
Broad-Spectrum Small-Molecule Inhibitors of the SARS-CoV-2 Spike—ACE2 Protein–Protein Interaction from a Chemical Space of Privileged Protein Binders



Abstract
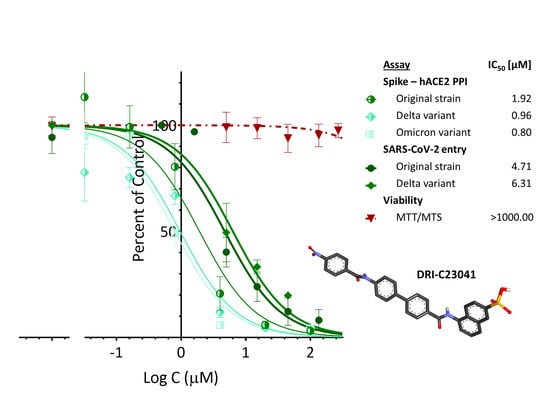
:
{kind=link}
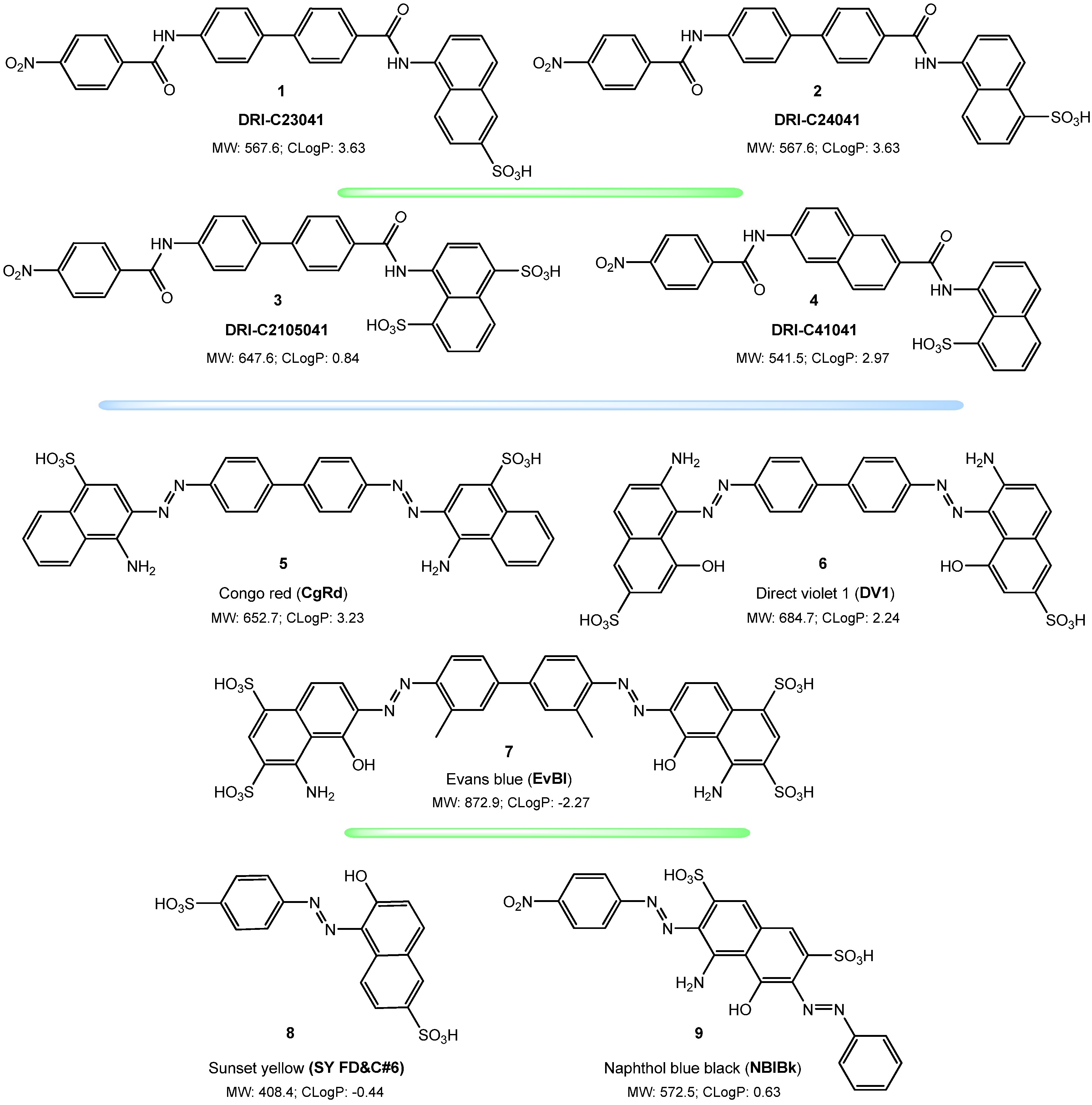{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
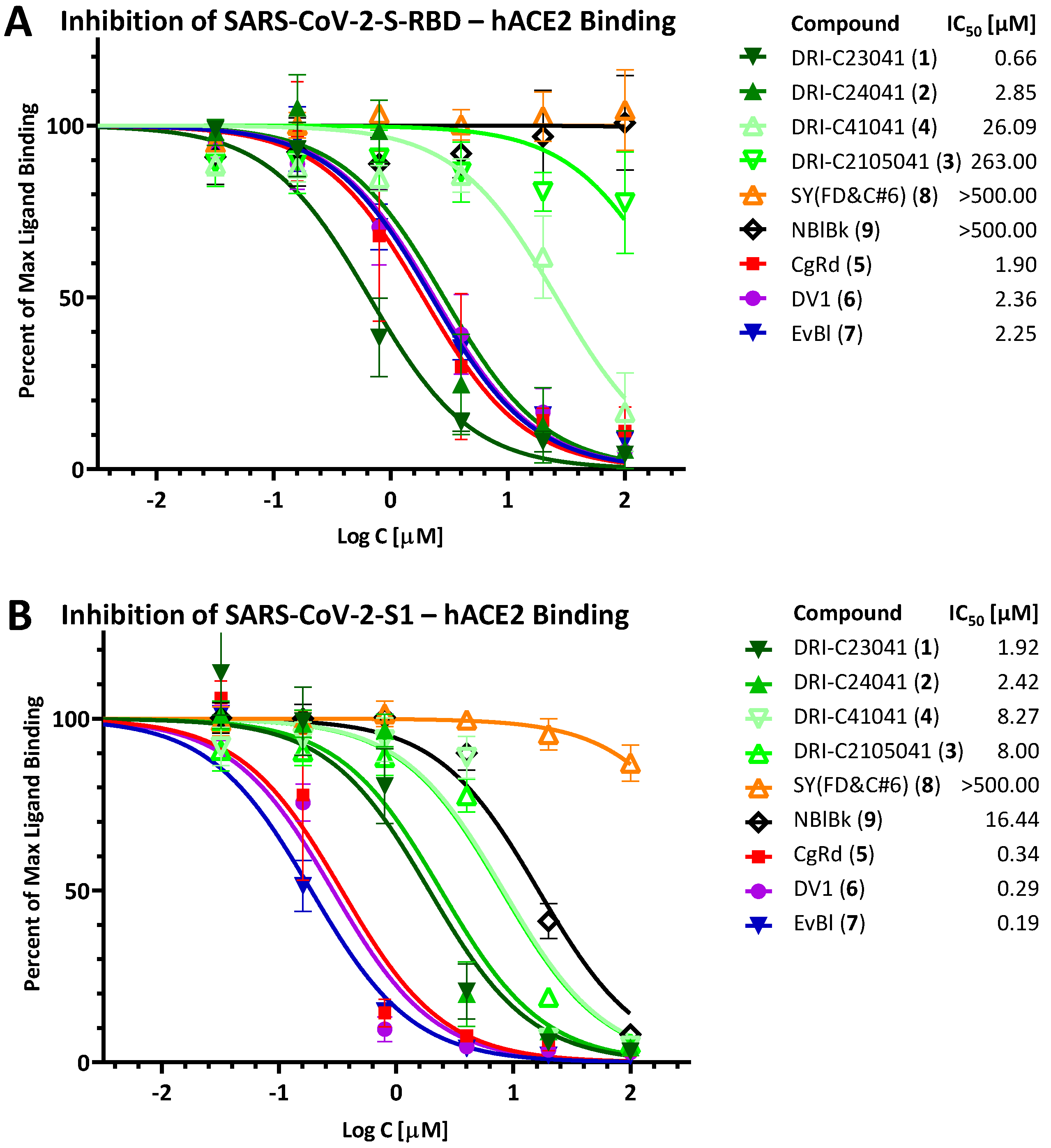
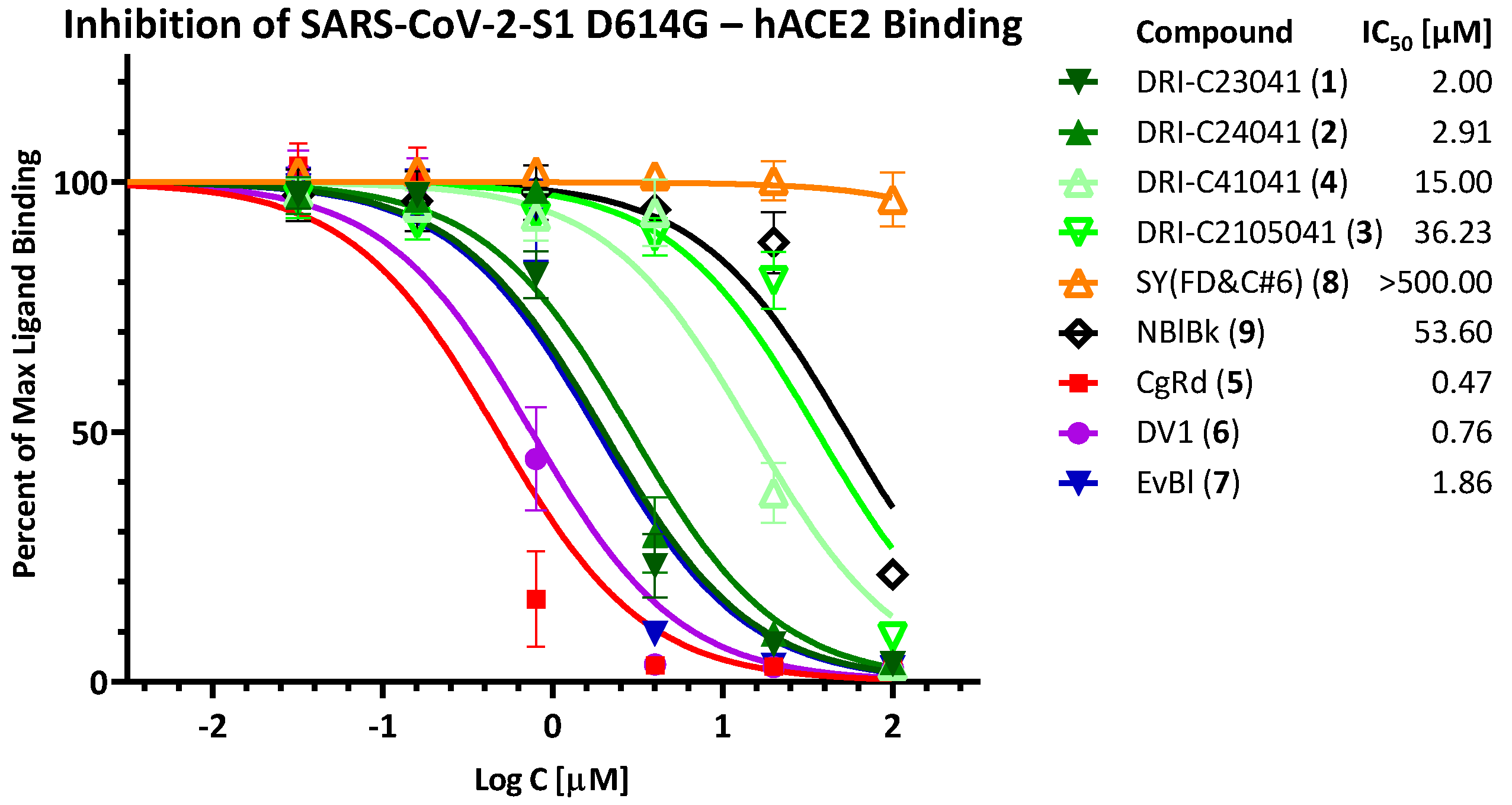
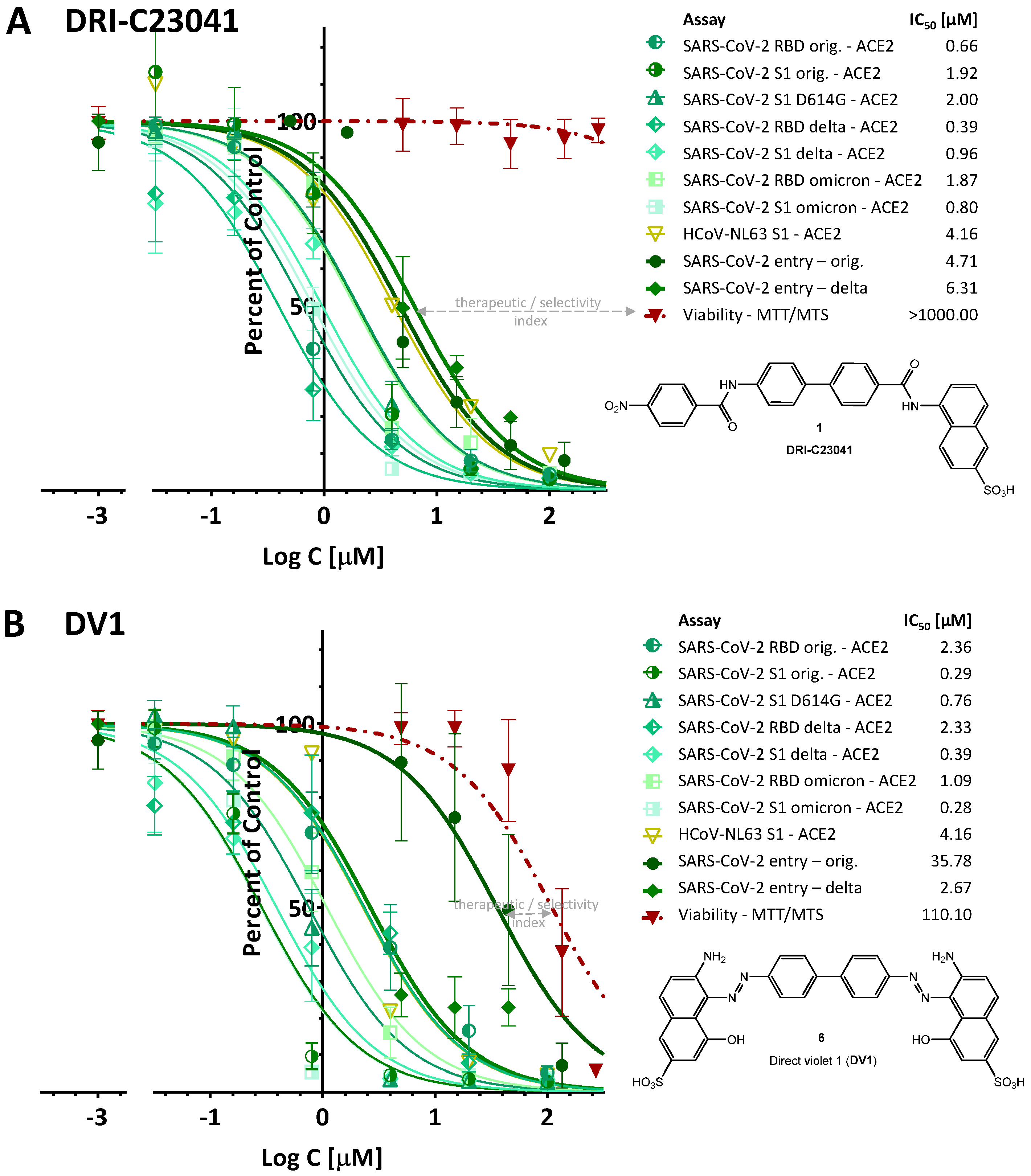
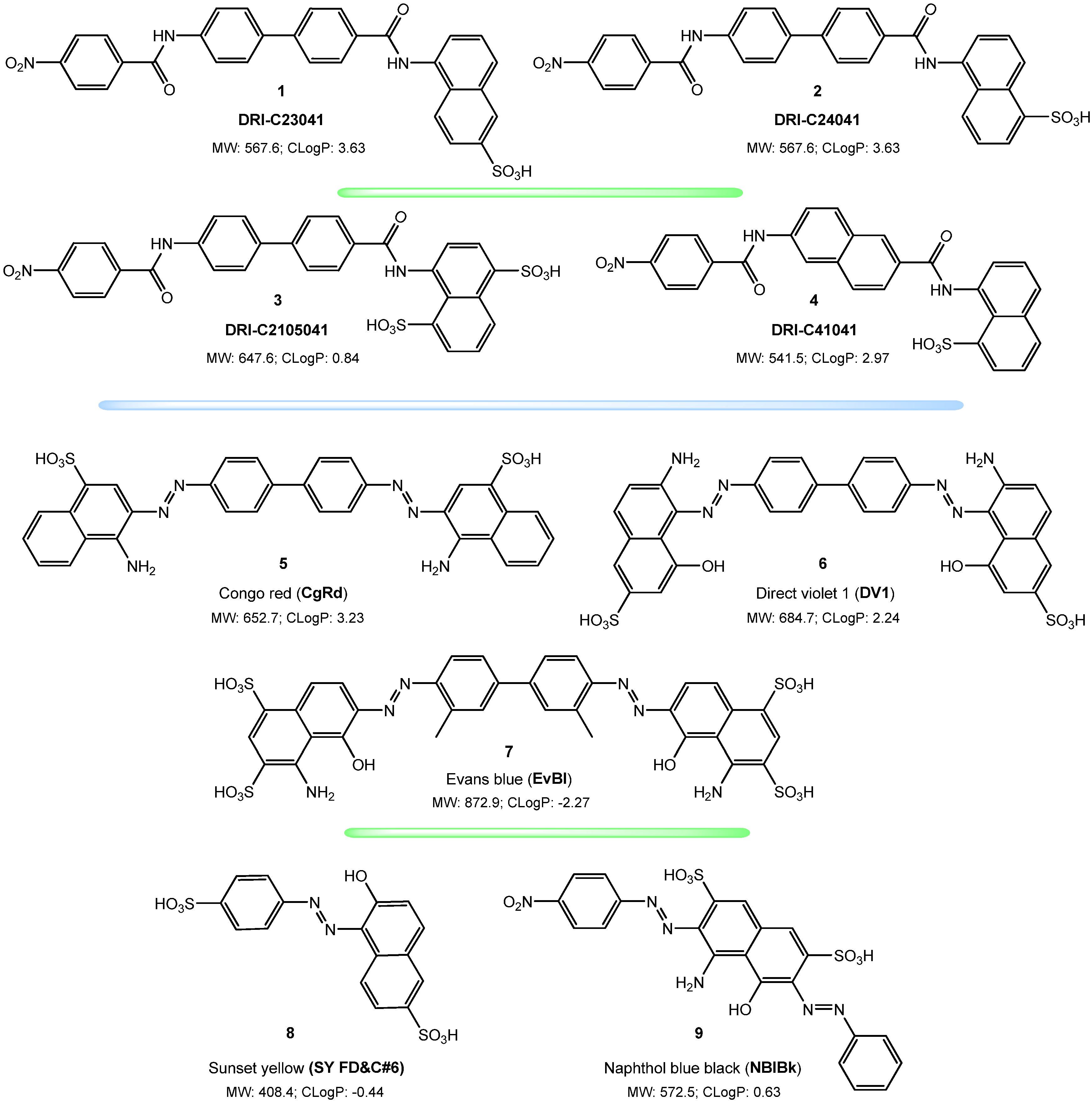
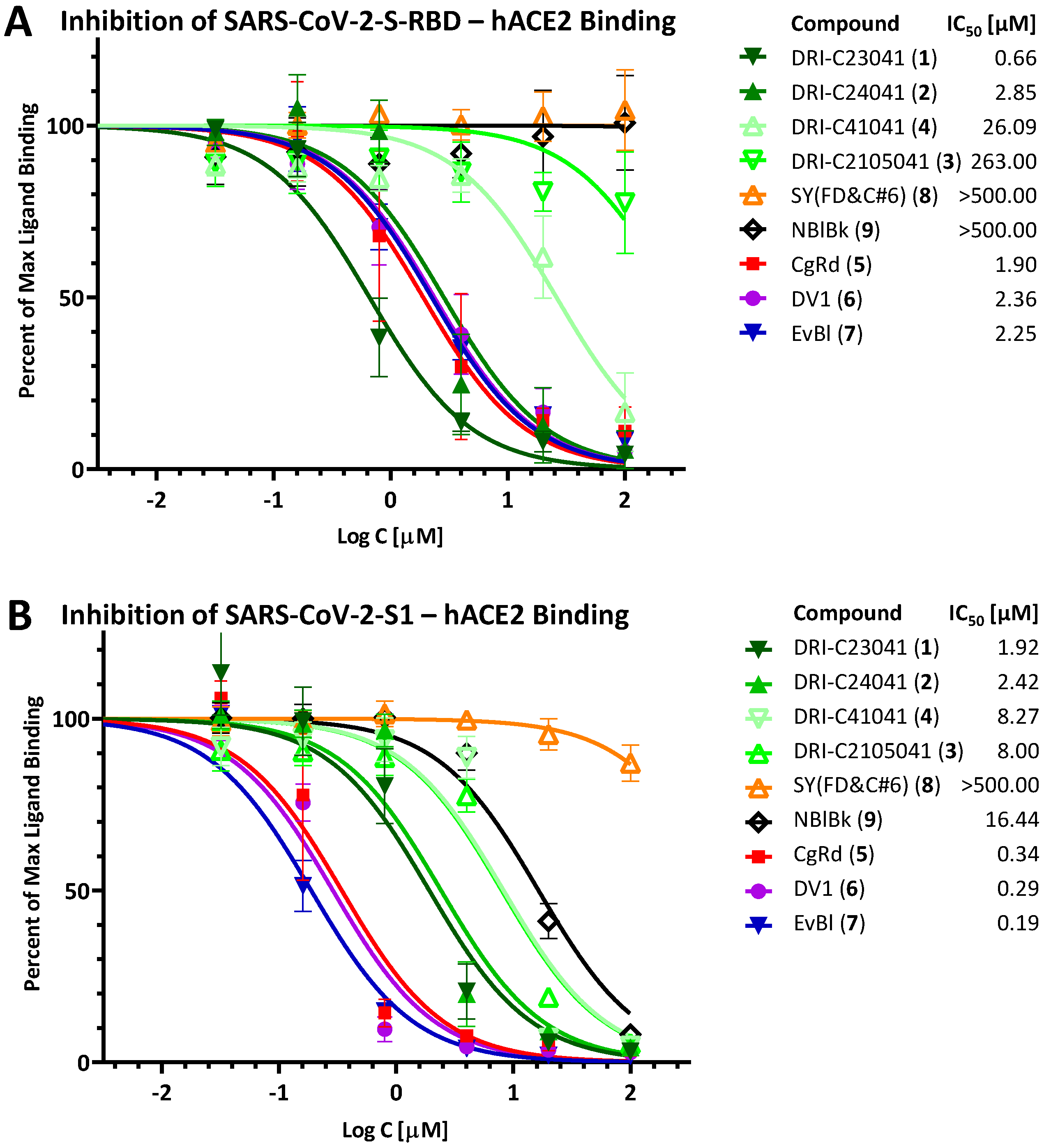
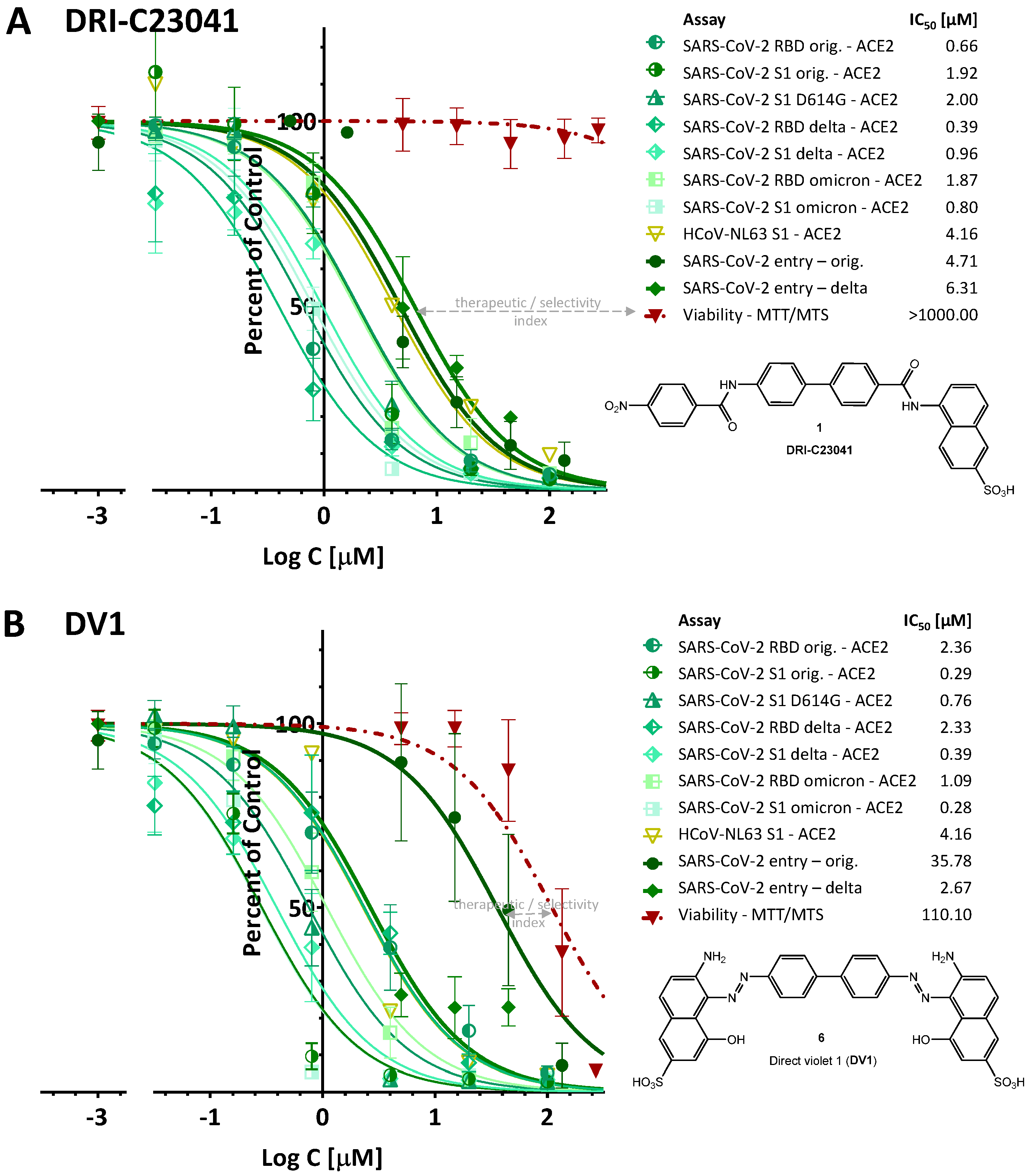
2.1. Inhibition of SARS-CoV-2 Spike—hACE2 PPI (Original Strain)
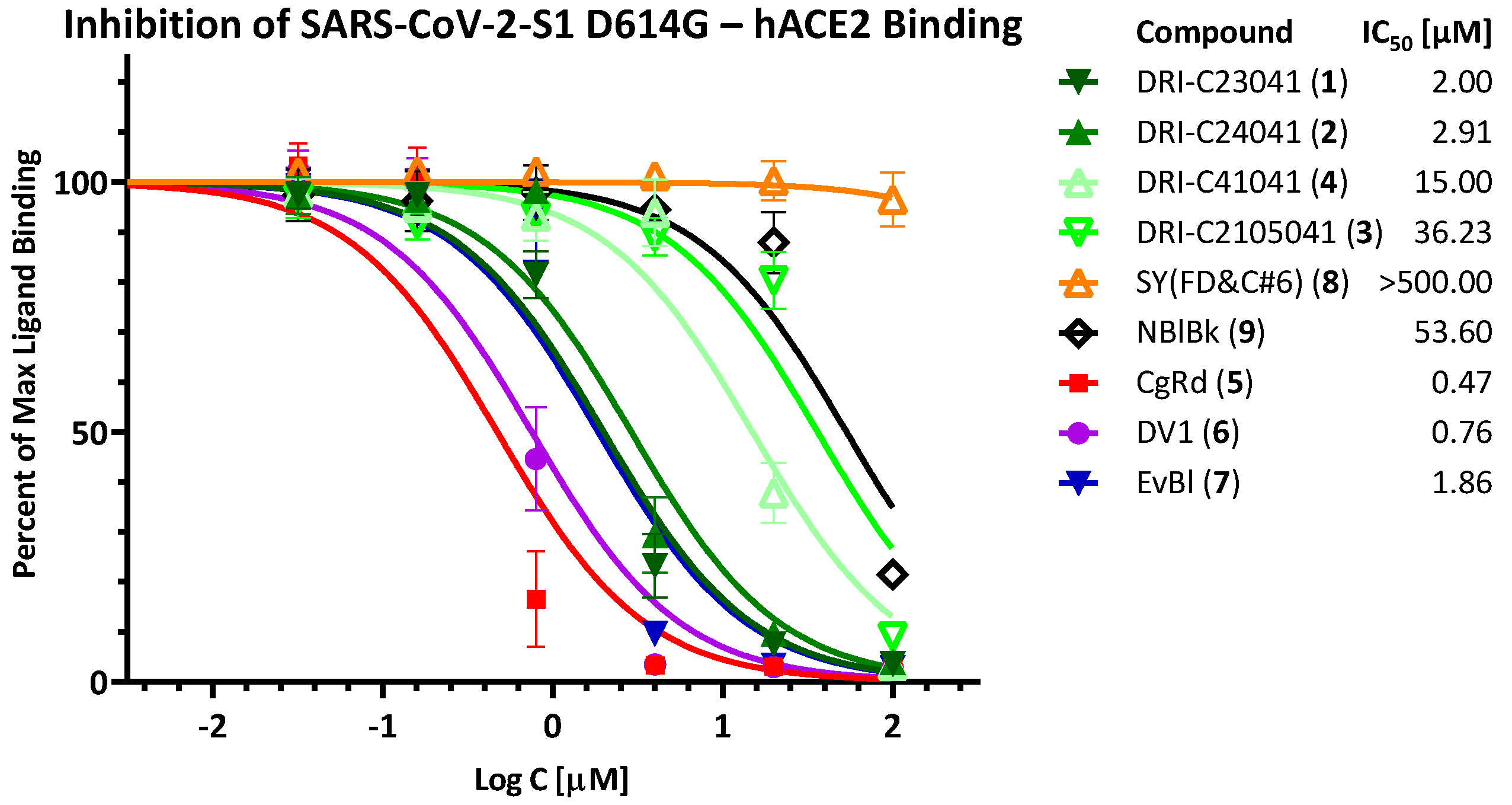
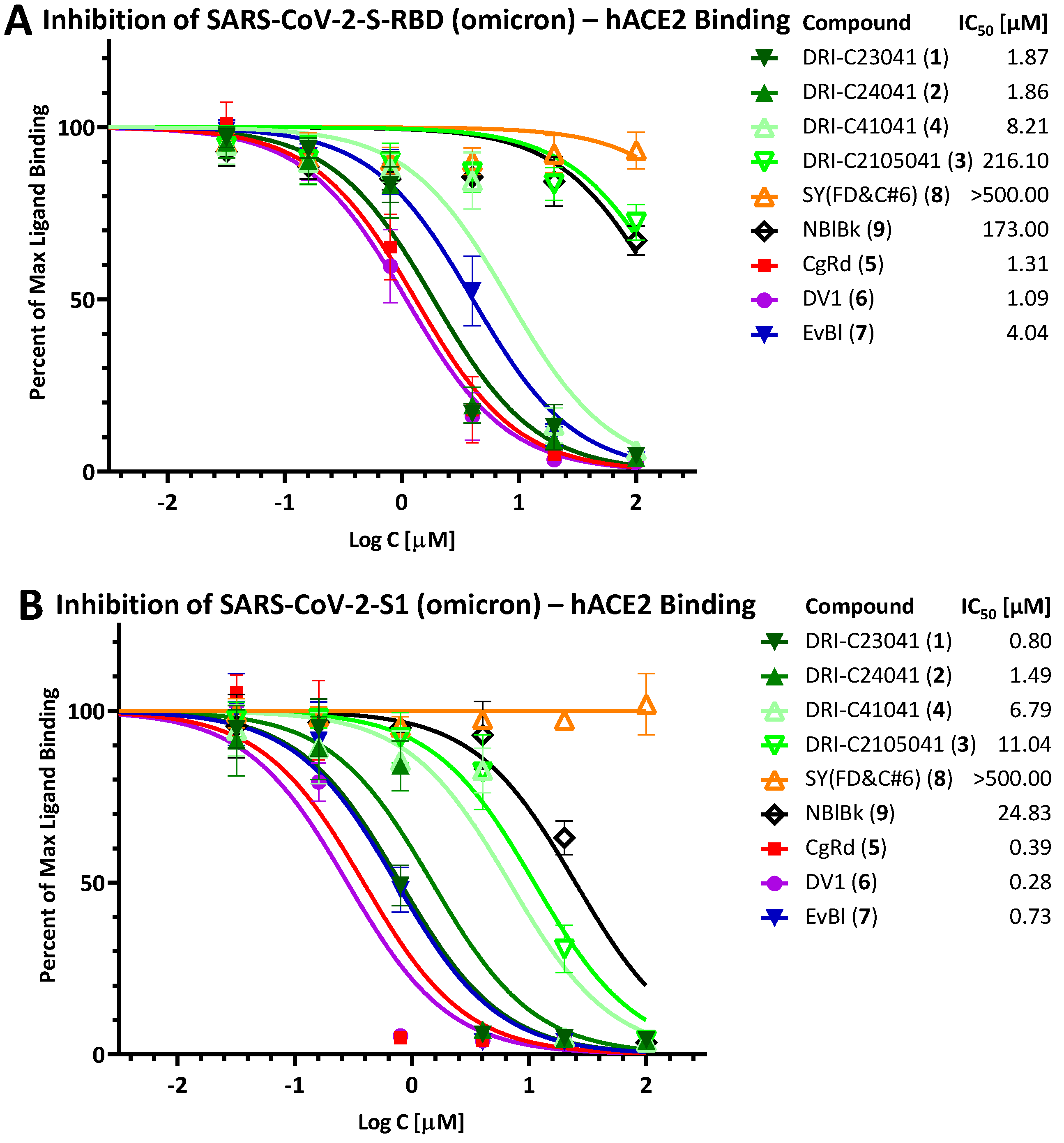
2.2. Inhibition of SARS-CoV-2 Spike—hACE2 PPI (Variants of Concern including Delta and Omicron)
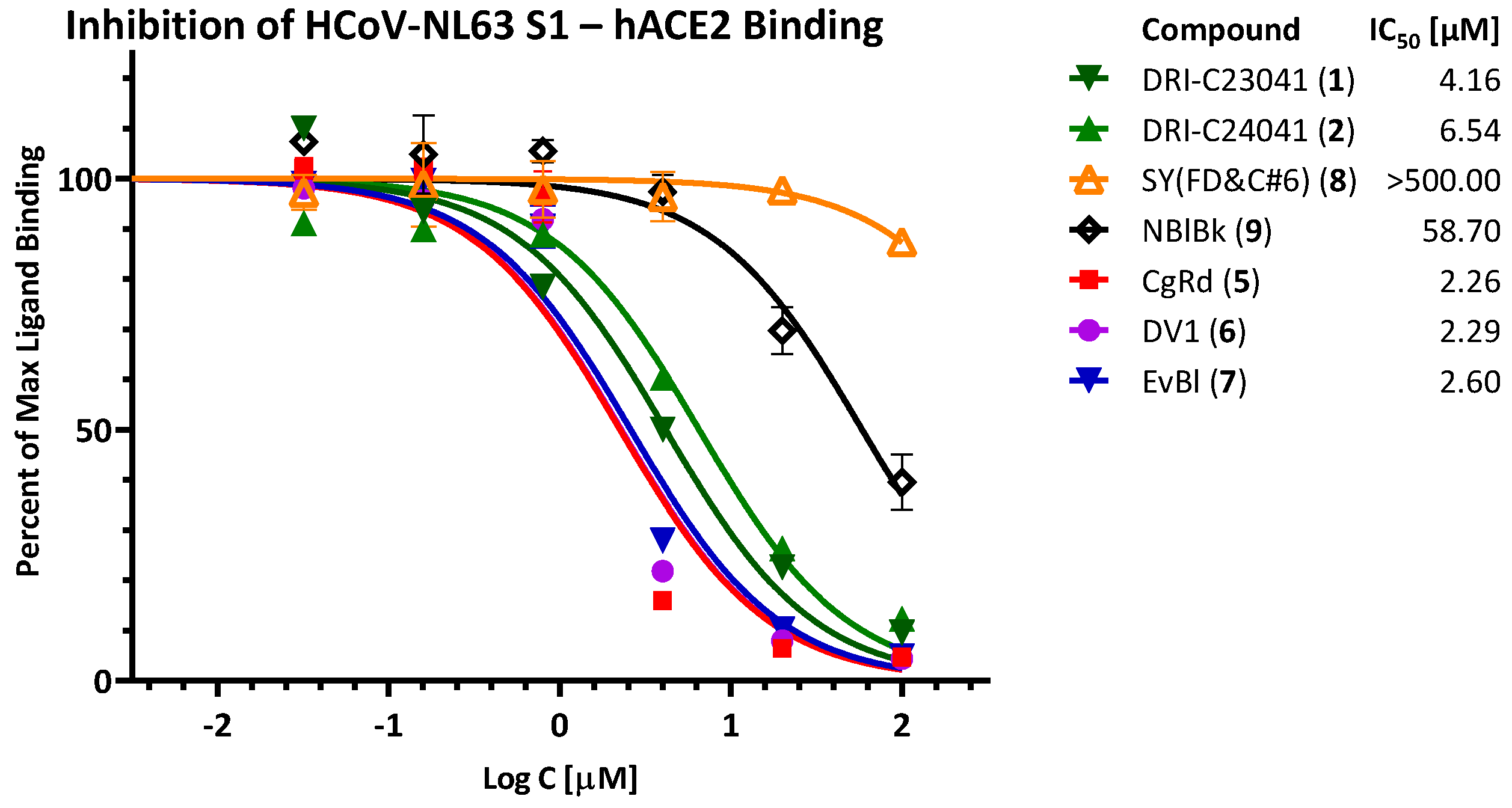
2.3. Inhibition of HCoV-NL63 S—hACE2 PPI
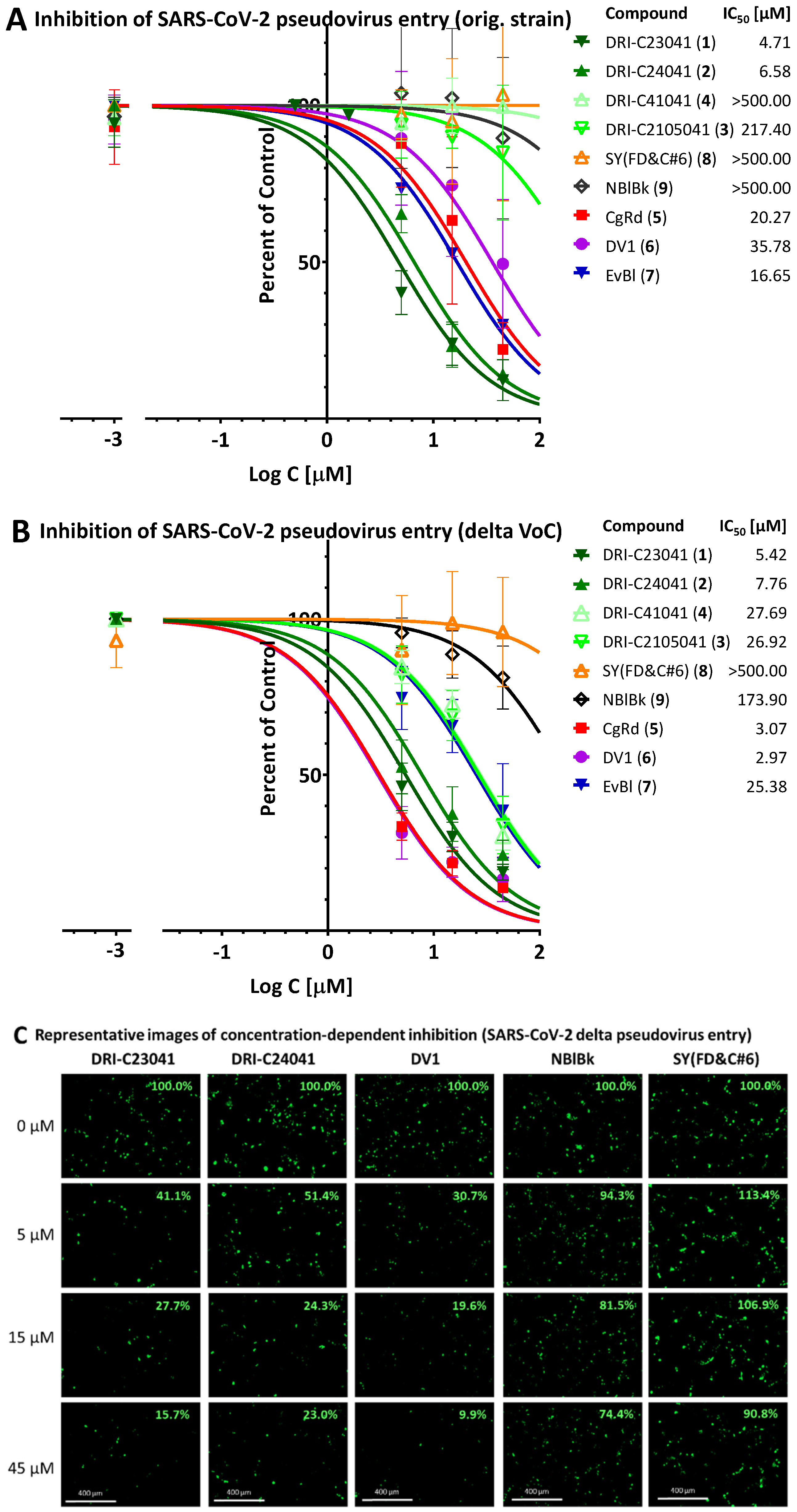
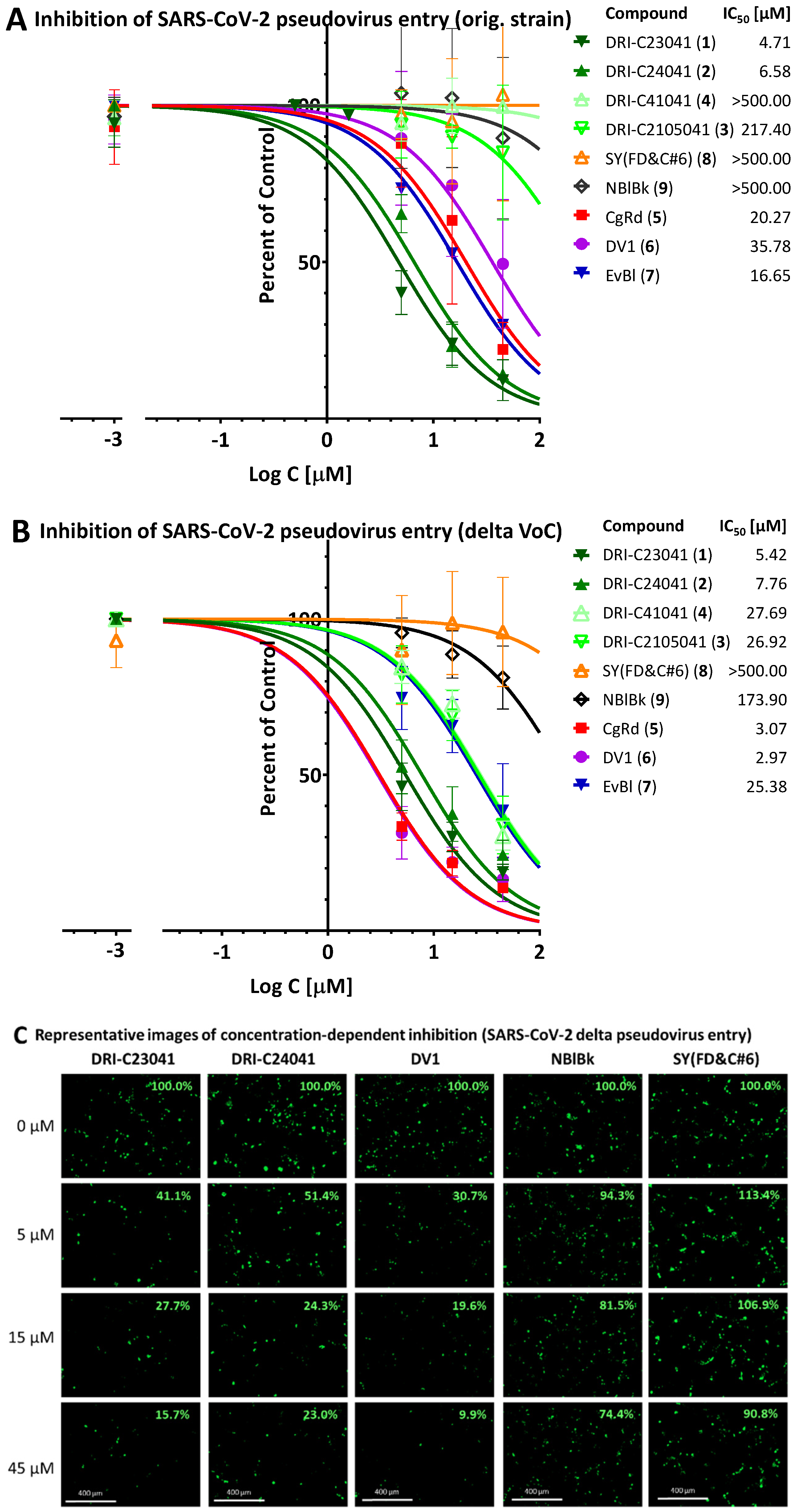
2.4. Inhibition of SARS-CoV-2 Pseudovirus Entry (Original Strain and Delta VoC)
3. Discussion
4. Materials and Methods
4.1. Binding Assays
4.2. SARS-CoV-2 Pseudovirus Assays
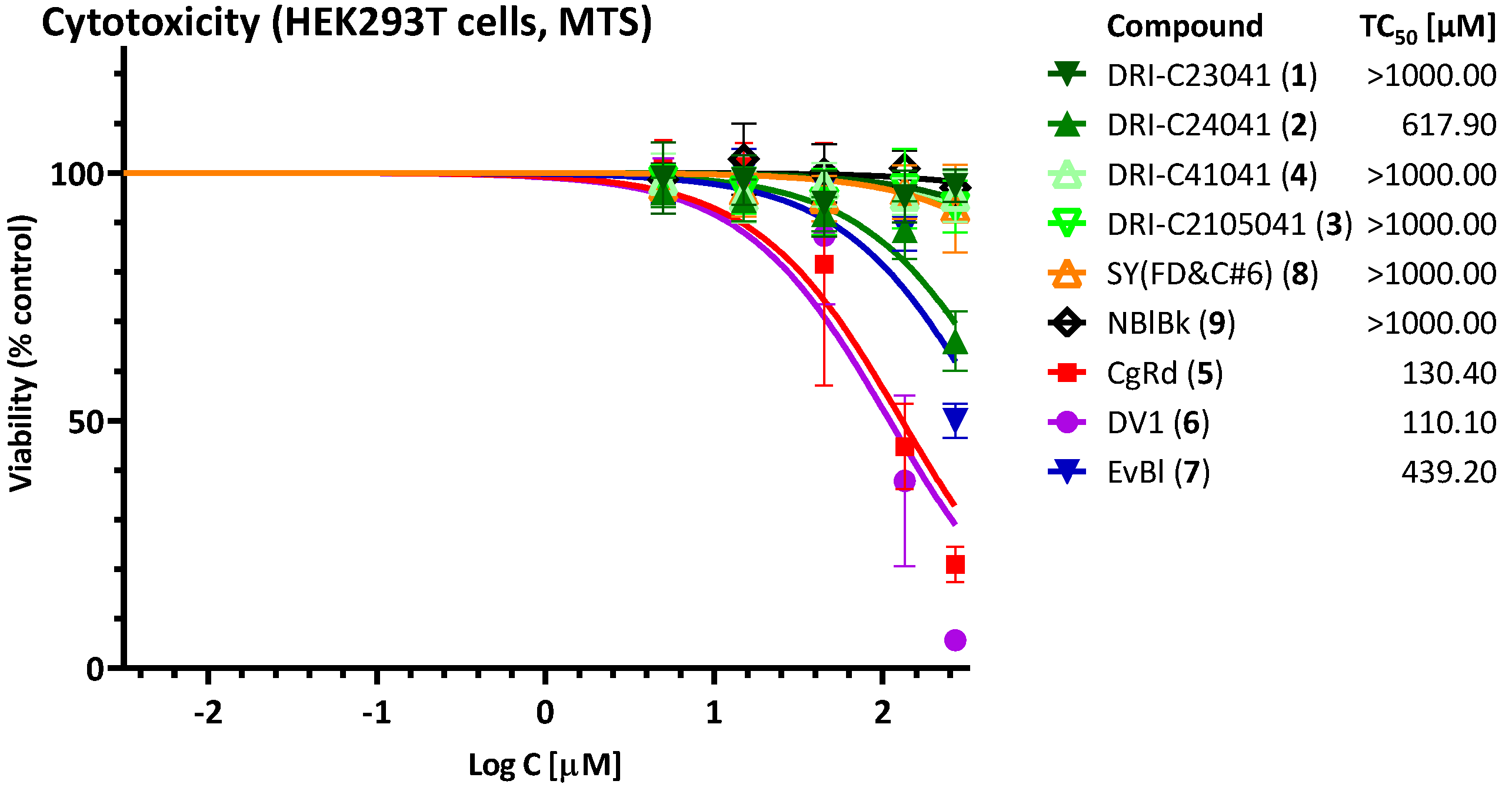
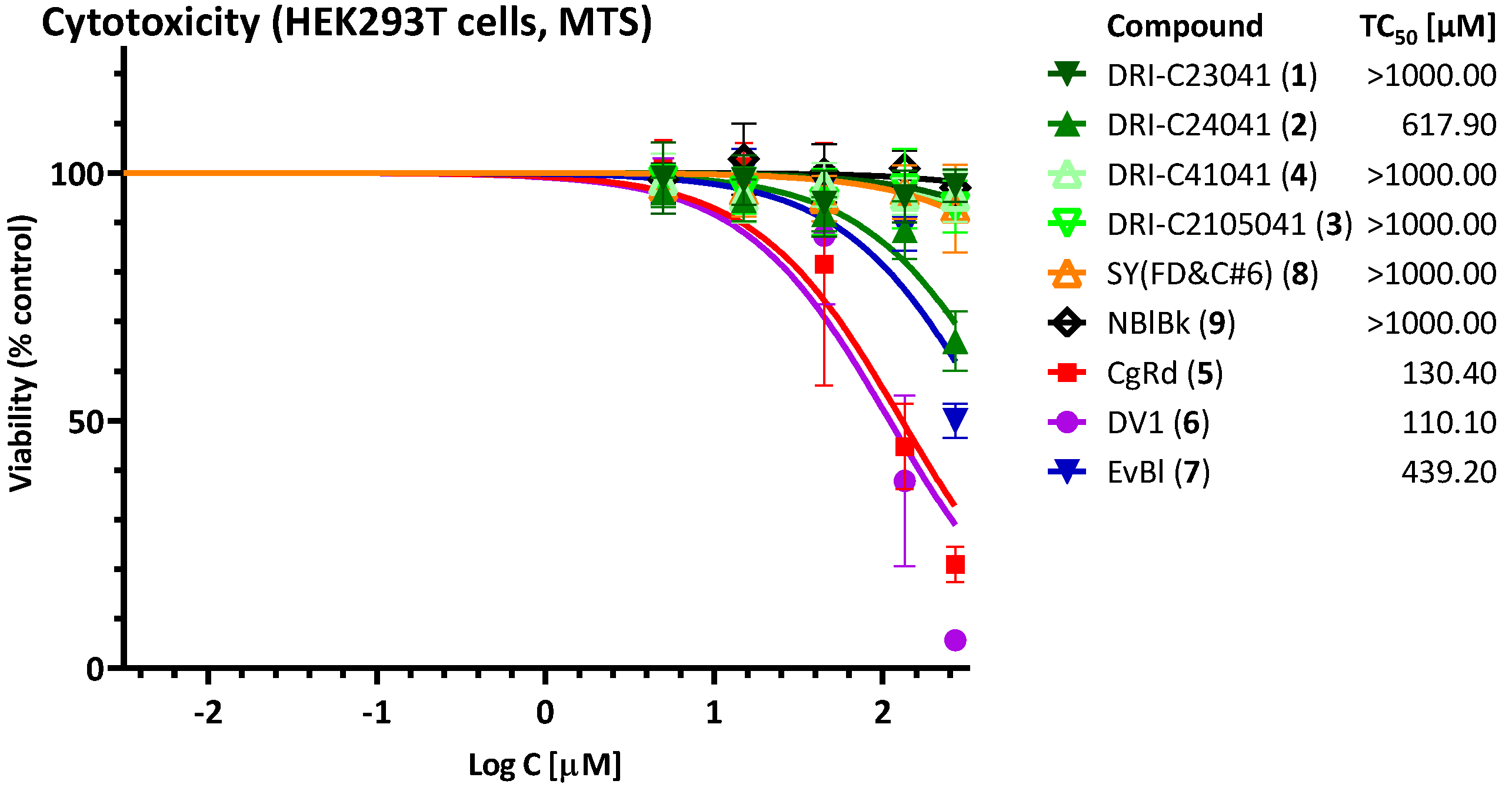
4.3. Cytotoxicity Assay
4.4. Statistical Analysis and Data Fitting
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin converting enzyme 2 |
| CgRd | Congo red |
| CoV | coronavirus |
| DV1 | direct violet 1 |
| EvBl | Evans blue |
| NBlBk | naphthol blue black |
| PPI | protein–protein interaction |
| RBD | receptor binding domain |
| SARS | severe acute respiratory syndrome |
