
Comparison of the Anti-Tumour Activity of the Somatostatin Receptor (SST) Antagonist [177Lu]Lu-Satoreotide Tetraxetan and the Agonist [177Lu]Lu-DOTA-TATE in Mice Bearing AR42J SST2-Positive Tumours


 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. In Vitro
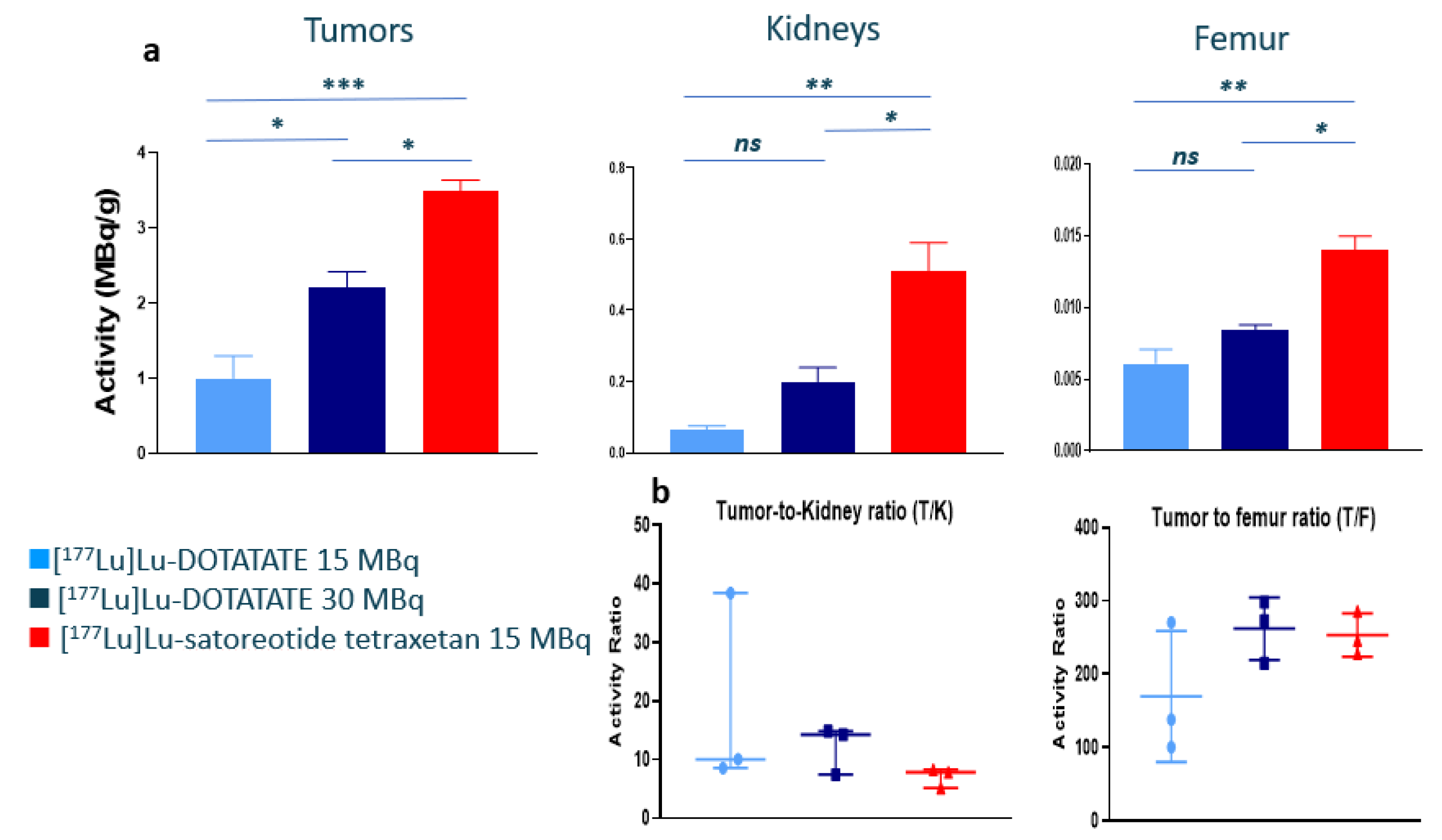
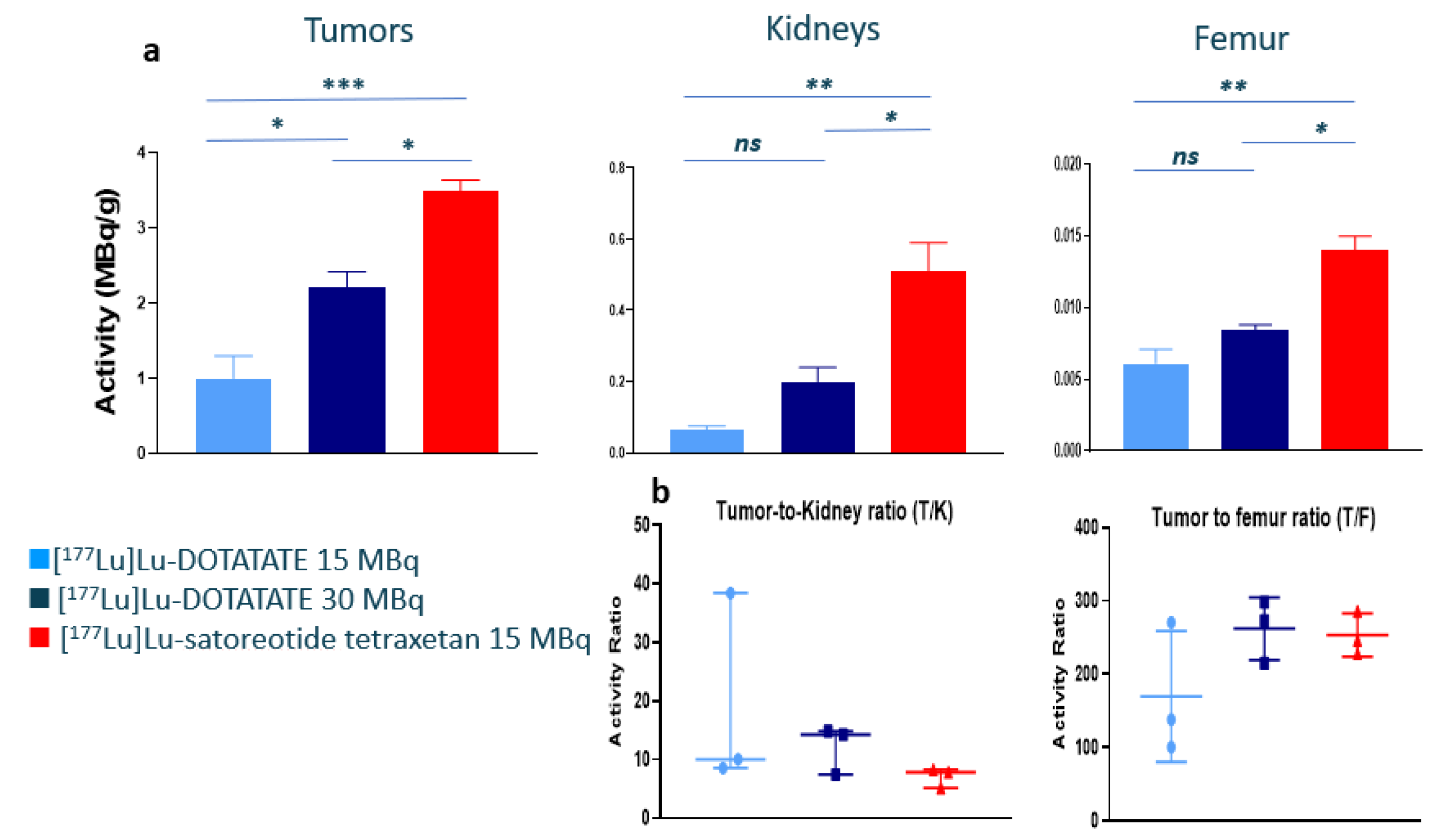
2.2. In Vivo Biodistribution of 177Lu-Satoreotide Tetraxetan versus [177Lu]Lu-DOTA-TATE
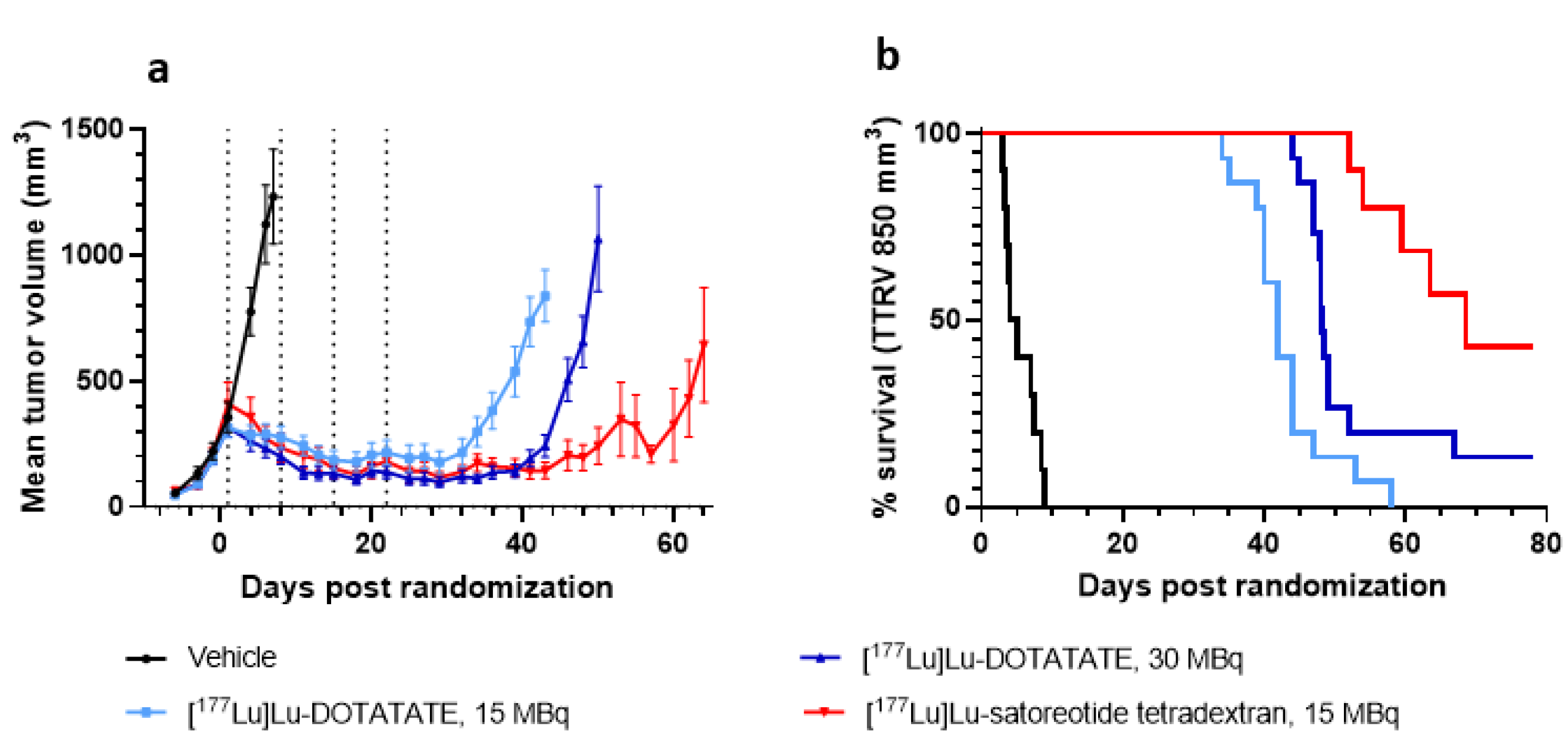
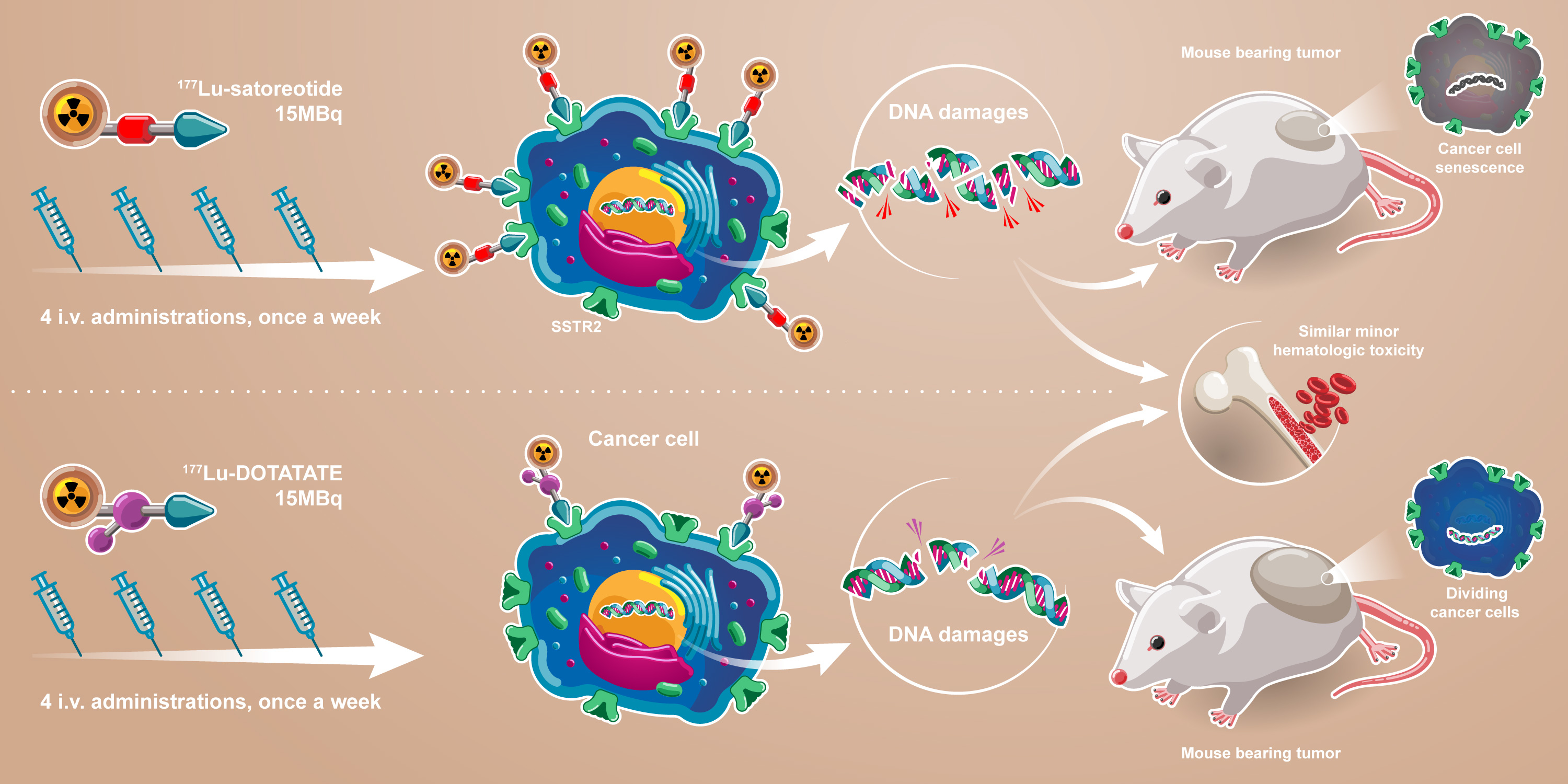
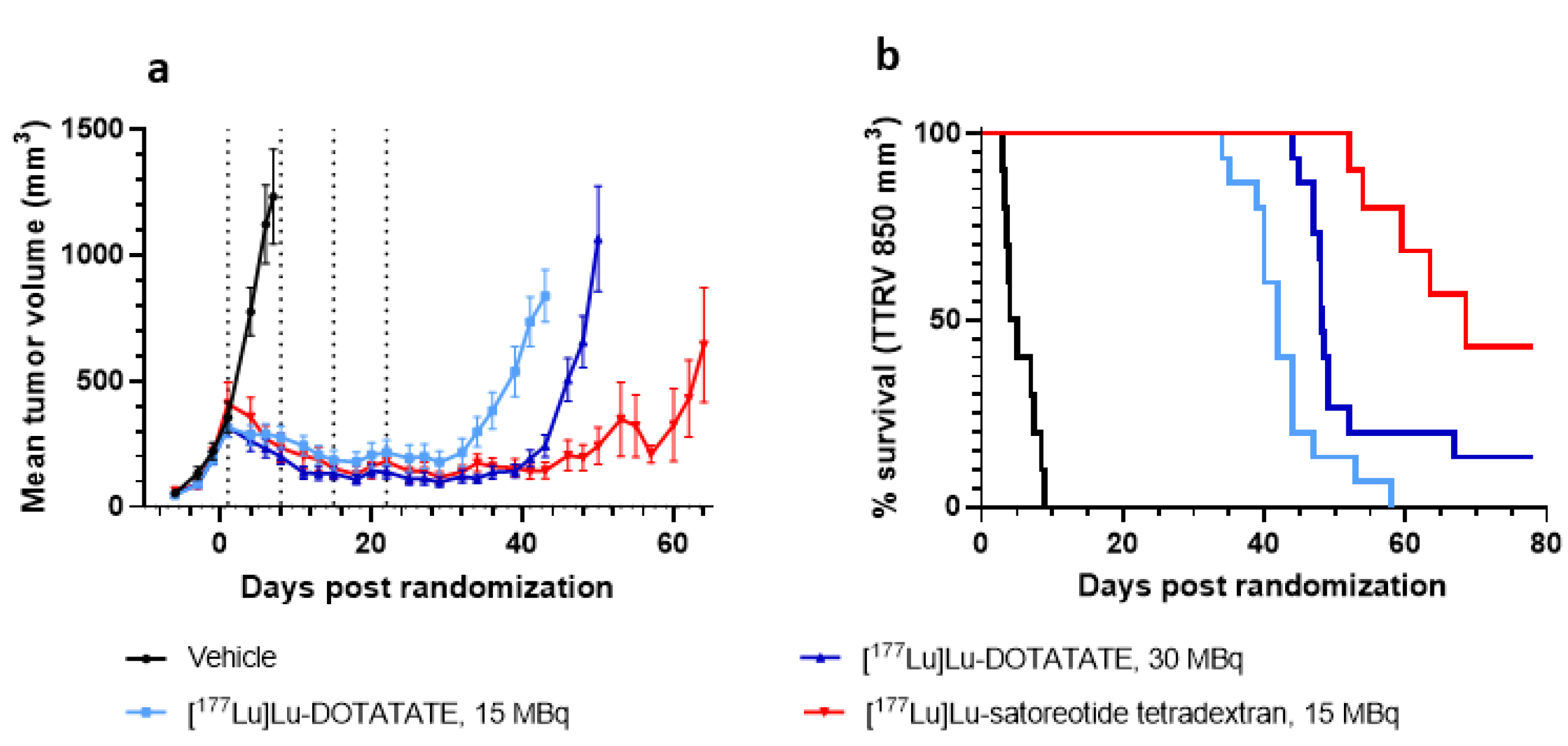
2.3. In Vivo Anti-Tumour Activity of [177Lu]Lu-Satoreotide Tetraxetan versus [177Lu]Lu-DOTA-TATE
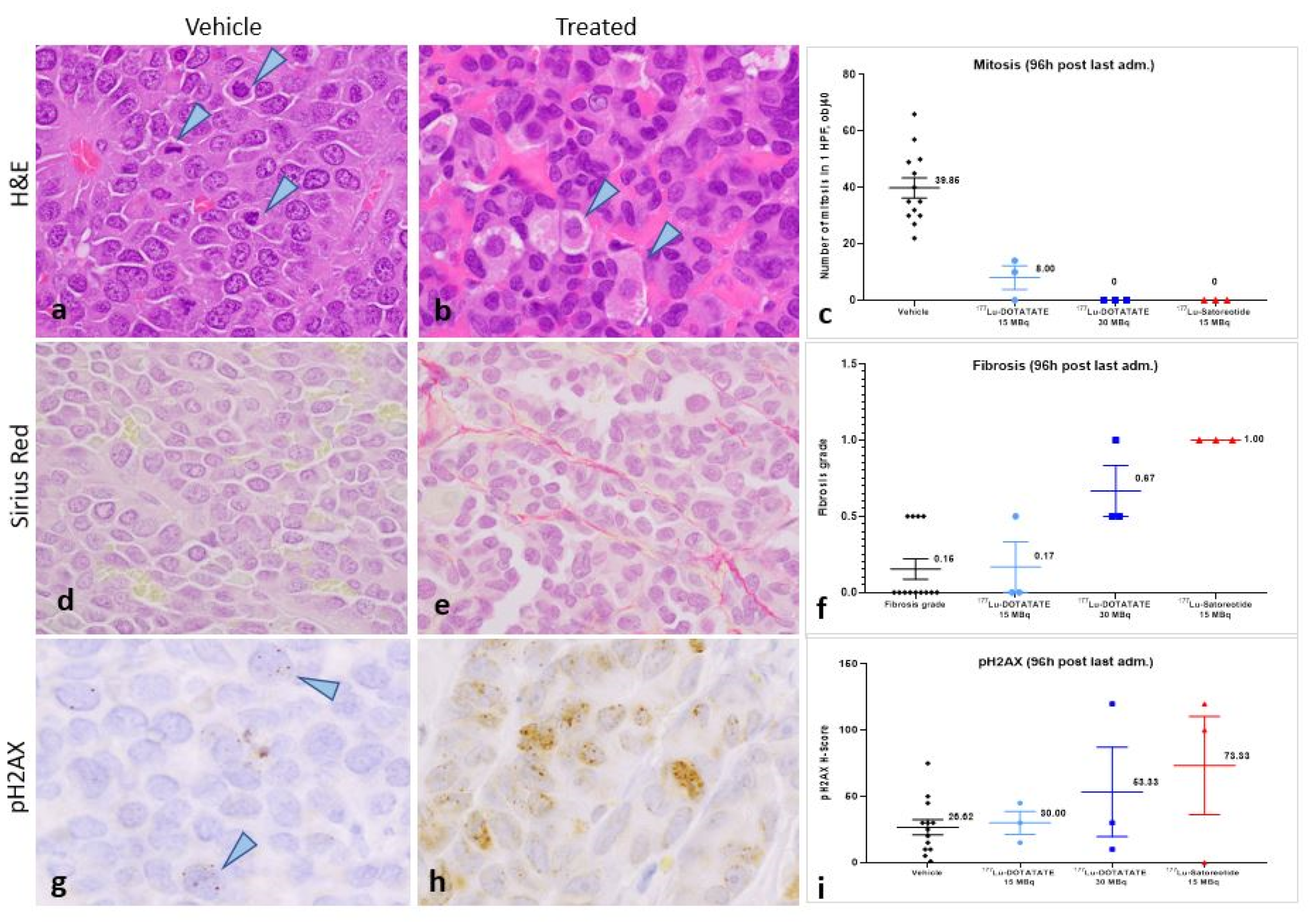
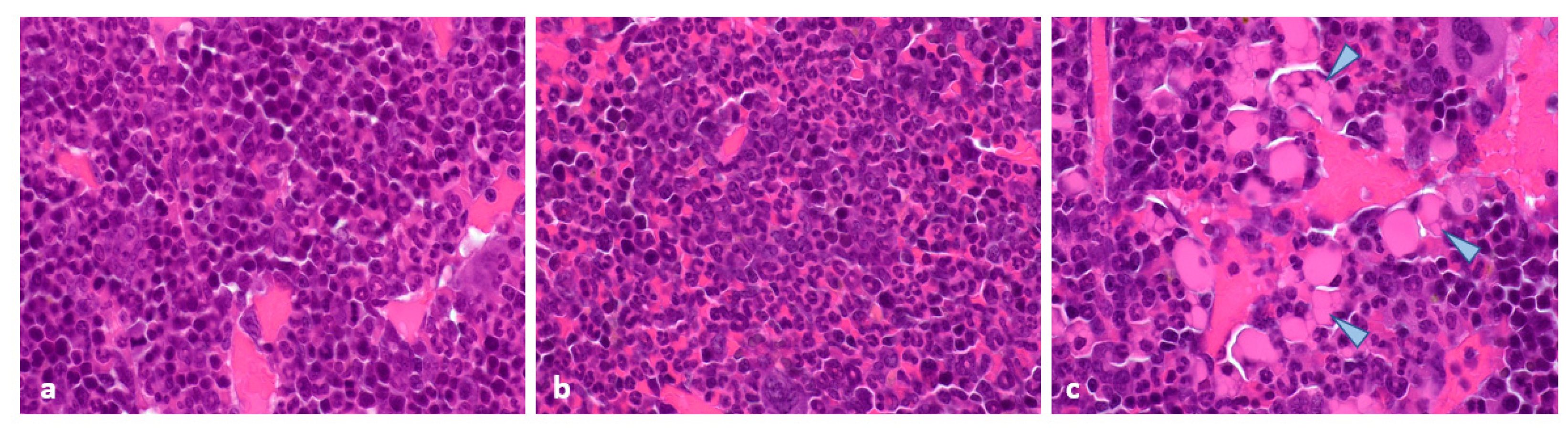
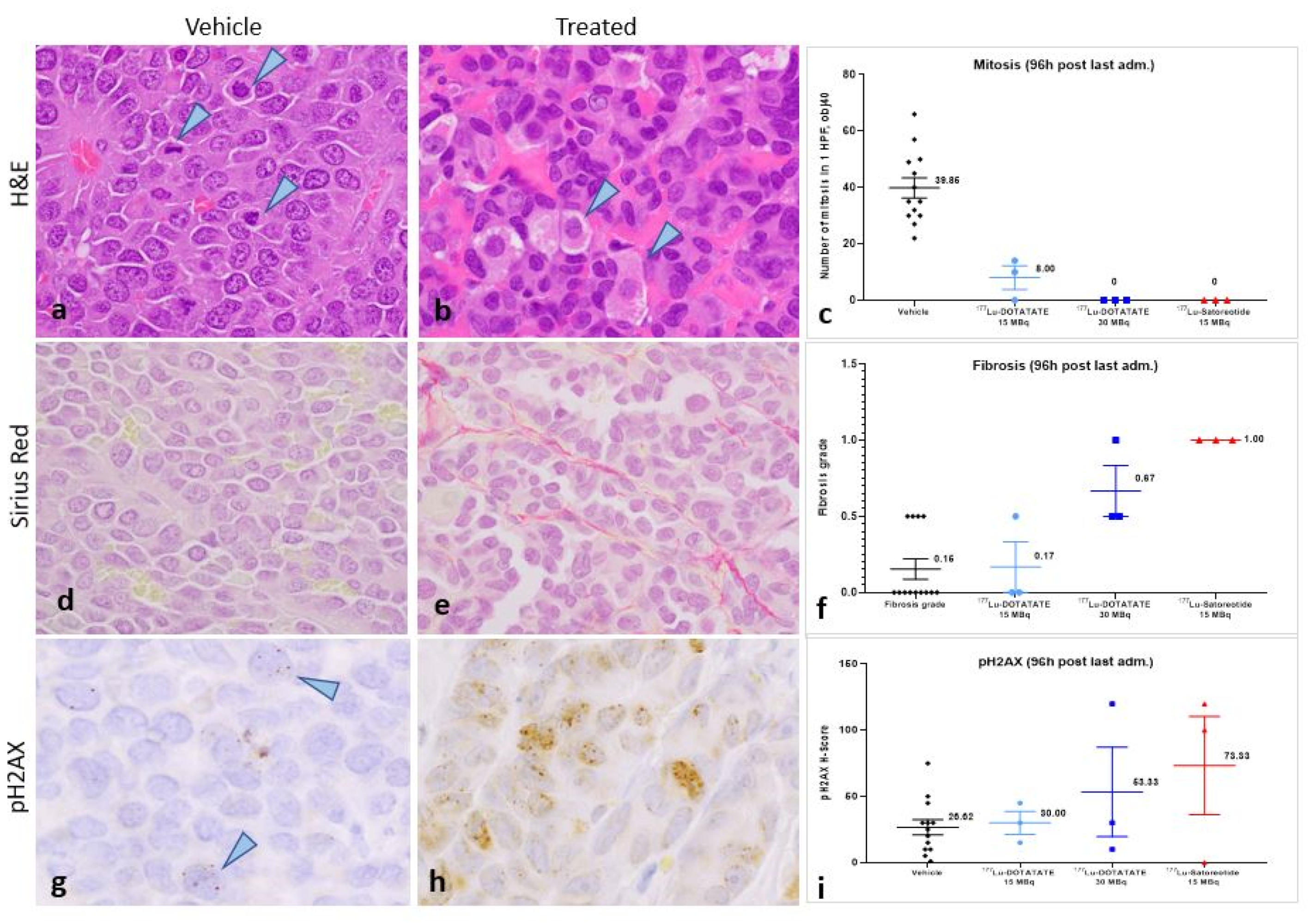
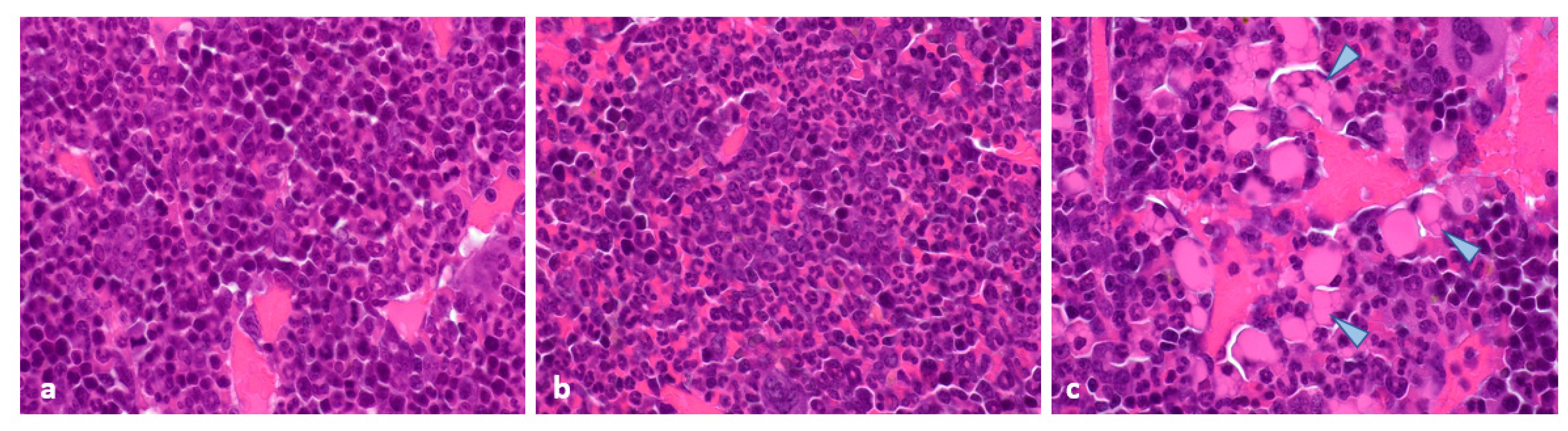
2.4. Histopathological and Immunohistochemical Analyses of Tumours at 96 Hours Post-Administration of Last Treatment
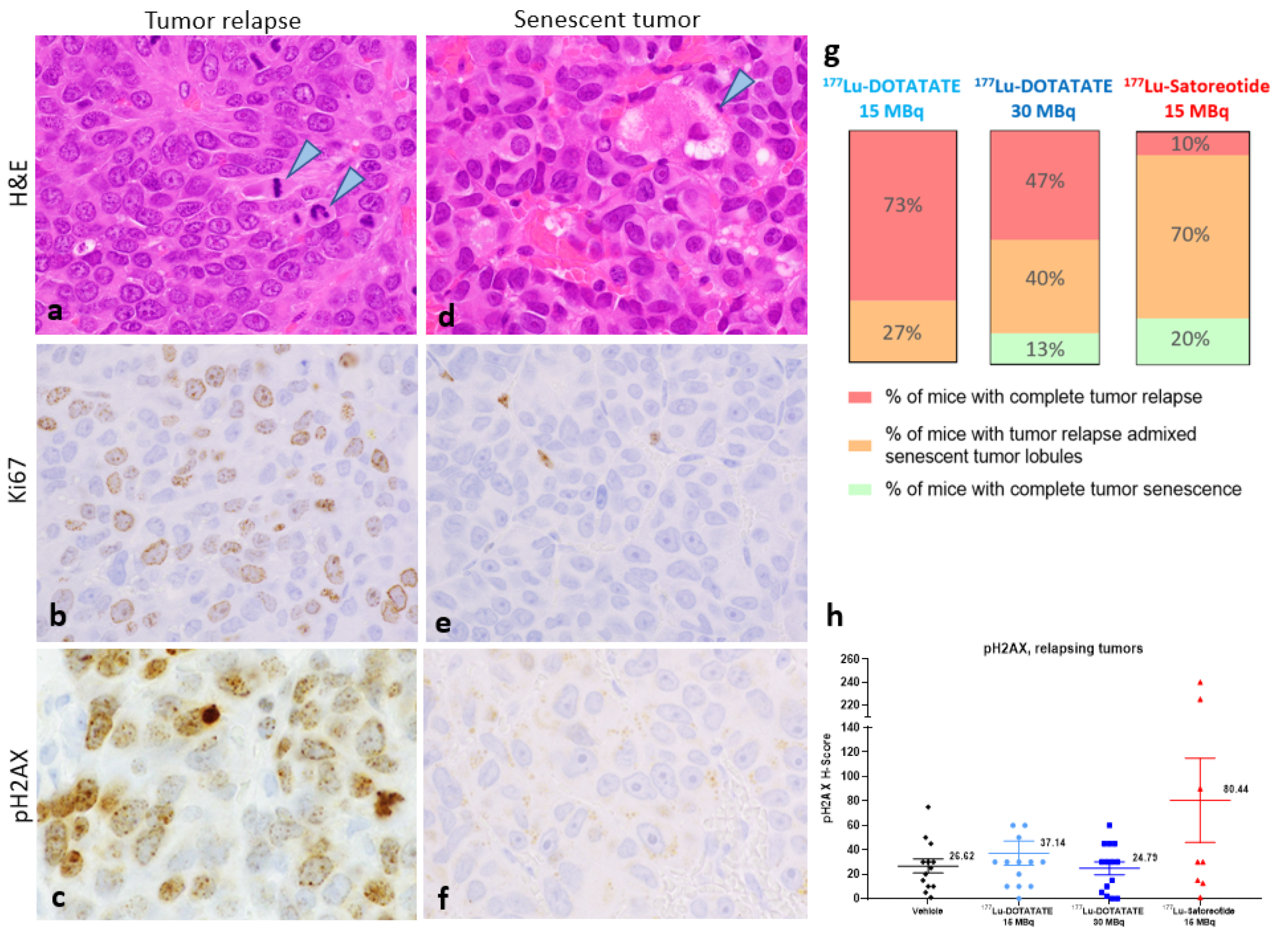
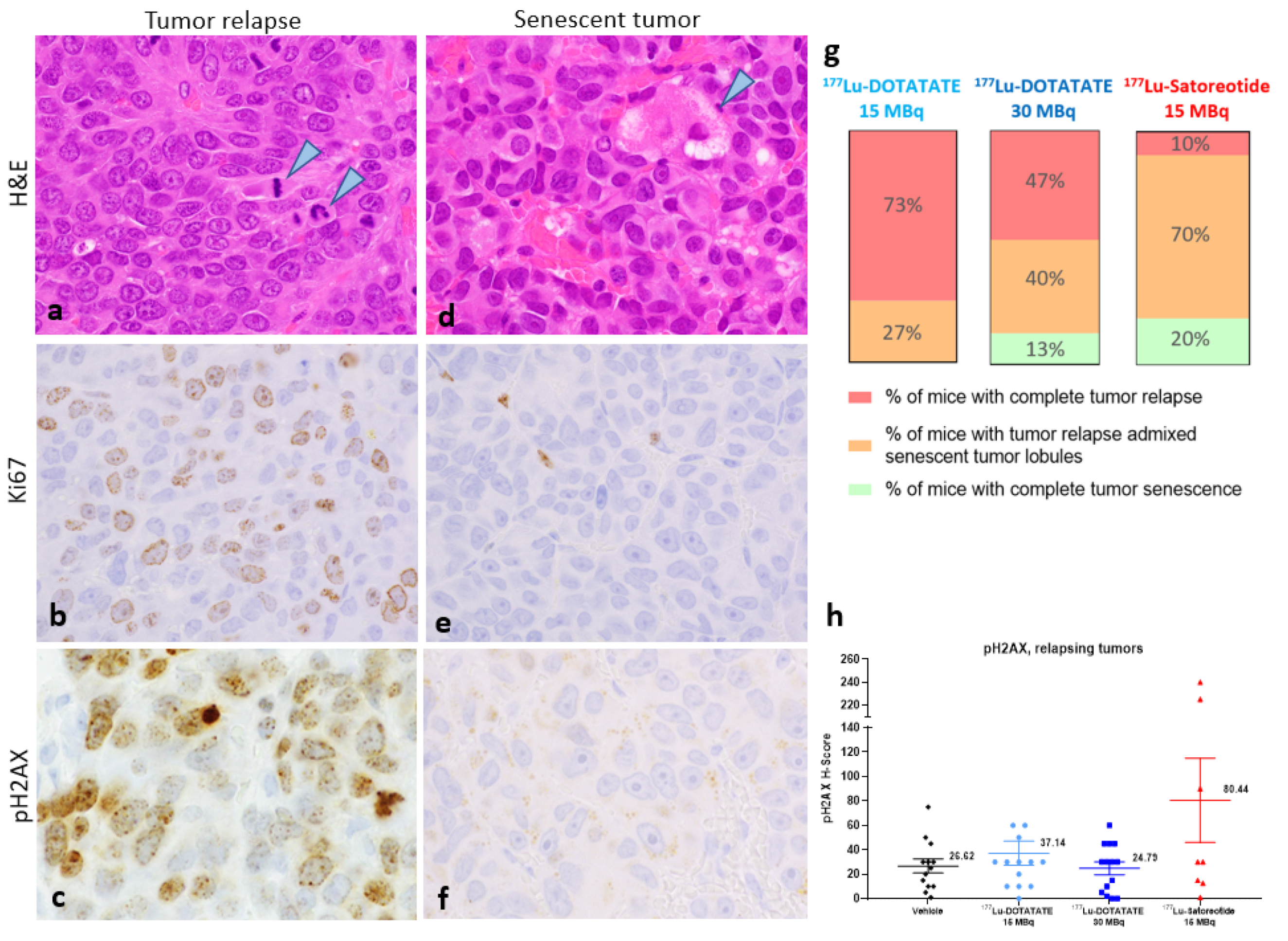
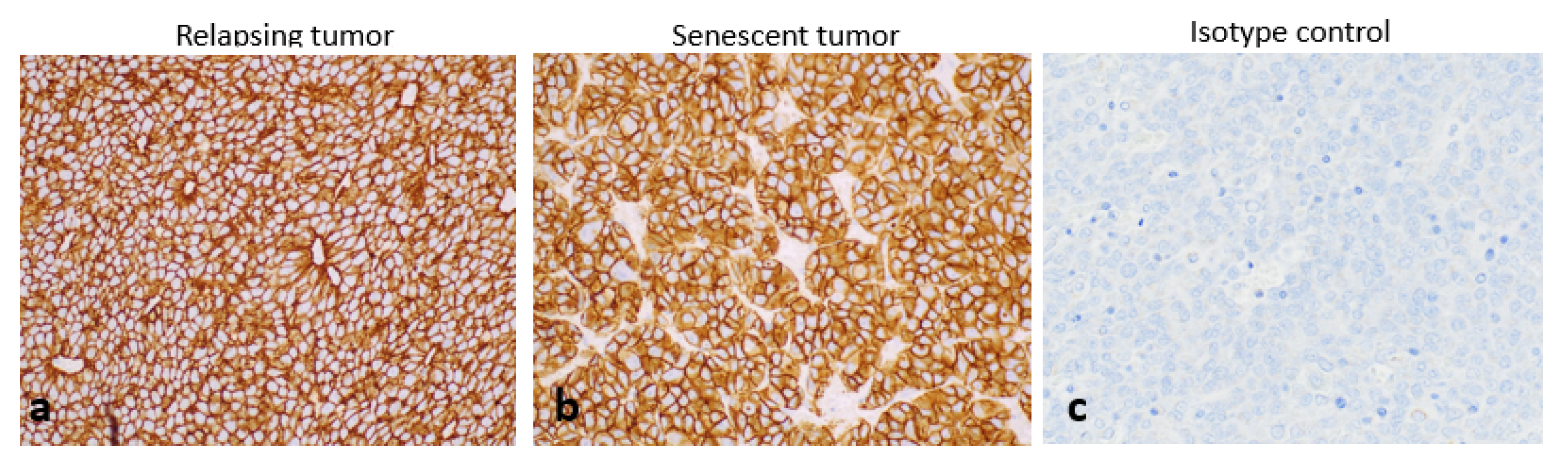
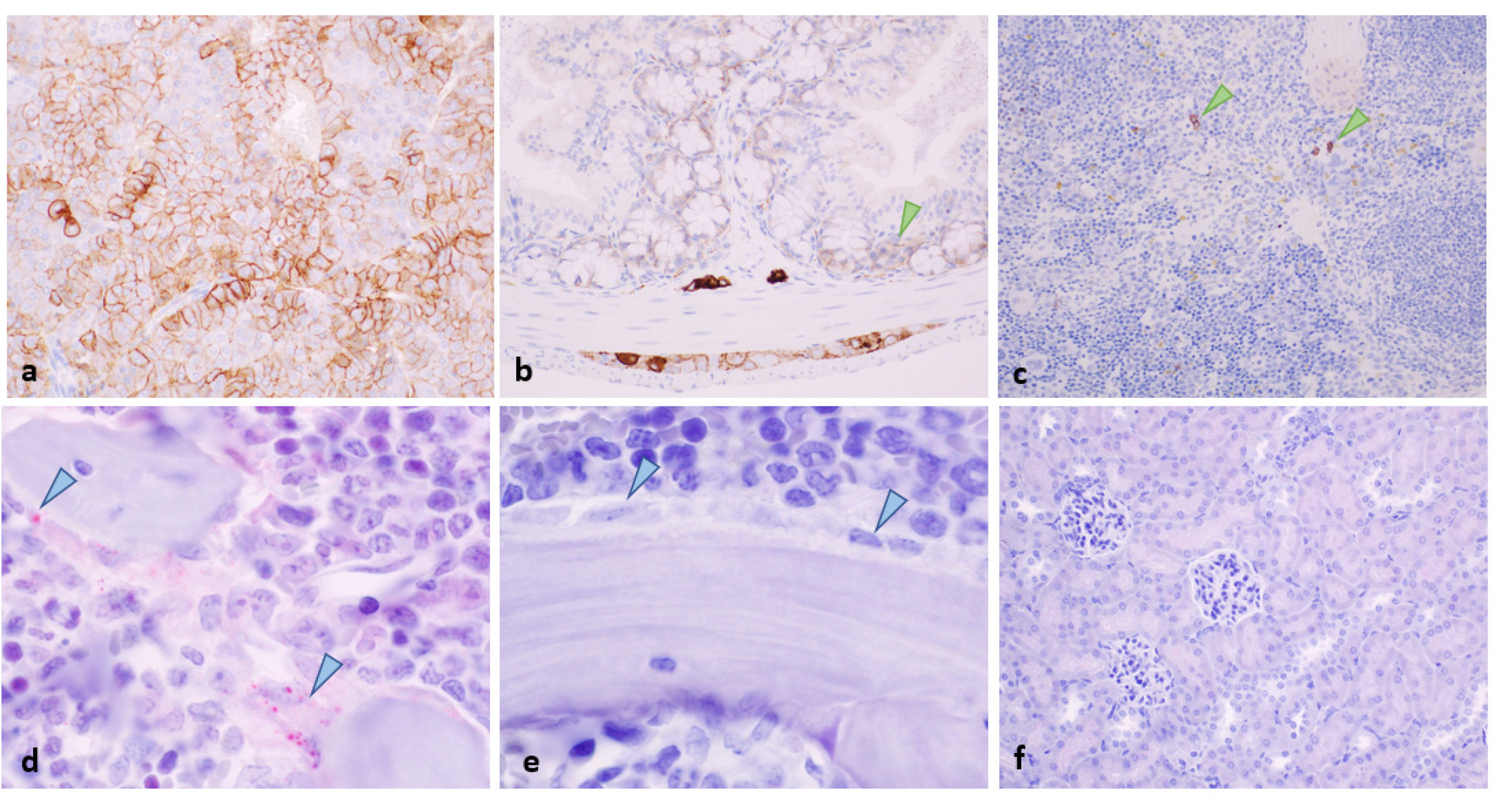
2.5. Histopathology and Immunohistochemistry of Tumours at the End of Study
