
Rational Strategy for Designing Peptidomimetic Small Molecules Based on Cyclic Peptides Targeting Protein–Protein Interaction between CTLA-4 and B7-1
 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
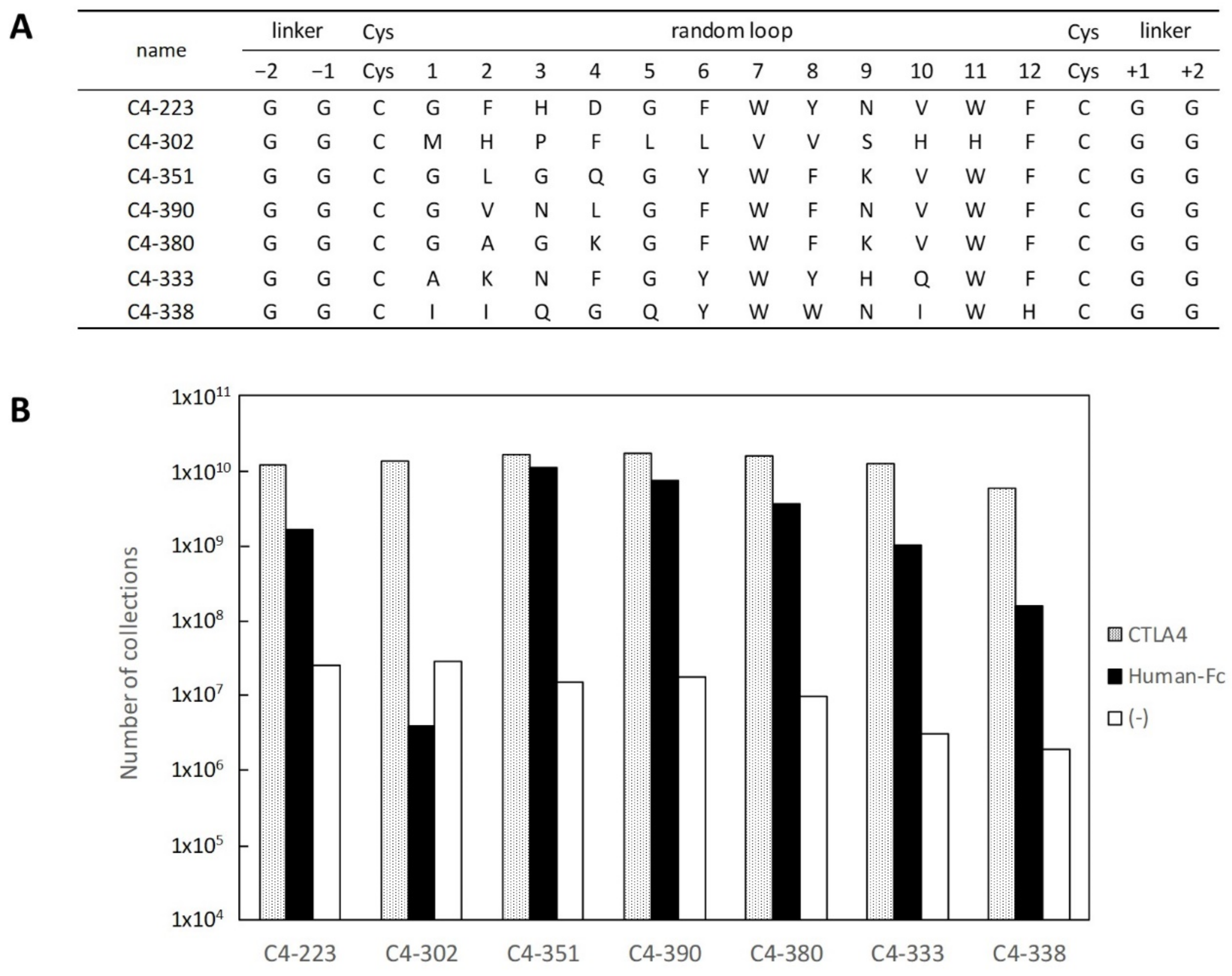