| SMI | small-molecule inhibitor |
| SY | sunset yellow FCF |
| VoC | variant of concern |
| WHO | World Health Organization |
References
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The science underlying COVID-19: Implications for the cardiovascular system. Circulation 2020, 142, 68–78. [Google Scholar]
- Lee, R.K.; Li, T.N.; Chang, S.Y.; Chao, T.L.; Kuo, C.H.; Pan, M.Y.; Chiou, Y.T.; Liao, K.J.; Yang, Y.; Wu, Y.H.; et al. Identification of entry inhibitors against delta and omicron variants of SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 4050. [Google Scholar]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar]
- Melby, T.; Westby, M. Inhibitors of viral entry. Handb. Exp. Pharmacol. 2009, 189, 177–202. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar]
- Matheson, N.J.; Lehner, P.J. How does SARS-CoV-2 cause COVID-19? Science 2020, 369, 510–511. [Google Scholar]
- Sivaraman, H.; Er, S.Y.; Choong, Y.K.; Gavor, E.; Sivaraman, J. Structural basis of the SARS-CoV-2/SARS-CoV receptor binding and small-molecule blockers as potential therapeutics. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 465–493. [Google Scholar]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramirez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar]
- Cannalire, R.; Stefanelli, I.; Cerchia, C.; Beccari, A.R.; Pelliccia, S.; Summa, V. SARS-CoV-2 entry inhibitors: Small molecules and peptides targeting virus or host cells. Int. J. Mol. Sci. 2020, 21, 5707. [Google Scholar]