2.6. Toxicity Evaluation
3. Discussion
4. Materials and Methods
4.1. Radioligands
4.2. Cell Culture and Membrane Preparation
4.3. Saturation Binding Assays
4.4. Animals and Ethics
4.5. In Vivo PRRT Studies
4.6. In Vivo Biodistribution Studies
4.7. Haematology, Histopathology and Immunohistochemistry
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, S.; Al-Toubah, T.; El-Haddad, G.; Strosberg, J. 177Lu-DOTA-TATE for the treatment of gastroenteropancreatic neuroendocrine tumours. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Macke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumours. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the therapeutic response to treatment with a 177Lu-labeled somatostatin receptor agonist and antagonist in preclinical models. J. Nucl. Med. 2016, 57, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Exner, S.; Groetzinger, C.; Prasad, S.; Konietschke, F.; Beindorff, N.; Kuehl, A.A.; Prasad, V.; Brenner, W.; Koziolek, E.J. Multimodal imaging of 2-cycle PRRT with 177Lu-DOTA-JR11 and 177Lu-DOTA-TOC in an orthotopic neuroendocrine xenograft tumour mouse model. J. Nucl. Med. 2021, 62, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: A pilot study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gönen, M.; Lohrmann, C.; et al. Phase I trial of well-differentiated neuroendocrine tumours (NETs) with radiolabeled somatostatin antagonist 177Lu-satoreotide tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Q.; Cai, L.; Xie, Y.; Chen, Y. The efficacy of 177Lu-DOTA-TATE peptide receptor radionuclide therapy (PRRT) in patients with metastatic neuroendocrine tumours: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2020, 146, 1533–1543. [Google Scholar] [CrossRef]
- Borgna, F.; Haller, S.; Rodriguez, J.M.M.; Ginj, M.; Grundler, P.V.; Zeevaart, J.R.; Köster, U.; Schibli, R.; van der Meulen, N.P.; Müller, C. Combination of terbium-161 with somatostatin receptor antagonists—a potential paradigm shift for the treatment of neuroendocrine neoplasms. EJNMMI 2022, 49, 1113–1126. [Google Scholar] [CrossRef]
- Mansi, R.; Plas, P.; Vauquelin, G.; Fani, M. Distinct in vitro binding profile of the somatostatin receptor subtype 2 antagonist [177Lu]Lu-OPS201 compared to the agonist [177Lu]Lu-DOTA-TATE. Pharmaceuticals 2021, 14, 1265. [Google Scholar] [CrossRef]
