; Silva, A.D.; Ho, R.T.; Mills, G.B.; Coleman, R.L.; Janku, F.; Matei, D. Safety and Early Efficacy Signals for Coti-2, an Orally Available Small Molecule Targeting P53, in a Phase I Trial of Recurrent Gynecologic Cancer. Cancer Res. 2018, 78, 615. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the Tumor Suppressor Function to Mutant P53 by a Low-Molecular-Weight Compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. Apr-246 Reactivates Mutant P53 by Targeting Cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Prima-1 Reactivates Mutant P53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsen, L.; Rozenberg, H.; Shakked, Z. Structural Basis of Reactivation of Oncogenic P53 Mutants by a Small Molecule: Methylene Quinuclidinone (Mq). Nat. Commun. 2021, 12, 7057. [Google Scholar] [CrossRef]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maiga, S.; Lode, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. Prima-1met Induces Myeloma Cell Death Independent of P53 by Impairing the Gsh/Ros Balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the System Xc(-)/Glutathione Axis Selectively Targets Cancers with Mutant-P53 Accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; Koropatnick, J. Coti-2, a Novel Small Molecule That Is Active against Multiple Human Cancer Cell Lines in Vitro and in Vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.L.; Osman, A.A.; Patel, A.A.; Tanaka, N.; Tang, L.; Zhou, G.; Myers, J.N. A Novel Third Generation Thiosemicarbazone, Coti-2, Is Highly Effective in Killing Head and Neck Squamous Cell Carcinomas (Hnscc) Bearing a Variety of Tp53 Mutations. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 942. [Google Scholar] [CrossRef]
- Wang, G.; Fersht, A.R. Multisite Aggregation of P53 and Implications for Drug Rescue. Proc. Natl. Acad. Sci. USA 2017, 114, E2634–E2643. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A Structure-Guided Molecular Chaperone Approach for Restoring the Transcriptional Activity of the P53 Cancer Mutant Y220c. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural Basis for Understanding Oncogenic P53 Mutations and Designing Rescue Drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted Rescue of a Destabilized Mutant of P53 by an in Silico Screened Drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef]
- Wilcken, R.; Liu, X.R.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant P53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small Molecule Induced Reactivation of Mutant P53 in Cancer Cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Bauer, M.R.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Roy, D.H.; Joerger, A.C.; Fersht, A.R. Aminobenzothiazole Derivatives Stabilize the Thermolabile P53 Cancer Mutant Y220c and Show Anticancer Activity in P53-Y220c Cell Lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild Alkylating Agents with Anticancer Activity toward P53-Compromised Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [PubMed]