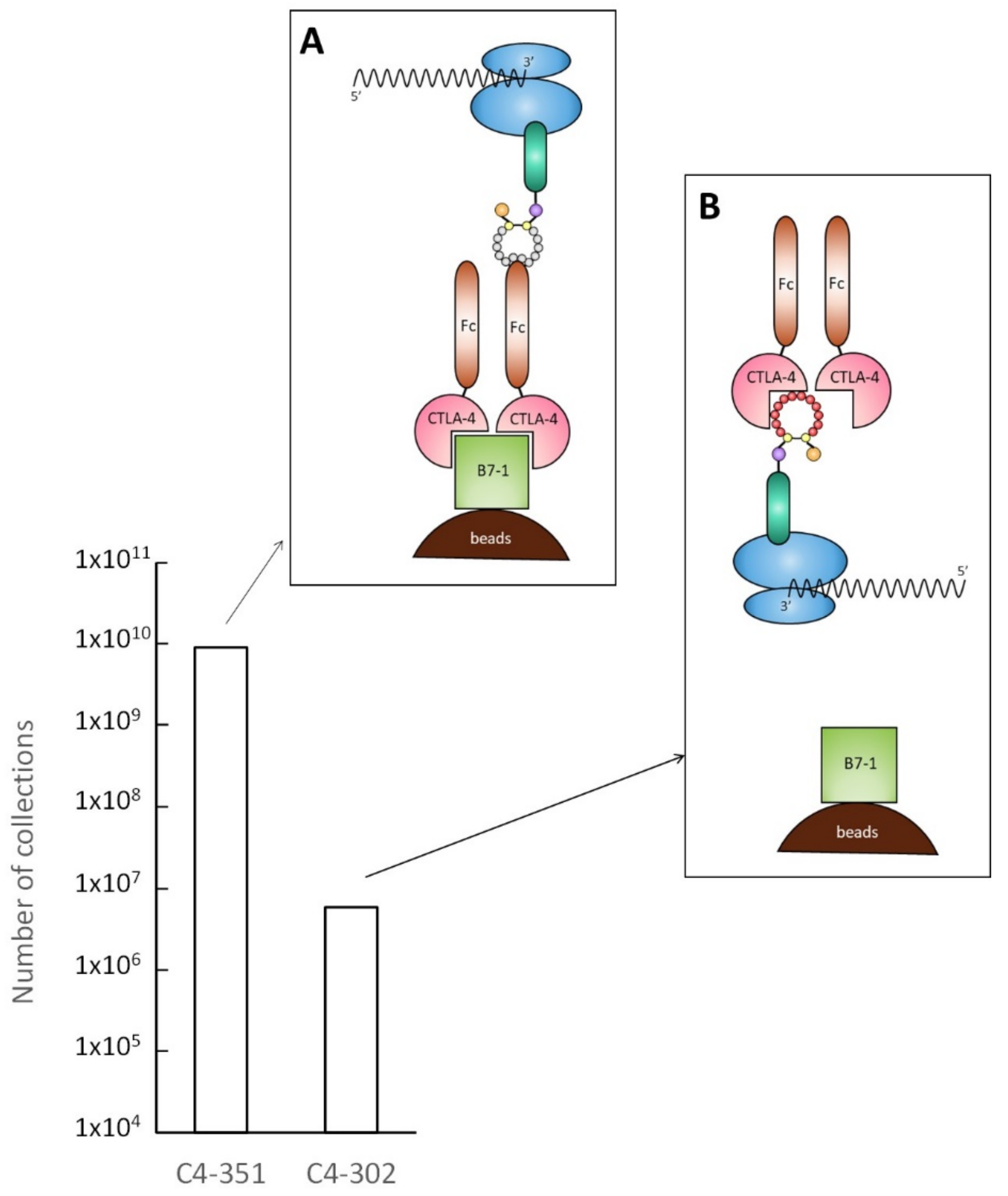
2.1. Selection and Affinity Maturation of Cyclic Peptide Ligands against CTLA-4
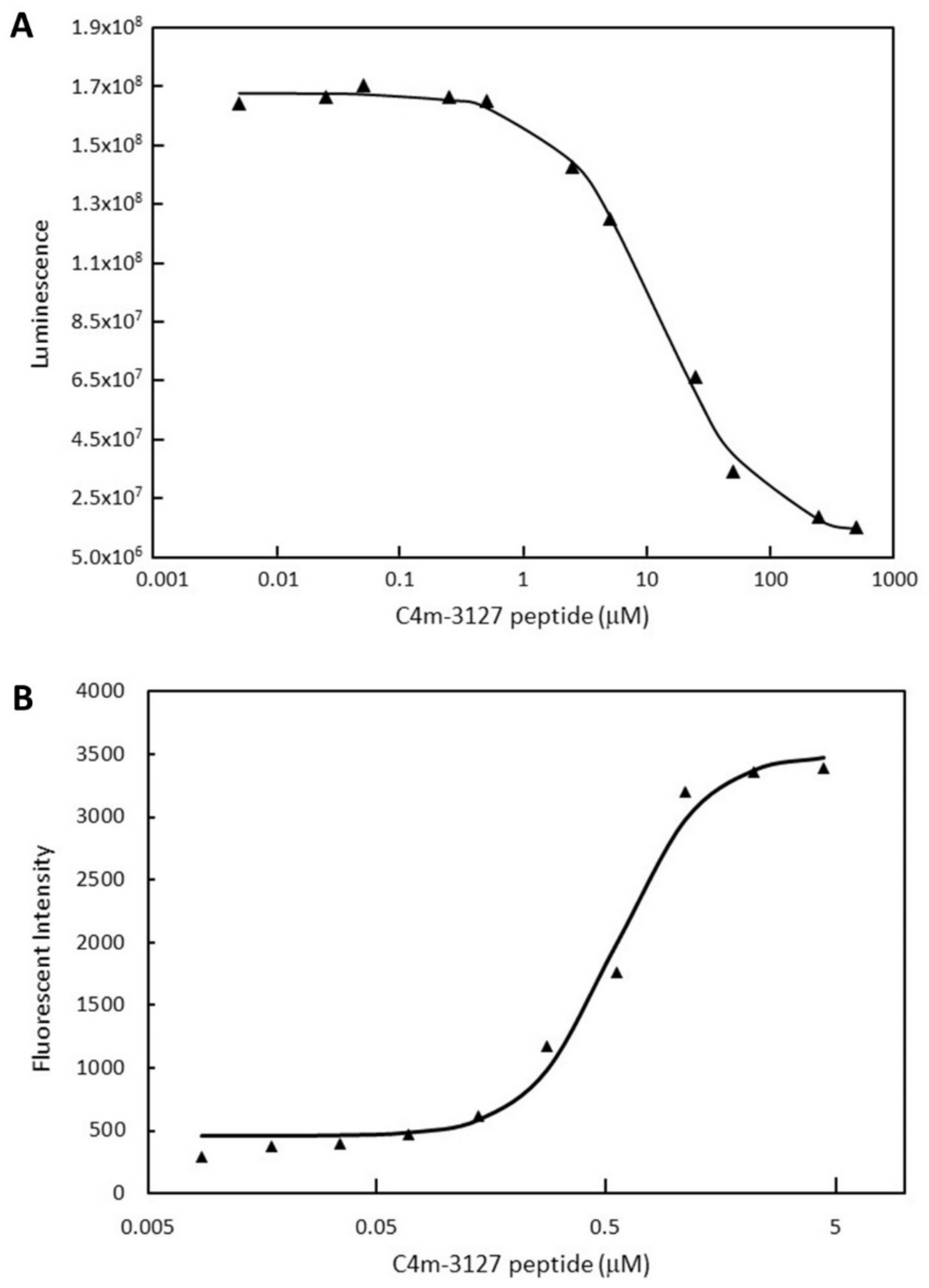
2.2. Characterization of Selected Cyclic Peptide
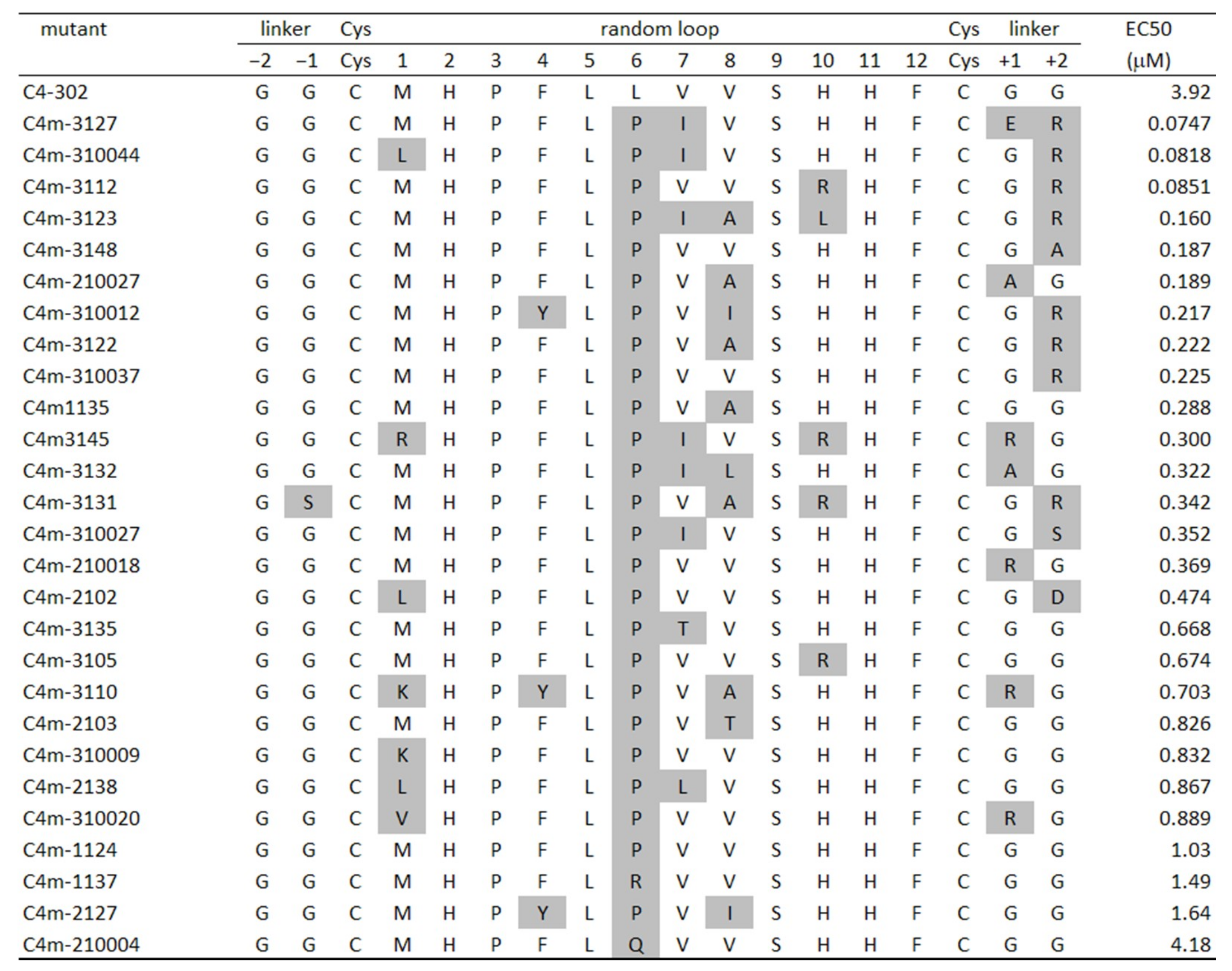