- Cescato, R.; Loesch, K.; Waser, B.; Mäcke, H.; Rivier, J.; Reubi, J.; Schonbrunn, A. Agonist-biased signaling at the sst2A receptor: The multi-somatostatin analogs KE108 and SOM230 activate and antagonize distinct signaling pathways. Mol. Endocrinol. 2010, 24, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.; Fahrer, J.; Maus, S.; Morgenstern, A.; Bruchertseifer, F.; Venkatachalam, S.; Fottner, C.; Weber, M.M.; Huelsenbeck, J.; Schreckenberger, M.; et al. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS ONE 2014, 9, e88239. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Lobachevsky, P.; Jackson, P.; Thompson, M.; Martin, O.A.; Hicks, R.J. Analysis of 177Lu-DOTA-octreotate therapy-induced DNA damage in peripheral blood lymphocytes of patients with neuroendocrine tumours. J. Nucl. Med. 2015, 56, 505–511. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Sundlöv, A.; Sjögreen-Gleisner, K.; Tennvall, J.; Dahl, L.; Svensson, J.; Åkesson, A.; Bernhardt, P.; Lindgren, O. Pituitary function after high-dose 177Lu-DOTA-TATE therapy and long-term follow-up. Neuroendocrinology 2021, 111, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-J.; Fennell, M.; et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 2020, 181, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Ansquer, C.; Lenzo, N.P.; Grønbæk, H.; Haug, A.; Navalkissoor, S.; Beauregard, J.M.; Germann, N.; McEwan, S.; Wild, D.; et al. An international open-label study on safety and efficacy of 177Lu-satoreotide tetraxetan in somatostatin receptor positive neuroendocrine tumours (NETs): An interim analysis [ESMO abstract 1160O]. Ann. Oncol. 2020, 31 (Suppl. S4), S771. [Google Scholar] [CrossRef]
- Cullinane, C.; Jeffery, C.M.; Roselt, P.D.; van Dam, E.M.; Jackson, S.; Kuan, K.; Jackson, P.; Binns, D.; van Zuylekom, J.; Harris, M.J.; et al. Peptide receptor radionuclide therapy with 67Cu-CuSarTATE is highly efficacious against a somatostatin-positive neuroendocrine tumour model. J. Nucl. Med. 2020, 61, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.P.; Mansi, R.; McDougall, L.; Kaufmann, J.