- Puzio-Kuter, A.M.; Mulligan, C.; Russo, B.; Wiebesiek, A.; Xu, L.; Yang, H.; Vu, B.; Dumble, M. Abstract 1295: Small Molecule Reactivators of Y220c Mutant P53 Modulate Tumor Infiltrating Leukocytes and Synergize with Immune Checkpoint Inhibitors. Cancer Res. 2022, 82, 1295. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target Binding | Cell Potency | In Vivo | Development Stage | PDB | Reference |
|---|---|---|---|---|---|---|
 | MDM2 HTRF IC50 = 18 nM | wt-p53 cells (HCT-116, SJSA-1, RKO) Average MTT IC50 = 400 nM | SJSA-1, Tumor regression with over 100 mg/kg PO, QD | Clinical trials | 4IPF | [36] |
 | MDM2 Ki = 0.88 nM | wt-p53 cells SJSA-1 IC50 = 92 nM RS4;11 IC50 = 89 nM HCT-116 IC50 = 200 nM | SJSA-1 and RS4;11 100% Tumor regression with 100 mg/kg PO, QD | Clinical trials | 5TRF | [37] |
 | MDM2 HTRF IC50 = 6 nM | wt-p53 cells (HCT-116, SJSA-1, RKO) Average MTT IC50 = 30 nM | SJSA-1 Tumor regression with 50 mg/kg PO, QD | Clinical trials | Analog co-crystal 4JSC | [38] |
 | MDM2 HTRF IC50 = 0.6 nM | wt-p53 cells SJSA-1 EdU assay IC50 = 9.1 nM | SJSA-1 Complete Tumor regression with 60 mg/kg PO, QD | Clinical trials | Analog co-crystal 4OAS | [39,40] |
 | hMDM2 TR-FRET IC50 = 1.7 nM hMDM4 TR-FRET IC50 = 2000 nM | wt-p53 cells SJSA-1 IC50 = 353 nM HCT116 IC50 = 454 nM | SJSA-1 Complete Tumor regression with 70 mg/kg PO, 3QW >85% regression 30 mg/kg PO, QD | Clinical trials | 4ZYF | [41] |
 | Not disclosed | Not disclosed | 10-fold better than NVP-CGM097 | Clinical trials | 5OC8 | [42] |
 | Not disclosed | wt-p53 cells SK-N-SH IC50 = 22 nM SH-SY5Y IC50 = 18 nM IMR32 IC50 = 53 nM IMR5 IC50 = 26 nM LAN5 IC50 = 44 nM | Wt-TP53 neuroblastoma TGI and prolonged survival | Clinical trials | None | [43] |
 | MDM2 IC50 = 0.0048 µM | wt-p53 cells SJSA-1 IC50 = 60 nM HCT116 IC50 = 104 nM | SJSA-1 Complete Tumor regression with over 100 mg/kg PO, QD | Clinical trials | None | [44] |
 | MDM2 IC50 = 4 nM | wt-p53 cells SJSA-1 IC50 = 471 nM | SJSA-1 Tumor regression with 25 mg/kg PO, QD or 100 mg/kg PO, single dose | BI-907828 in clinical trials | 5LAZ | [45] |
 | MDM2 IC50 = 7 nM | wt-p53 cells SJSA-1 IC50 = 80 nM | SJSA-1 TGI = 92% with 50 mg/kg PO, QD 3 days for 4 weeks, or Tumor regression = 60% with 200 mg/kg PO, QD 24 days | NO | 5HMH | [46] |
 | MDM2 Kd = 3.8 nM | wt-p53 cells median IC50 = 70 nM | Extended survival and tumor regression in various in vivo models | Clinical trials | None | [47,48] |
 | MDM2 TR-FRET IC50 = 0.65 nM | wt-p53 cells HCT116 IC50 = 122 nM | SJSA-1 Tumor regression with 100 mg/kg PO, QD 7 days | Preclinical | 7NA2 | [49] |
| PROTAC–MDM2 Degrader | Degradation | Cell Potency | In Vivo | Development Stage | Reference |
|---|---|---|---|---|---|
 | DC50 < 1 nM (DC60 = 1 nM) | RS4;11 IC50 = 2.8 nM | NA | Discovery | [82] |
 | DC50 < 1 nM (DC80 = 1 nM) | RS4;11 IC50 = 1.5 nM | RS4;11 complete tumor regression with 25 mg/kg qd (1–5) or 50 mg/kg qd (1, 3, 5) | Discovery | [82] |
| (Undisclosed structure) AA-265 | DC50 < 1 nM | RS4;11 IC50 = 0.72 nM | Unpublished | Clinical candidate | |
 | DC50 = 22.6 nM | RS4;11 IC50 = 3.2 nM | NA | Discovery | [84] |
 | MDM2 DC50 = 4.1 nM p53 DC50 = 29 nM | RS4;11 IC50 = 1.2 nM | NA | Discovery | [85] |