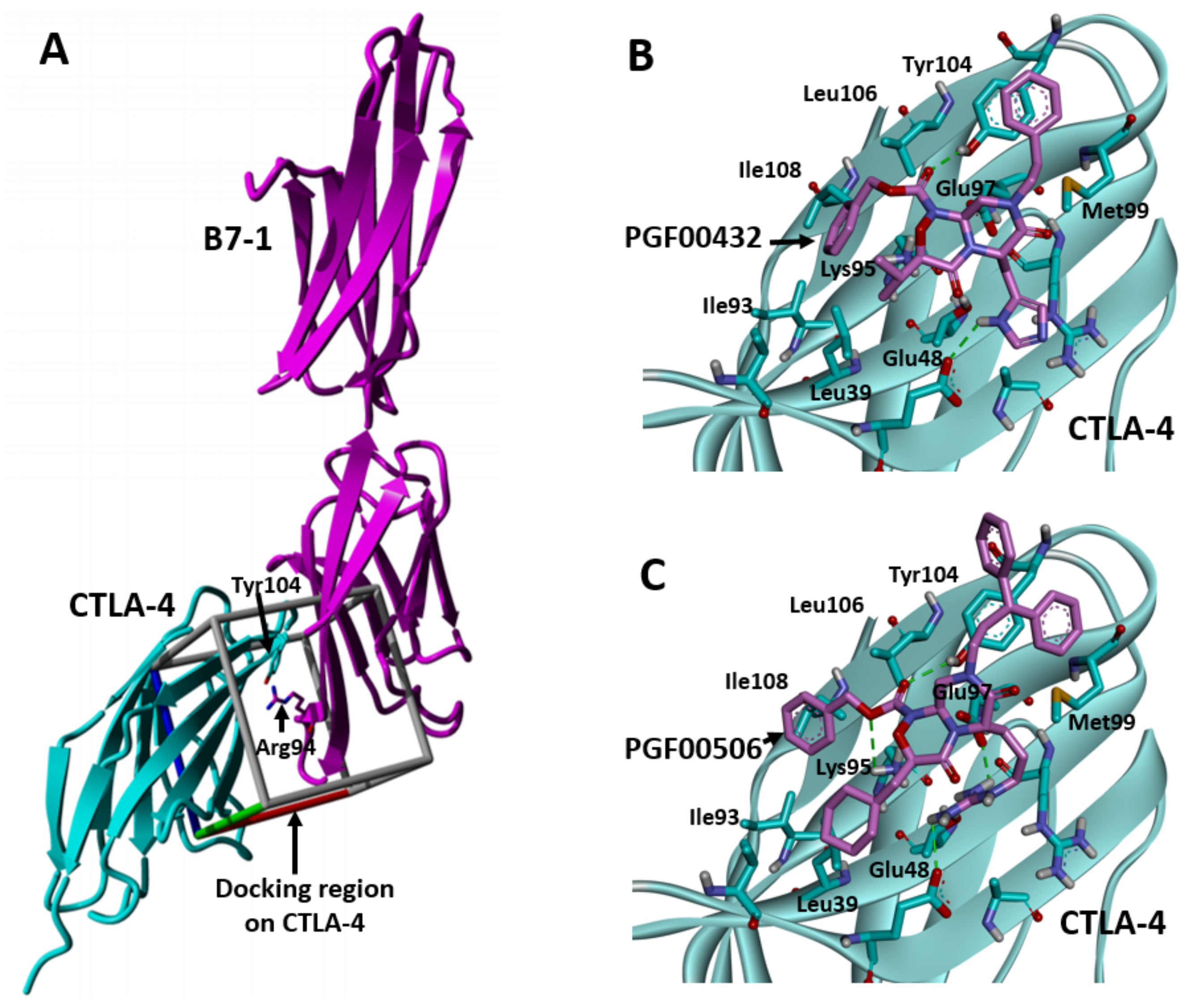
2.3. Identification of Amino Acids Involved in Binding with CTLA-4
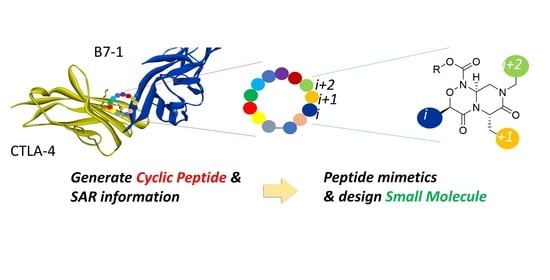
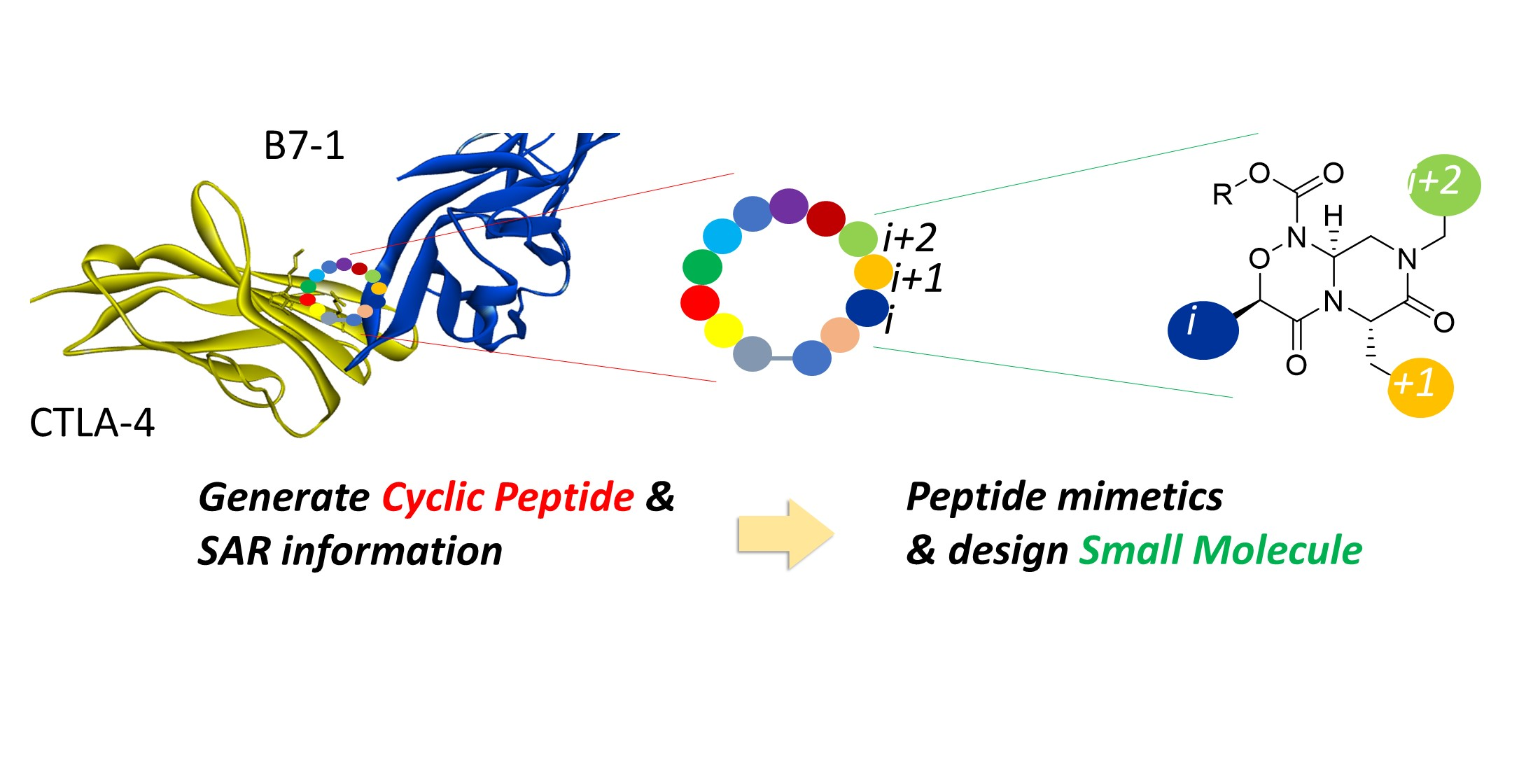
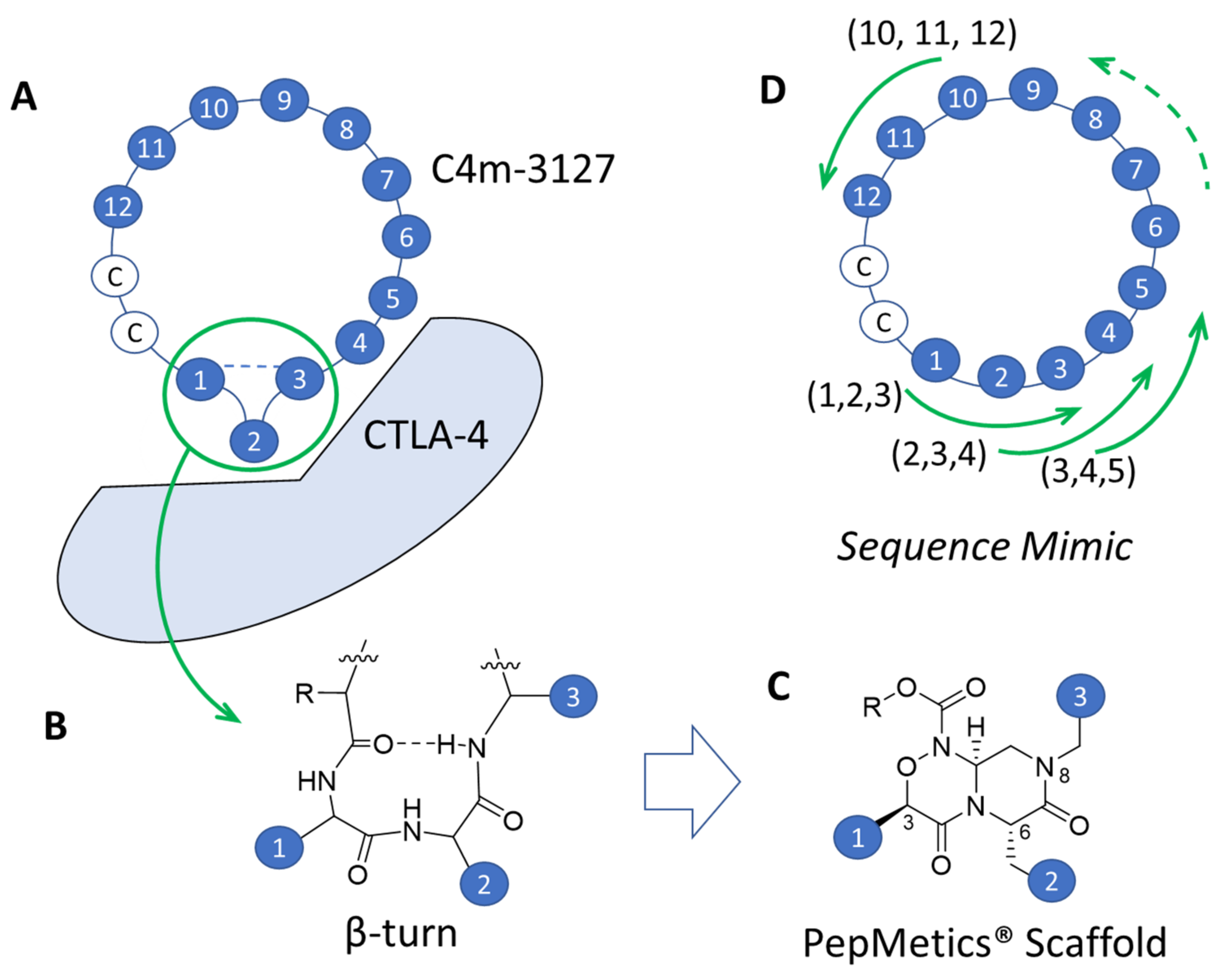
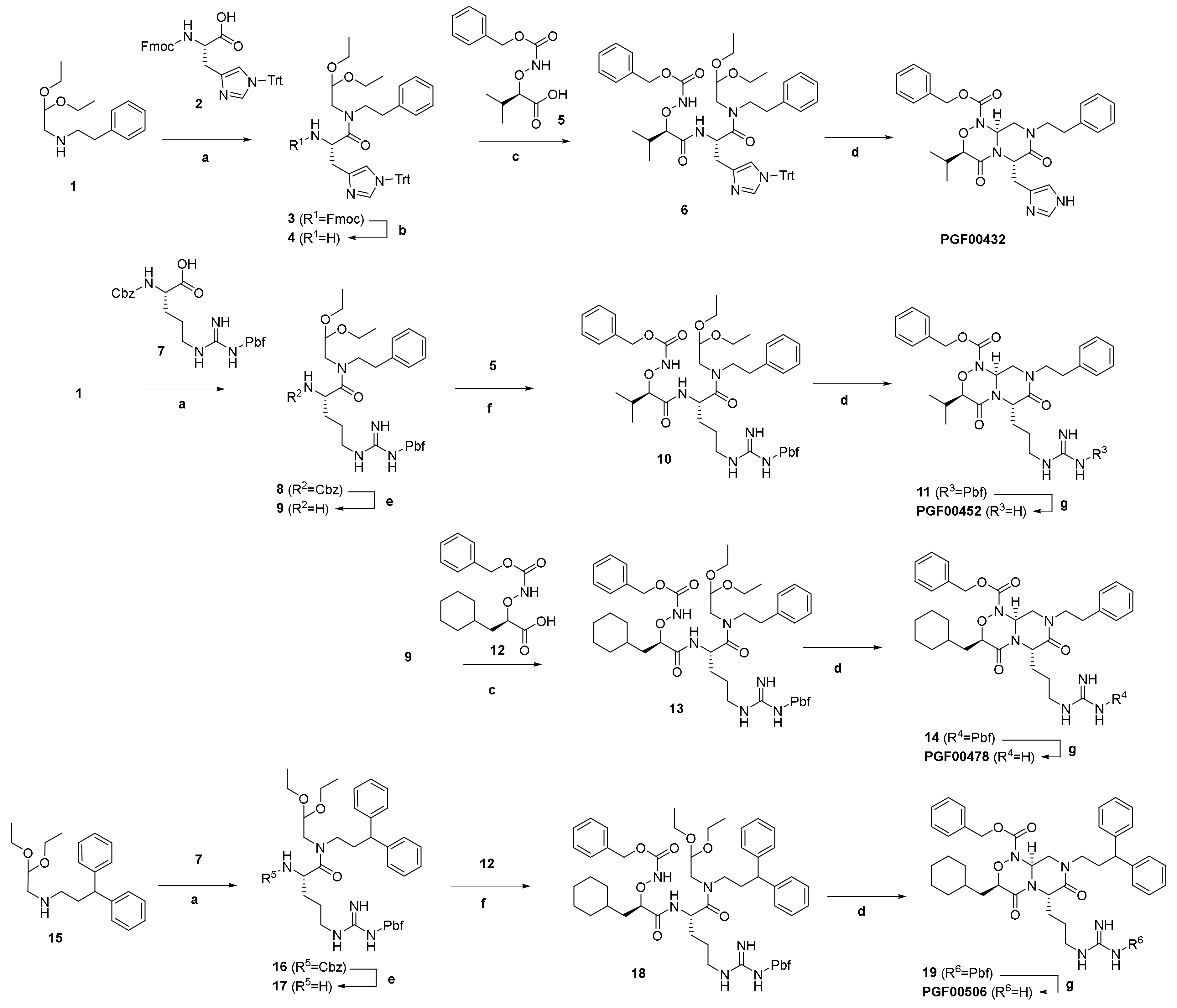