- Buchwald, P. Developing small-molecule inhibitors of protein-protein interactions involved in viral entry as potential antivirals for COVID-19. Front. Drug Discov. 2022, 2, 898035. [Google Scholar]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2022, 376, 1327–1332. [Google Scholar]
- Riccio, A.; Santopolo, S.; Rossi, A.; Piacentini, S.; Rossignol, J.F.; Santoro, M.G. Impairment of SARS-CoV-2 spike glycoprotein maturation and fusion activity by nitazoxanide: An effect independent of spike variants emergence. Cell. Mol. Life Sci. 2022, 79, 227. [Google Scholar]
- Petrou, A.; Zagaliotis, P.; Theodoroula, N.F.; Mystridis, G.A.; Vizirianakis, I.S.; Walsh, T.J.; Geronikaki, A. Thiazole/thiadiazole/benzothiazole based thiazolidin-4-one derivatives as potential inhibitors of main protease of SARS-CoV-2. Molecules 2022, 27, 2180. [Google Scholar]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Lambson, B.E.; Vermeulen, M.; van den Berg, K.; Rossouw, T.; Boswell, M.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226. [Google Scholar]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar]
- Kupferschmidt, K. Evolving threat. Science 2021, 373, 844–849. [Google Scholar]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 vaccines against the B.1.617.2 (delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar]
- Khan, A.; Randhawa, A.W.; Balouch, A.R.; Mukhtar, N.; Sayaf, A.M.; Suleman, M.; Khan, T.; Ali, S.; Ali, S.S.; Wang, Y.; et al. Blocking key mutated hotspot residues in the RBD of the omicron variant (B.1.1.529) with medicinal compounds to disrupt the RBD-hACE2 complex using molecular screening and simulation approaches. RSC Adv. 2022, 12, 7318–7327. [Google Scholar]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar]