| (Undisclosed structure) KT-253 | DC50 = 0.4 nM | RS4;11 IC50 = 0.3 nM | RS4;11 complete tumor regression with single dose at 1 mg/kg, or 3 mg/kg, or 10 mg/kg | Clinical candidate | [87] |
| (Undisclosed structure) XY-27 | Undisclosed | Undisclosed | No data | Discovery | [86] |
| MDMX Inhibitors | Target Binding | Cell Potency | In Vivo | Development Stage | PDB | Reference |
|---|---|---|---|---|---|---|
 | MDM2 IC50 = 7.7 nM MDMX IC50 = 24.7 nM | MOLM13 (24 h) IC50 = 1.4 µM MOLM14 (24 h) IC50 = 1.4 µM ML2 (24 h) IC50 = 7.9 µM OCI/AML5 (24 h) IC50 = >10 µM OCI/AML5 (24 h) IC50 = 3.6 µM | Improved survival in AML Xenograft models | Clinical trials | NA | [98] |
 | MDM2 Ki = 0.9 nM MDMX Ki = 6.8 nM | SJSA-1 IC50 = 50 nM (1% FBS) IC50 = 600 nM (10% FBS) | SJSA-1 (iv) 15 mg/kg or 30 mg/kg achieved TGI = 61% MCF-7 (iv) 20 mg/kg TGI = 63% and 30 mg/kg TGI = 87% | NO | 4N5T | [97] |
 | MDM2 IC50 = 33 nM MDMX IC50 = 41 nM | Poor solubility | NA | NO | 3U15 | [8] |
 | MDM2 IC50 = 17 nM MDMX IC50 = 24 nM | Active in WT-p53 cells MCF-7, HCT-116, RKO; Inactive in MT-p53 cells SW480, MDA-MB435 | NA | NO | NA | [8] |
 | MDM2 FP IC50 = 109 nM MDMX FP IC50 = 19,700 nM | NA | NA | NO | 3LBJ | [101] |
 | MDM2 TR-FRET IC50 < 0.1 nM MDMX TR-FRET IC50 = 17 nM | NA | NA | NO | 6Q9S | [99] |
| Mutant p53 Reactivators | Target Binding | Cell Potency | In Vivo | Development Stage | PDB | Reference |
|---|---|---|---|---|---|---|
 | Covalent binding to Cys124, Cys229, Cys277 | Saos-2 (His-273 mutant p53) PRIMA-1 IC50 = 14 µM, APR246 IC50 = 9 µM; H1299 (His-175 mutant p53) PRIMA-1 IC50 = 24 µM, APR246 IC50 = 19 µM | PRIMA-1: Saos-2-His-273 xenograft IV, BID, 3 days at 20 mg/kg or 100 mg/kg achieve 90% and 98% TGI, respectively | APR-246 in clinical trials | NA | [125,128] |
 | NA | nanomolar (nM) inhibition of proliferation after 72 h treatment of a diverse group of cancer cell lines, regardless of tissue origin and genetic background | Significant TGI in SHP-77 (Human SCLC) xenograft at 3 mg/kg IV; and in OVCAR3 (Human Ovarian Carcinoma) xenografts 20 mg/kg IV or 75 mg/kg PO | Clinical trials | NA | [134] |
 | Mutant p53 Y220C PK083 Kd = 125 µM PK9328 Kd = 1.7 µM | PK9328 100% viability loss at 7.5 µM in HUH7 cells ~50% viability loss at 7.5 µM in HUH7 p53 KO cells | NO | Discovery | PK083 PDB: 2VUK PK9328 PDB: 6GGF | [137,139] |
 | Mutant p53 Y220C Kd = 9.7 µM | 50 µM induced apoptosis onset in NUGC-3 (Y220C mutant p53) and No apoptosis in NUGC-4 (wt-p53) | NO | Discovery | 4AGQ | [140] |
 | Covalent binding Cys182, Cys277 | PK11007: Viability reduction at 15 to 30 µM in mutant p53 cell lines MKN1(V143A), HUH-1(Y220C), NUGC-3(Y220C), and SW480(R273H/P309S) and wt-p53 HUH-6, NUGC-4, and WI38 cells were less sensitive; reducing viability between 60 and 120 µM also has p53-independent activity | NO | Discovery | PK11000 PDB: 5LAP | [143] |
 | Mutant p53 Y220C Kd = 140 µM | PK7088 at 200 µM induces apoptosis in NUGC-3, HUH-7 (Y220C mutant p53 cells), and minimal effect in NUGC-4, HUH-6 (wt-p53) | NO | Discovery | PK7242 PDB: 3ZME | [141] |
