
Recent Advances in Oral Peptide or Protein-Based Drug Liposomes


Abstract
:1. Introduction
2. Properties of Protein and Peptide Drugs
3. Phospholipid Materials Suitable for the Oral Administration of Liposomes
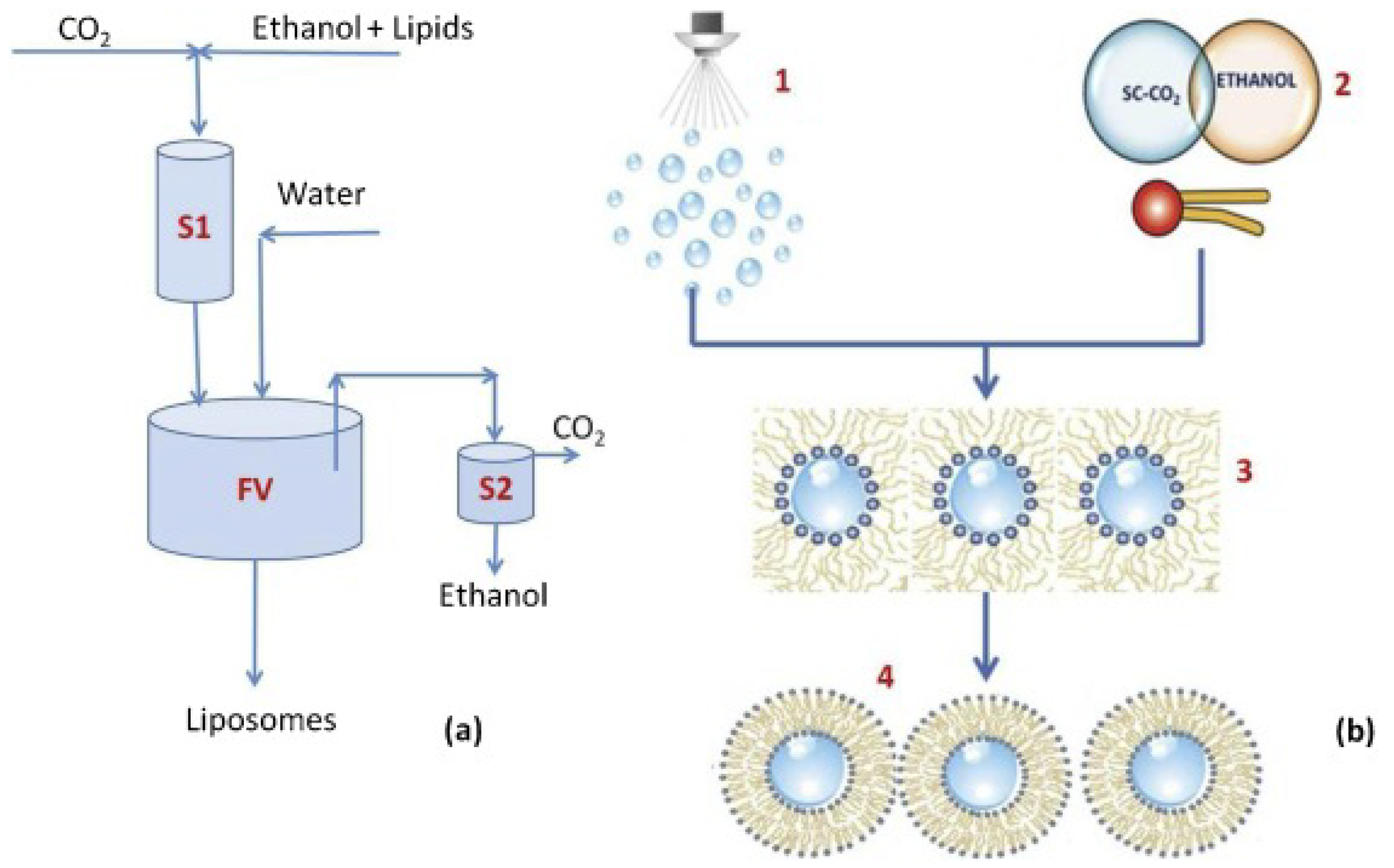
4. Preparation Methods for Polypeptide Liposomes
5. Stability Strategy
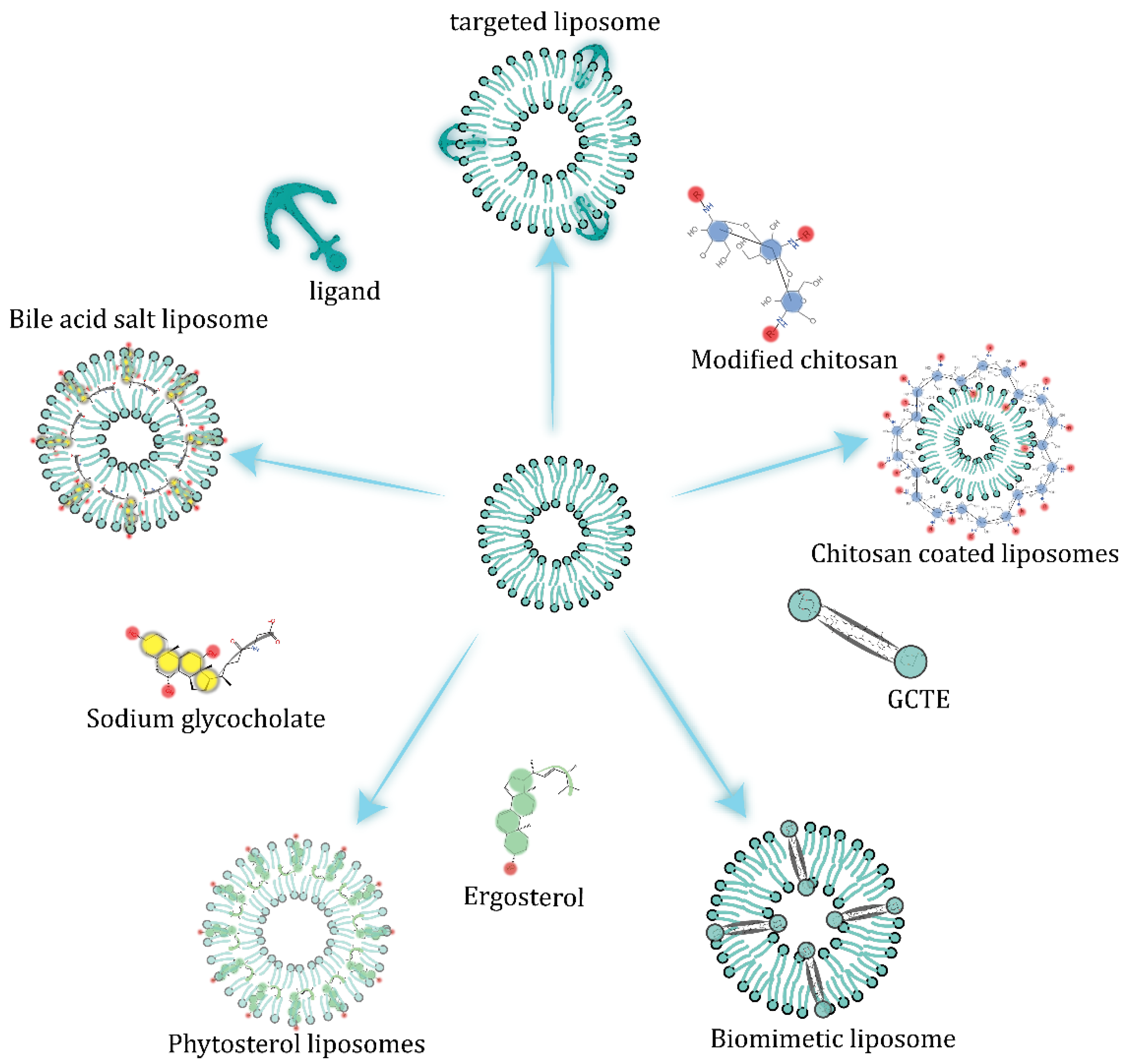
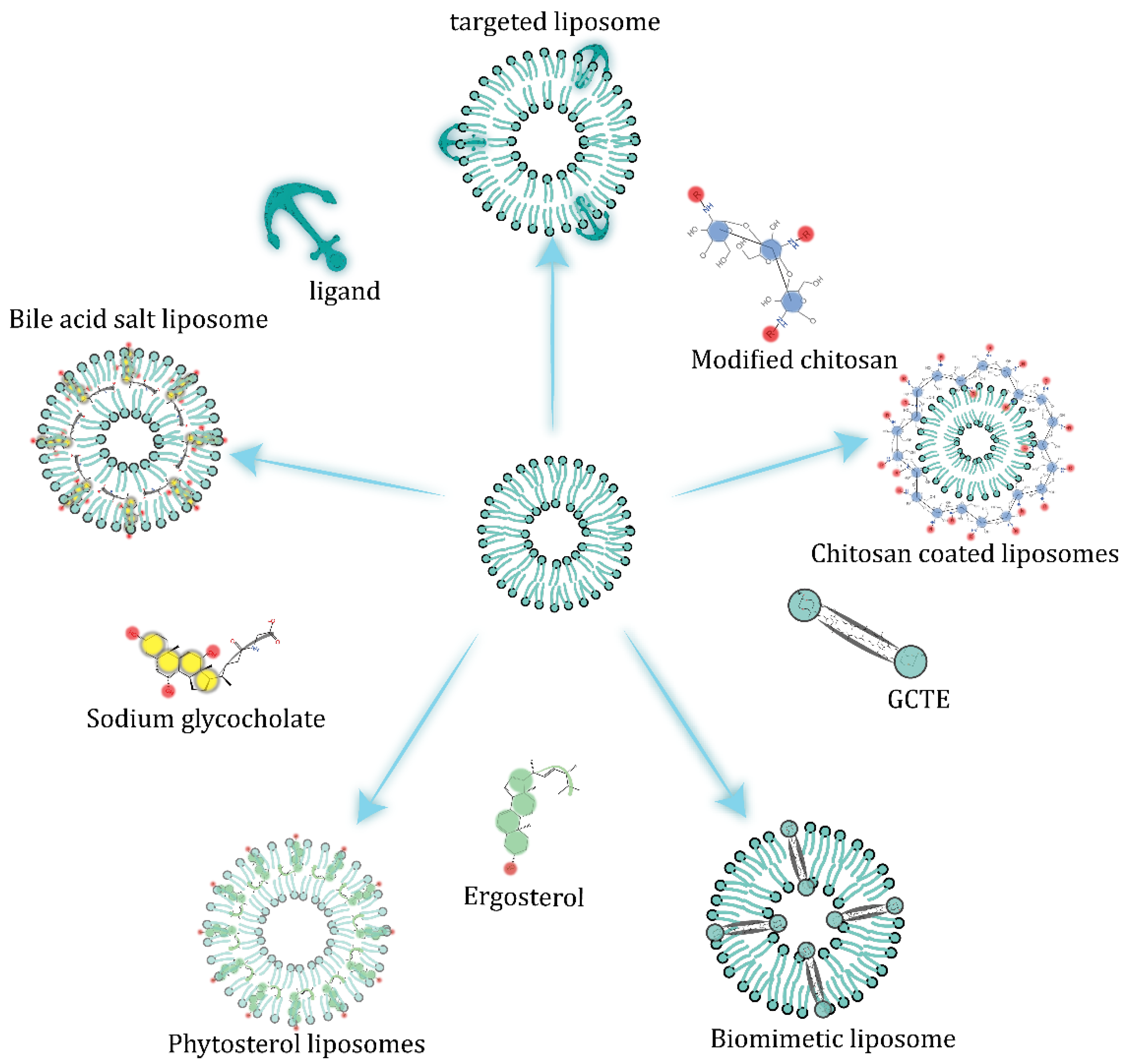
5.1. Alteration of Liposome Membrane Composition
5.2. Embedded Bile Salts
5.3. Surface-Coating Strategy
5.4. Diversified Dosage Forms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Phospholipid | Strategy to Protect Liposomes from the Damage of GIT | Properties | EE (%) ± SD | MD (nm) ± SD | Zeta (mv) ± SD | Ref |
|---|---|---|---|---|---|---|---|
| Myrcludex B | EPC | GCTE, which is resistant to hydrolysis and oxidation, was embedded in the phospholipid bilayer | At least 7% of the initial dose of Myrcludex B was absorbed, with a 3.5-fold increase in oral effectiveness | 65.7 ± 2.9 | 140.7 ± 4.3 | −4.2 ± 0.5 | [25] |
| rhINS | SPC DPPG Chol | Phytosterols with stronger interactions with phospholipids were used instead of cholesterol | After 4 h in SGF, Er-lip retained more than 70% of the insulin; the plasma glucose level could be reduced to about 60% of the initial value and kept low for 8 h | 30 ± 2.0 | 157.1 ± 0.4 | −60.5 ± 9.8 | [41] |