2.4. Strategy for Transformation of Cyclic Peptides to Small Molecules
2.5. SBDD Approach
2.6. Competitive ELISA
3. Discussion
4. Materials and Methods
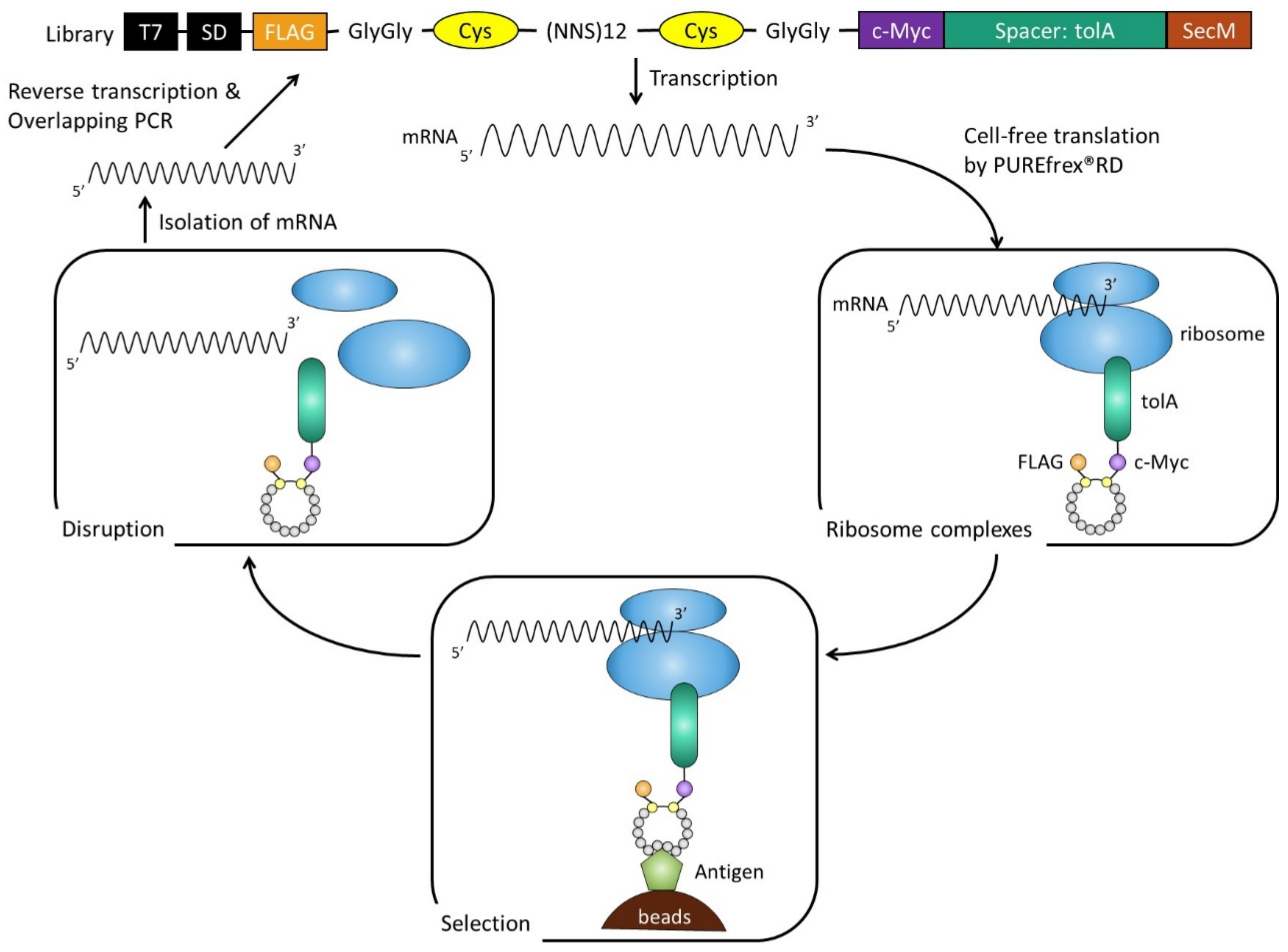
4.1. Construction of Cyclic Peptide Library
4.2. RD Selection and Affinity Maturation
4.3. Cloning and Inhibitory Activity Evaluation
4.4. Overexpression and Purification of Cyclic Peptide–MBP Fusion
4.5. ELISA-Based Determination of EC50 Value
4.6. Affinity Measurement of Synthetic Cyclic Peptide
4.7. Chemistry
4.7.1. General Methods
4.7.2. Abbreviations
- HATU: [dimethylamino(triazolo [4,5-b]pyridin-3-yloxy)methylene]-dimethyl-ammonium;hexafluorophosphate
- DMT-MM: 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium chloride
- DBU: 1,8-diazabicyclo [5.4.0]-7-undecene
- DIEA: N,N-diisopropylethylamine
- Trt: triphenylmethyl
- Pbf: 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- Fmoc: 9-fluorenylmethyloxycarbonyl
- The synthetic routes and conditions are shown in Scheme 1.