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, pharmacokinetics, and dosimetry of 177Lu-, 90Y-, and 111In-labeled somatostatin receptor antagonist OPS201 in comparison to the agonist 177Lu-DOTA-TATE: The mass effect. J. Nucl. Med. 2017, 58, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Périer, C.; Martin, V.; Cornet, S.; Favre-Guilmard, C.; Rocher, M.; Bindler, J.; Wagner, S.; Andriambeloson, E.; Rudkin, B.B.; Marty, R.; et al. Recombinant botulinum neurotoxin serotype A1 in vivo characterization. Pharmacol. Res. Perspect. 2021, 9, e00857. [Google Scholar] [CrossRef]
- Lezmi, S.; Rokh, N.; Saint-Macary, G.; Pino, M.; Sallez, V.; Thevenard, F.; Roome, N.; Rosolen, S. Chloroquine causes similar electroretinogram modifications, neuronal phospholipidosis and marked impairment of synaptic vesicle transport in albino and pigmented rats. Toxicology 2013, 308, 50–59. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organs | [177Lu]Lu-DOTA-TATE 15 MBq | [177Lu]Lu-DOTA-TATE 30 MBq | [177Lu]Lu-Satoreotide Tetraxetan 15 MBq |
|---|---|---|---|
| Tumour | 5.6 (1.7) | 5.8 (0.58) | 20 (0.80) |
| Kidneys | 0.36 (0.067) | 0.51 (0.11) | 2.9 (0.44) |
| Adrenals | 0.59 (0.082) | 0.60 (0.048) | 0.70 (0.34) |