- Taha, H.R.; Keewan, N.; Slati, F.; Al-Sawalha, N.A. Remdesivir: A closer look at its effect in COVID-19 pandemic. Pharmacology 2021, 106, 462–468. [Google Scholar]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernandez Garcia, C.; Kieny, M.P.; et al. Repurposed antiviral drugs for COVID-19-Interim WHO Solidarity trial results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar]
- Zhao, L.; Li, S.; Zhong, W. Mechanism of action of small-molecule agents in ongoing clinical trials for SARS-CoV-2: A review. Front. Pharmacol. 2022, 13, 840639. [Google Scholar]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar]
- Lamb, Y.N. Nirmatrelvir plus ritonavir: First approval. Drugs 2022, 82, 585–591. [Google Scholar]
- Song, Y.; Margolles-Clark, E.; Bayer, A.; Buchwald, P. Small-molecule modulators of the OX40–OX40L costimulatory protein–protein interaction. Br. J. Pharmacol. 2014, 171, 4955–4969. [Google Scholar]
- Chen, J.; Song, Y.; Bojadzic, D.; Tamayo-Garcia, A.; Landin, A.M.; Blomberg, B.B.; Buchwald, P. Small-molecule inhibitors of the CD40-CD40L costimulatory protein-protein interaction. J. Med. Chem. 2017, 60, 8906–8922. [Google Scholar]
- Bojadzic, D.; Chen, J.; Alcazar, O.; Buchwald, P. Design, synthesis, and evaluation of novel immunomodulatory small molecules targeting the CD40-CD154 costimulatory protein-protein interaction. Molecules 2018, 23, 1153. [Google Scholar]