4.7.3. Synthesis of PGF00432
4.7.4. Synthesis of PGF00452
4.7.5. Synthesis of PGF00478
4.7.6. Synthesis of PGF00506
4.8. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug. Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Dariavach, P.; Mattei, M.G.; Golstein, P.; Lefranc, M.P. Human Ig superfamily CTLA-4 gene: Chromosomal localization and identity of protein sequence between murine and human CTLA-4 cytoplasmic domains. Eur. J. Immunol. 1988, 18, 1901–1905. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Perry, E.T. Discovery of Small Molecule Inhibitors of Immune Checkpoint Proteins. Ph.D. Thesis, Vanderbilt University, Nashville, TN, USA, 15 March 2019. Available online: https://etd.library.vanderbilt.edu/etd-03152019-112318 (accessed on 10 May 2019).
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef]
- Dreier, B.; Pluckthun, A. Ribosome Display: A technology for selecting and evolving proteins from large libraries. Methods Mol. Biol. 2011, 687, 283–306. [Google Scholar] [CrossRef]
- Malde, A.K.; Hill, T.A.; Iyer, A.; Fairlie, D.P. Crystal Structures of Protein-Bound Cyclic Peptides. Chem. Rev. 2019, 119, 9861–9914. [Google Scholar] [CrossRef]
- Roxin, Á.; Zheng, G. Flexible or Fixed: A Comparative Review of Linear and Cyclic Cancer-Targeting Peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef]
- Brueckner, A.C.; Deng, Q.; Cleves, A.E.; Lesburg, C.A.; Alvarez, J.C.; Reibarkh, M.Y.; Sherer, E.C.; Jain, A.N. Conformational Strain of Macrocyclic Peptides in Ligand–Receptor Complexes Based on Advanced Refinement of Bound-State Conformers. J. Med. Chem. 2021, 64, 3282–3298. [Google Scholar] [CrossRef]
- Takashima, H.; Yoshimori, A.; Honda, E.; Taguri, T.; Ozawa, J.; Kasai, M.; Shuto, S.; Takehara, D. Visualized and Quantitative Conformational Analysis of Peptidomimetics. ACS Omega 2021, 6, 26601–26612. [Google Scholar] [CrossRef]
- Podlesnykh, S.V.; Abramova, K.E.; Gordeeva, A.; Khlebnikov, A.I.; Chapoval, A.I. Peptide Blocking CTLA-4 and B7-1 Interaction. Molecules 2021, 26, 253. [Google Scholar] [CrossRef]
- Yoshida, S.; Uehara, S.; Kondo, N.; Takahashi, Y.; Yamamoto, S.; Kameda, A.; Kawagoe, S.; Inoue, N.; Yamada, M.; Yoshimura, N.; et al. Peptide-to-Small molecule: A Pharmacophore-Guided Small molecule Lead Generation Strategy from High-Affinity Macrocyclic Peptides. J. Med. Chem. 2022, 65, 10655–10673. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein synthesis by pure translation systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef]
- Ohashi, H.; Kanamori, T.; Osada, E.; Akbar, B.K.; Ueda, T. Peptide screening using pure ribosome display. Method Mol. Biol. 2012, 805, 251–259. [Google Scholar] [CrossRef]
- Zhao, H.; Giver, L.; Zhao, Z.; Affholter, J.A.; Arnold, F.H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 1998, 16, 258–261. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Stamper, C.C.; Zhang, Y.; Tobin, J.F.; Erbe, D.V.; Ikemizu, S.; Davis, S.J.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Sonnen, A.F.; George, R.; Dessailly, B.H.; Stagg, L.J.; Evans, E.J.; Orengo, C.A.; Stuart, D.I.; Ladbury, J.E.; Ikemizu, S.; et al. Rigid-body ligand recognition drives cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor triggering. J. Biol. Chem. 2011, 286, 6685–6696. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuihiji, K.; Honda, E.; Kojoh, K.; Katoh, S.; Taguri, T.; Yoshimori, A.; Takashima, H. Rational Strategy for Designing Peptidomimetic Small Molecules Based on Cyclic Peptides Targeting Protein–Protein Interaction between CTLA-4 and B7-1. Pharmaceuticals 2022, 15, 1506. https://doi.org/10.3390/ph15121506
Tsuihiji K, Honda E, Kojoh K, Katoh S, Taguri T, Yoshimori A, Takashima H. Rational Strategy for Designing Peptidomimetic Small Molecules Based on Cyclic Peptides Targeting Protein–Protein Interaction between CTLA-4 and B7-1. Pharmaceuticals. 2022; 15(12):1506. https://doi.org/10.3390/ph15121506
Chicago/Turabian StyleTsuihiji, Kumiko, Eiji Honda, Kanehisa Kojoh, Shizue Katoh, Tomonori Taguri, Atsushi Yoshimori, and Hajime Takashima. 2022. "Rational Strategy for Designing Peptidomimetic Small Molecules Based on Cyclic Peptides Targeting Protein–Protein Interaction between CTLA-4 and B7-1" Pharmaceuticals 15, no. 12: 1506. https://doi.org/10.3390/ph15121506
APA StyleTsuihiji, K., Honda, E., Kojoh, K., Katoh, S., Taguri, T., Yoshimori, A., & Takashima, H. (2022). Rational Strategy for Designing Peptidomimetic Small Molecules Based on Cyclic Peptides Targeting Protein–Protein Interaction between CTLA-4 and B7-1. Pharmaceuticals, 15(12), 1506. https://doi.org/10.3390/ph15121506