| rhINS | SPC | GCA was able to reduce the degradation of liposomes in GIT and promote the internalization of lipid particles | High oral bioavailability of 11.0%, with a mild and lasting hypoglycemic effect | 35 ± 2.1 | 358 ± 28.0 | - | [42] |
| Calcitonin | PC DSPG Chol | Surface-modified CPPs and TMC promoted the cellular uptake of liposomes | Effectively enhanced the oral absorption of calcitonin | 80 ± 2.0 | 118 ± 18.0 | −27.1 ± 5.8 | [47] |
| FID | DPPC DPPE-MCC | Chitosan coating with thiol group modification enhanced the adhesion and permeability of liposomes and inhibited the degradation of lipid membranes by enzymes | Papp was 2.8–3.4 times stronger than the initial value | - | 702.6 ± 138.0 | 8.62 ± 1.4 | [44] |
| Insulin | DPPC | Silica coating isolated liposomes from digestive enzymes | Silica coating was able to reduce the lipolysis rate and continuously release the drug for up to 8 h | 70.0 | 297 ± 0.4 | −15 ± 4.0 | [45] |
| Bee venom | PC | Liposomes were encapsulated into Eudragit S100-coated calcium alginate gel microspheres to slow the drug leakage of liposomes at non-specific sites | Liposomes completed drug release at the colon and maintained structural integrity in Git | 95.36 ± 0.3 | 2.05 ± 0.7 mm | - | [46] |
| rhINS | E-PC | Liposomes with chitosan coating were encapsulated into double extrusion by “two-step” microfluidic technology | Exhibited the characteristics of pH-responsive release and accelerated the intracellular internalization of encapsulated insulin | 91 ± 4.0% | 19 ± 1.0 μm | - | [11] |
6. Receptor-Mediated Transportation across Enterocytes
7. Small Intestine-Lymphatic Circulation Prevents the First-Pass Effect
8. Inhibition of P-gp and CYP3A4
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rader, A.F.B.; Weinmuller, M.; Reichart, F.; Schumacher-Klinger, A.; Merzbach, S.; Gilon, C.; Hoffman, A.; Kessler, H. Orally Active Peptides: Is There a Magic Bullet? Angew. Chem. Int. Ed. 2018, 57, 14414–14438. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Kang, W.R.; Li, W.Q.; Chen, S.M.; Gao, Y.F. Oral delivery of protein and peptide drugs: From non-specific formulation approaches to intestinal cell targeting strategies. Theranostics 2022, 12, 1419–1439. [Google Scholar] [CrossRef] [PubMed]