| Femur | 0.034 (0.0028) | 0.022 (0.0011) | 0.08 (0.0081) |
| Spleen | 0.018 (0.0014) | 0.015 (0.0015) | 0.072 (0.0076) |
| Tail | 0.055 (0.012) | 0.042 (0.010) | 0.082 (0.0080) |
| Groups | Vehicle | [177Lu]Lu-DOTA-TATE 15 MBq | [177Lu]Lu-DOTA-TATE 30 MBq | [177Lu]Lu-Satoreotide Tetraxetan 15 MBq |
|---|---|---|---|---|
| Time to reach tumour volume ≥850 mm3, days | 4.3 (2.5–7.7) | 43 (40–45) | 48 (47–49) | 68 (51–not reached at end of study) |
| Comparisons * between groups of time to reach tumour volume ≥850 mm3 | ||||
| Vehicle | Reference | p < 0.0001 | p < 0.0001 | p < 0.0001 |
| 177Lu-DOTA-TATE 15 MBq | - | Reference | p = 0.0036 | p < 0.0001 |
| 177Lu-DOTA-TATE 30 MBq | - | - | Reference | p = 0.0044 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plas, P.; Limana, L.; Carré, D.; Thiongane, A.; Raguin, O.; Mansi, R.; Meyer-Losic, F.; Lezmi, S. Comparison of the Anti-Tumour Activity of the Somatostatin Receptor (SST) Antagonist [177Lu]Lu-Satoreotide Tetraxetan and the Agonist [177Lu]Lu-DOTA-TATE in Mice Bearing AR42J SST2-Positive Tumours. Pharmaceuticals 2022, 15, 1085. https://doi.org/10.3390/ph15091085