- Bojadzic, D.; Alcazar, O.; Chen, J.; Chuang, S.T.; Condor Capcha, J.M.; Shehadeh, L.A.; Buchwald, P. Small-molecule inhibitors of the coronavirus spike—ACE2 protein-protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect. Dis. 2021, 7, 1519–1534. [Google Scholar]
- Bojadzic, D.; Alcazar, O.; Buchwald, P. Methylene blue inhibits the SARS-CoV-2 spike—ACE2 protein-protein interaction—A mechanism that can contribute to its antiviral activity against COVID-19. Front. Pharmacol. 2021, 11, 600372. [Google Scholar]
- Chuang, S.T.; Papp, H.; Kuczmog, A.; Eells, R.; Condor Capcha, J.M.; Shehadeh, L.A.; Jakab, F.; Buchwald, P. Methylene blue is a nonspecific protein–protein interaction inhibitor with potential for repurposing as an antiviral for COVID-19. Pharmaceuticals 2022, 15, 621. [Google Scholar]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar]
- Bojadzic, D.; Buchwald, P. Toward small-molecule inhibition of protein-protein interactions: General aspects and recent progress in targeting costimulatory and coinhibitory (immune checkpoint) interactions. Curr. Top. Med. Chem. 2018, 18, 674–699. [Google Scholar]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar]
- Gadek, T.R.; Burdick, D.J.; McDowell, R.S.; Stanley, M.S.; Marsters, J.C., Jr.; Paris, K.J.; Oare, D.A.; Reynolds, M.E.; Ladner, C.; Zioncheck, K.A.; et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 2002, 295, 1086–1089. [Google Scholar]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 attachment: The discovery and development of temsavir and its prodrug fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar]
- Giannoukakis, N.; Phillips, B.; Trucco, M. Toward a cure for type 1 diabetes mellitus: Diabetes-suppressive dendritic cells and beyond. Pediatr. Diabetes 2008, 9, 4–13. [Google Scholar]