 | MB725, Kd = not soluble MB710, Kd = 4 µM | IC50s Y220C mutant p53 NUGC3 MB725 = 10 µM, MB710 = 90 µM; BXPC3 MB725 = 18 µM, MB710 = ND; HUH7 MB725 = 10 µM, MB710 = ND; and in wt-p53 NUGC4, WI38 MB725, M710 >120 µM; HUH6 MB725, MB710 = ND; and in R273C mutant p53 SW1088 MB725, MB710 >120 µM | NO | Discovery | MB710 PDB: 5O1I | [142] |
| PC14586 (Undisclosed structure) | Y220C binder | Lymphoma and sarcoma cell lines generated from HUPKI-Y220C mouse tumors were sensitive to Y220C p53 reactivator PC14586 IC50 ~192–722 nM | HUPKI Y220C mutant sarcoma cell xenograft 100 mg/kg QD antitumor activity with two cures and synergy with 50 mg/kg PC14586 QD + 200 µg anti-PD-1 Q3D | Clinical trials | NA | [144] |
| Drug | Target | Development Phase | Combination | Disease | NCT Number | Status |
|---|---|---|---|---|---|---|
| RG7112 | MDM2–p53 inhibitor | Phase I | Myelogenous Leukemia, Chronic Neoplasms, Myelogenous Leukemia, Acute | NCT01677780 | Completed | |
| Phase I | Neoplasms | NCT01164033 | Completed | |||
| Phase I | Doxorubicin | Sarcoma | NCT01605526 | Completed | ||
| Phase I | Sarcoma | NCT01143740 | Completed | |||
| Phase I | Hematologic Neoplasms | NCT00623870 | Completed | |||
| Phase I | Neoplasms | NCT00559533 | Completed | |||
| Phase I | Cytarabine | Acute Myelogenous Leukemia | NCT01635296 | |||
| MI773 | MDM2–p53 inhibitor | Phase I | Neoplasm Malignant | NCT01636479 | Completed | |
| Phase I | Pimasertib | Neoplasm Malignant | NCT01985191 | Completed | ||
| RG7388 (Idasanutlin) | MDM2–p53 inhibitor | Phase I | Pegasys | Polycythemia Vera, Essential Thrombocythemia | NCT02407080 | Completed |
| Phase III | Cytarabine Placebo | Leukemia, Myeloid, Acute | NCT02545283 | Terminated | ||
| Phase II | Polycythemia Vera | NCT03287245 | Terminated | |||
| Phase I | Placebo [13C]-radiolabeled Idasanutlin [14C]-radiolabeled Idasanutlin | Solid Tumors | NCT02828930 | Completed | ||
| Phase I | Solid Tumors | NCT03362723 | Completed | |||
| Phase I, II | Cytarabine Daunorubicin Allogeneic Hematopoietic Stem Cell Transplant (Allo-HSCT) | Acute Myeloid Leukemia | NCT03850535 | Terminated | ||
| Phase I, II | Venetoclax Cyclophosphamide Topotecan Fludarabine Cytarabine Intrathecal Chemotherapy | Acute Myeloid Leukemia (AML), Acute Lymphoblastic Leukemia (ALL), Neuroblastoma, Solid Tumors | NCT04029688 | Recruiting | ||
| Phase I, II | Dexamethasone Ixazomib Citrate | Loss of Chromosome 17p, Recurrent Plasma Cell Myeloma | NCT02633059 | Active, not recruiting | ||
| Phase I, II | Obinutuzumab Venetoclax Rituximab | Follicular Lymphoma, Lymphoma, Large B Cell, Diffuse | NCT03135262 | Terminated | ||
| Phase I, II | Obinutuzumab Rituximab | Non-Hodgkin Lymphoma | NCT02624986 | Terminated | ||
| Phase I | Cobimetinib Venetoclax | Leukemia, Myeloid, Acute | NCT02670044 | Completed | ||
| Phase I, II | Atezolizumab Cobimetinib | Stage III, IIIA, IIIB, IIIC, IV Breast Cancer, Estrogen Receptor-positive, HER2/Neu Negative | NCT03566485 | Terminated | ||
| Phase II | Entrectinib Alectinib Atezolizumab Ipatasertib Trastuzumab emtansine Inavolisib Belvarafenib Pralsetinib | Solid Tumors | NCT04589845 | Recruiting | ||