Plas P, Limana L, Carré D, Thiongane A, Raguin O, Mansi R, Meyer-Losic F, Lezmi S. Comparison of the Anti-Tumour Activity of the Somatostatin Receptor (SST) Antagonist [177Lu]Lu-Satoreotide Tetraxetan and the Agonist [177Lu]Lu-DOTA-TATE in Mice Bearing AR42J SST2-Positive Tumours. Pharmaceuticals. 2022; 15(9):1085. https://doi.org/10.3390/ph15091085
Chicago/Turabian StylePlas, Pascale, Lorenzo Limana, Denis Carré, Amath Thiongane, Olivier Raguin, Rosalba Mansi, Florence Meyer-Losic, and Stéphane Lezmi. 2022. "Comparison of the Anti-Tumour Activity of the Somatostatin Receptor (SST) Antagonist [177Lu]Lu-Satoreotide Tetraxetan and the Agonist [177Lu]Lu-DOTA-TATE in Mice Bearing AR42J SST2-Positive Tumours" Pharmaceuticals 15, no. 9: 1085. https://doi.org/10.3390/ph15091085
APA StylePlas, P., Limana, L., Carré, D., Thiongane, A., Raguin, O., Mansi, R., Meyer-Losic, F., & Lezmi, S. (2022). Comparison of the Anti-Tumour Activity of the Somatostatin Receptor (SST) Antagonist [177Lu]Lu-Satoreotide Tetraxetan and the Agonist [177Lu]Lu-DOTA-TATE in Mice Bearing AR42J SST2-Positive Tumours. Pharmaceuticals, 15(9), 1085. https://doi.org/10.3390/ph15091085