- Cochrane, G.M.; Horne, R.; Chanez, P. Compliance in asthma. Respir. Med. 1999, 93, 763–769. [Google Scholar]
- Moia, M.; Mantovani, L.G.; Carpenedo, M.; Scalone, L.; Monzini, M.S.; Cesana, G.; Mannucci, P.M. Patient preferences and willingness to pay for different options of anticoagulant therapy. Intern. Emerg. Med. 2013, 8, 237–243. [Google Scholar]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 entry: Current and future opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar]
- Hunger, K. Industrial Dyes. Chemistry, Properties, Applications, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003; p. 660. [Google Scholar]
- Fletcher, S.; Hamilton, A.D. Targeting protein-protein interactions by rational design: Mimicry of protein surfaces. J. R. Soc. Interface 2006, 3, 215–233. [Google Scholar]
- Che, Y.; Brooks, B.R.; Marshall, G.R. Development of small molecules designed to modulate protein-protein interactions. J. Comput.-Aided Mol. Des. 2006, 20, 109–130. [Google Scholar]
- Hershberger, S.J.; Lee, S.G.; Chmielewski, J. Scaffolds for blocking protein-protein interactions. Curr. Top. Med. Chem. 2007, 7, 928–942. [Google Scholar]
- Cagno, V.; Medaglia, C.; Cerny, A.; Cerny, T.; Zwygart, A.C.; Cerny, E.; Tapparel, C. Methylene blue has a potent antiviral activity against SARS-CoV-2 and H1N1 influenza virus in the absence of UV-activation in vitro. Sci. Rep. 2021, 11, 14295. [Google Scholar]