| Phase I, II | Regorafenib Atezolizumab Imprime PGG Bevacizumab Isatuximab Selicrelumab AB928 Genetic: LOAd703 | Colorectal Cancer | NCT03555149 | Active, not recruiting | ||
| Phase I, II | APG101 Alectinib Atezolizumab Vismodegib Temsirolimus Palbociclib | Glioblastoma, Adult | NCT03158389 | Recruiting | ||
| AMG232 | MDM2–p53 inhibitor | Phase I | Advanced Malignancy, Advanced Solid Tumors, Glioblastoma, Multiple Myeloma | NCT01723020 | Completed | |
| Phase I | Trametinib | Advanced Malignancy, Acute Myeloid Leukemia | NCT02016729 | Completed | ||
| Phase I | Trametinib Dabrafenib | Advanced Malignancy, Advanced Solid Tumors, Melanoma | NCT02110355 | Completed | ||
| Phase I | Navtemadlin Radiation: Radiation Therapy | Glioblastoma, Gliosarcoma, MGMT-Unmethylated Glioblastoma, Recurrent Glioblastoma | NCT03107780 | Suspended | ||
| Phase I | Carfilzomib Dexamethasone Dexamethasone Sodium Phosphate Lenalidomide Navtemadlin | Plasmacytoma, Recurrent Plasma Cell Myeloma, Refractory Plasma Cell Myeloma | NCT03031730 | Recruiting | ||
| Phase I | Decitabine Navtemadlin | Acute Myeloid Leukemia, Recurrent Acute Myeloid Leukemia, Refractory Acute Myeloid Leukemia, Secondary Acute Myeloid Leukemia | NCT03041688 | Suspended | ||
| Phase I | Navtemadlin Radiation: Radiation Therapy | Resectable Soft Tissue Sarcoma, Soft Tissue Sarcoma | NCT03217266 | Active, not recruiting | ||
| Phase I | Cytarabine Idarubicin Hydrochloride Navtemadlin | Acute Myeloid Leukemia, Acute Myeloid Leukemia Arising From Previous Myelodysplastic Syndrome | NCT04190550 | Recruiting | ||
| NVPCGM097 | MDM2–p53 inhibitor | Phase I | Solid Tumor With p53 Wild-Type Status | NCT01760525 | Completed | |
| NVPHDM201 | MDM2–p53 inhibitor | Phase I, II | Pazopanib | Advanced Soft-Tissue Sarcoma, Metastatic Soft-Tissue Sarcoma | NCT05180695 | Recruiting |
| Phase I | Midostaurin | AML, Adult | NCT04496999 | Recruiting | ||
| Phase I | Trametinib | Colorectal Cancer, Advanced Cancer, Metastatic Cancer | NCT03714958 | Recruiting | ||
| Phase I | LEE011 | Liposarcoma | NCT02343172 | Completed | ||
| Phase I | Ancillary treatment | Advanced Solid and Hematological WT-TP53 Tumors | NCT02143635 | Completed | ||
| Phase I | MBG453 Venetoclax | Acute Myeloid Leukemia (AML), High-Risk Myelodysplastic Syndrome (MDS) | NCT03940352 | Recruiting | ||
| Phase I, II | cytarabine anthracycline midostaurin liposomal cytarabine/daunorubicin posaconazole midazolam | Leukemia, Myeloid, Acute | NCT03760445 | Withdrawn | ||
| Phase I | LXS196 | Uveal Melanoma | NCT02601378 | Terminated | ||
| Phase I, II | Siremadlin | Acute Myeloid Leukemia, Allogeneic Stem Cell Transplantation | NCT05447663 | Not yet recruiting | ||
| Phase I, II | siremadlin venetoclax azacitidine | Acute Myeloid Leukemia | NCT05155709 | Recruiting | ||
| Phase I, II | Ruxolitinib Siremadlin Crizanlizumab Sabatolimab LTT462 NIS793 | Myelofibrosis | NCT04097821 | Active, not recruiting | ||
| Phase II | Ribociclib Cabozantinib Alectinib Regorafenib Trametinib Dabrafenib | Malignant Solid Tumor | NCT04116541 | Recruiting | ||
| Phase I | PDR001 LCL161 Everolimus Panobinostat QBM076 | Colorectal Cancer, Non-Small Cell Lung Carcinoma (Adenocarcinoma), Triple Negative Breast Cancer, Renal Cell Carcinoma | NCT02890069 | Completed | ||
| DS3032 (Milademetan) | MDM2–p53 inhibitor | Phase I | Myeloma | NCT02579824 | Terminated | |
| Phase I, II | Cytarabine Venetoclax | Acute Myeloid Leukemia, Recurrent Acute Myeloid Leukemia, Refractory Acute Myeloid Leukemia | NCT03634228 | Completed | ||
| Phase I | Acute Myeloid Leukemia | NCT03671564 | Completed | |||
| Phase I | AZA | Acute Myelogenous Leukemia, Myelodysplastic Syndrome | NCT02319369 | Terminated | ||
| Phase I | Quizartinib | Acute Myeloid Leukemia | NCT03552029 | Terminated | ||
| Early Phase I | Itraconazole Posaconazole | Pharmacokinetics | NCT03614455 | Completed | ||
| Phase I | Advanced Solid Tumor, Lymphoma | NCT01877382 | Completed | |||
| Early Phase I | Food Effects on Pharmacokinetics | NCT03647202 | Completed | |||
| APG115 (Alrizomadlin) | MDM2–p53 inhibitor | Phase I | Patients With Advanced Solid Tumor or Lymphoma | NCT02935907 | Completed | |
| Phase II | APG-2575 | T-Prolymphocytic Leukemia | NCT04496349 | Recruiting | ||
| Phase I, II | Toripalimab | Liposarcoma, Advanced Solid Tumor | NCT04785196 | Recruiting | ||
| Phase I, II | 5-azacitidine | Acute Myeloid Leukemia, Chronic (AML), Myelomonocytic Leukemia (CMML), Myelodysplastic Syndromes, High-Risk Myelodysplastic Syndrome, MDS | NCT04358393 | Recruiting | ||
| Phase I, II | Pembrolizumab | Unresectable or Metastatic Melanoma or Advanced Solid Tumors, Melanoma, Uveal Melanoma, P53 Mutation, MDM2 Gene Mutation, MPNST, Cutaneous Melanoma, Mucosal Melanoma, Malignant Peripheral Nerve Sheath Tumors | NCT03611868 | Recruiting | ||
| Phase I, II | Carboplatin | Malignant Salivary Gland Cancer, Salivary Gland Cancer | NCT03781986 | Recruiting | ||
| Phase I | Azacitidine Cytarabine | Acute Myeloid Leukemia (AML), Myelodysplastic Syndromes (MDS) | NCT04275518 | Recruiting | ||
| BI-907828 | MDM2–p53 inhibitor | Phase I | Rifampicin | Solid Tumors | NCT05372367 | Recruiting |
| MK-8242 | MDM2–p53 inhibitor | Phase I | Acute Myelogenous Leukemia (AML) | NCT01451437 | Terminated | |
| Phase I | Solid Tumors | NCT01463696 | Terminated | |||
| ALRN-6924 | MDM2/MDMX inhibitor | Phase I | cytarabine | Acute Myeloid Leukemia, Myelodysplastic Syndromes | NCT02909972 | Completed |
| Phase I | Paclitaxel | Advanced Malignant Solid Neoplasm, Anatomic Stage III Breast Cancer AJCC v8, Anatomic Stage IIIA Breast Cancer AJCC v8, Anatomic Stage IIIB Breast Cancer AJCC v8, Anatomic Stage IIIC Breast Cancer AJCC v8, Estrogen Receptor Positive, HER2/Neu Negative, Metastatic Malignant Solid Neoplasm, Prognostic Stage III Breast Cancer AJCC v8, Prognostic Stage IIIA Breast Cancer AJCC v8, and 5 more | NCT03725436 | Recruiting | ||
| Phase I, II | Solid Tumor, Lymphoma, Peripheral T-Cell Lymphoma | NCT02264613 | Completed | |||
| Phase I | Cytarabine | Leukemia, Brain Tumor, Solid Tumor, Lymphoma | NCT03654716 | Recruiting | ||