- Gendrot, M.; Andreani, J.; Duflot, I.; Boxberger, M.; Bideau, M.L.; Mosnier, J.; Jardot, P.; Fonta, I.; Rolland, C.; Bogreau, H.; et al. Methylene blue inhibits the replication of SARS-CoV-2 in vitro. Int. J. Antimicrob. Agents 2020, 56, 106202. [Google Scholar]
- Gendrot, M.; Jardot, P.; Delandre, O.; Boxberger, M.; Andreani, J.; Duflot, I.; Le Bideau, M.; Mosnier, J.; Fonta, I.; Hutter, S.; et al. In vitro evaluation of the antiviral activity of methylene blue alone or in combination against SARS-CoV-2. J. Clin. Med. 2021, 10, 3007. [Google Scholar]
- Murer, L.; Volle, R.; Andriasyan, V.; Petkidis, A.; Gomez-Gonzalez, A.; Yang, L.; Meili, N.; Suomalainen, M.; Bauer, M.; Sequeira, D.; et al. Identification of broad anti-coronavirus chemical agents for repurposing against SARS-CoV-2 and variants of concern. Curr. Res. Virol. Sci. 2022, 3, 100019. [Google Scholar]
- Day, C.J.; Bailly, B.; Guillon, P.; Dirr, L.; Jen, F.E.; Spillings, B.L.; Mak, J.; von Itzstein, M.; Haselhorst, T.; Jennings, M.P. Multidisciplinary approaches identify compounds that bind to human ACE2 or SARS-CoV-2 spike protein as candidates to block SARS-CoV-2-ACE2 receptor interactions. MBio 2021, 12, e03681-20. [Google Scholar]
- Fu, W.; Chen, Y.; Wang, K.; Hettinghouse, A.; Hu, W.; Wang, J.Q.; Lei, Z.N.; Chen, Z.S.; Stapleford, K.A.; Liu, C.J. Repurposing FDA-approved drugs for SARS-CoV-2 through an ELISA-based screening for the inhibition of RBD/ACE2 interaction. Protein Cell 2021, 12, 586–591. [Google Scholar]
- van Breemen, R.B.; Muchiri, R.N.; Bates, T.A.; Weinstein, J.B.; Leier, H.C.; Farley, S.; Tafesse, F.G. Cannabinoids block cellular entry of SARS-CoV-2 and the emerging variants. J. Nat. Prod. 2022, 85, 176–184. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar]
- DeGoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the rule of 5: Lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2017, 61, 2636–2651. [Google Scholar]
- Doak, B.C.; Kihlberg, J. Drug discovery beyond the rule of 5-opportunities and challenges. Expert Opin. Drug Discov. 2017, 12, 115–119. [Google Scholar]
- Mullard, A. Pioneering apoptosis-targeted cancer drug poised for FDA approval. Nat. Rev. Drug Discov. 2016, 15, 147–149. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, S.-T.; Buchwald, P. Broad-Spectrum Small-Molecule Inhibitors of the SARS-CoV-2 Spike—ACE2 Protein–Protein Interaction from a Chemical Space of Privileged Protein Binders. Pharmaceuticals 2022, 15, 1084. https://doi.org/10.3390/ph15091084
Chuang S-T, Buchwald P. Broad-Spectrum Small-Molecule Inhibitors of the SARS-CoV-2 Spike—ACE2 Protein–Protein Interaction from a Chemical Space of Privileged Protein Binders. Pharmaceuticals. 2022; 15(9):1084. https://doi.org/10.3390/ph15091084
Chicago/Turabian StyleChuang, Sung-Ting, and Peter Buchwald. 2022. "Broad-Spectrum Small-Molecule Inhibitors of the SARS-CoV-2 Spike—ACE2 Protein–Protein Interaction from a Chemical Space of Privileged Protein Binders" Pharmaceuticals 15, no. 9: 1084. https://doi.org/10.3390/ph15091084
APA StyleChuang, S.-T., & Buchwald, P. (2022). Broad-Spectrum Small-Molecule Inhibitors of the SARS-CoV-2 Spike—ACE2 Protein–Protein Interaction from a Chemical Space of Privileged Protein Binders. Pharmaceuticals, 15(9), 1084. https://doi.org/10.3390/ph15091084