| Phase I | Carboplatin, Pemetrexed, Placebo, Topotecan | Non-Small Cell Lung Cancer, Small-Cell Lung Cancer | NCT04022876 | Active, not recruiting | ||
| APR-246 (eprenetapopt) | Mutant p53 activator | Phase I, II | Oesophageal Carcinoma | NCT02999893 | Terminated | |
| Phase I | Venetoclax, Azacitidine | Myeloid Malignancy | NCT04214860 | Completed | ||
| Phase II | Acute Myeloid Leukemia or Myelodysplastic Syndromes | NCT03931291 | Completed | |||
| Phase I, II | Azacitidine | Myelodysplastic Syndrome With Gene Mutation, Acute Myeloid Leukemia With Gene Mutations, Myeloproliferative Neoplasm, Chronic Myelomonocytic Leukemia | NCT03588078 | Unknown | ||
| Phase I, II | Pembrolizumab | Bladder Cancer, Gastric Cancer, Non-Small Cell Lung Cancer, NSCLC, Urothelial Carcinoma, Advanced Solid Tumor | NCT04383938 | Completed | ||
| Phase I, II | + Acalabrutinib in CLL; + Venetoclax and Rituximab in CLL; (Acalabrutinib, OR, (Venetoclax +Rituximab)), in CLL and/or MCL and/or RT; Venetoclax + Rituximab in RT | Non-Hodgkin Lymphoma, Chronic Lymphocytic Leukemia, Mantle Cell Lymphoma | NCT04419389 | Suspended | ||
| Phase III | Azacitidine | MDS | NCT03745716 | Completed | ||
| Phase II | Pegylated Liposomal Doxorubicin Hydrochloride (PLD) | High-Grade Serous Ovarian Cancer | NCT03268382 | Completed | ||
| Phase I, II | Azacitidine | Myelodysplastic Syndrome, Acute Myeloid Leukemia, Myeloproliferative Neoplasm, Chronic Myelomonocytic Leukemia | NCT03072043 | Completed | ||
| Phase I | Hematologic Neoplasms, Prostatic Neoplasms | NCT00900614 | Completed | |||
| Phase I, II | Carboplatin and Pegylated Liposomal Doxorubicin Hydrochloride (PLD) | Platinum Sensitive Recurrent High-Grade Serous Ovarian Cancer With Mutated p53 | NCT02098343 | Completed | ||
| Phase I, II | Dabrafenib | Melanoma | NCT03391050 | Terminated | ||
| Phase II | Venetoclax | Recurrent Mantle Cell Lymphoma; Refractory Mantle Cell Lymphoma | NCT04990778 | Withdrawn | ||
| COTI-2 | Mutant p53 activator | Phase I | Cisplatin | Ovarian Cancer, Fallopian Tube Cancer, Endometrial Cancer, Cervical Cancer, Peritoneal Cancer, Head and Neck Cancer, HNSCC, Colorectal Cancer, Lung Cancer, Pancreatic Cancer | NCT02433626 | Unknown |
| PC14586 | Y220C Mutant p53 activator | Phase I, II | Advanced Solid Tumor, Advanced Malignant Neoplasm, Metastatic Cancer, Metastatic Solid Tumor | NCT04585750 | Recruiting | |
| Phase I | Healthy Volunteers | NCT05249348 | Recruiting | |||
| Phase I | Healthy Male Volunteers | NCT05523687 | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar, A.; Wang, S. Therapeutic Strategies to Activate p53. Pharmaceuticals 2023, 16, 24. https://doi.org/10.3390/ph16010024
Aguilar A, Wang S. Therapeutic Strategies to Activate p53. Pharmaceuticals. 2023; 16(1):24. https://doi.org/10.3390/ph16010024
Chicago/Turabian StyleAguilar, Angelo, and Shaomeng Wang. 2023. "Therapeutic Strategies to Activate p53" Pharmaceuticals 16, no. 1: 24. https://doi.org/10.3390/ph16010024
APA StyleAguilar, A., & Wang, S. (2023). Therapeutic Strategies to Activate p53. Pharmaceuticals, 16(1), 24. https://doi.org/10.3390/ph16010024