- Bonengel, S.; Jelkmann, M.; Abdulkarim, M.; Gumbleton, M.; Reinstadler, V.; Oberacher, H.; Prufert, F.; Bernkop-Schnurch, A. Impact of different hydrophobic ion pairs of octreotide on its oral bioavailability in pigs. J. Control. Release 2018, 273, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Martins, S.; Ferreira, D.; Souto, E.B. Oral insulin delivery by means of solid lipid nanoparticles. Int. J. Nanomed. 2007, 2, 743–749. [Google Scholar]
- Bai, Y.Y.; Zhang, Z.; Zhang, A.P.; Chen, L.; He, C.L.; Zhuang, X.L.; Chen, X.S. Novel thermo- and pH-responsive hydroxypropyl cellulose- and poly (L-glutamic acid)-based microgels for oral insulin controlled release. Carbohydr. Polym. 2012, 89, 1207–1214. [Google Scholar] [CrossRef]
- Karamanidou, T.; Karidi, K.; Bourganis, V.; Kontonikola, K.; Kammona, O.; Kiparissides, C. Effective incorporation of insulin in mucus permeating self-nanoemulsifying drug delivery systems. Eur. J. Pharm. Biopharm. 2015, 97, 223–229. [Google Scholar] [CrossRef]
- Gradauer, K.; Barthelmes, J.; Vonach, C.; Almer, G.; Mangge, H.; Teubl, B.; Roblegg, E.; Dunnhaupt, S.; Frohlich, E.; Bernkop-Schnurch, A.; et al. Liposomes coated with thiolated chitosan enhance oral peptide delivery to rats. J. Control. Release 2013, 172, 872–878. [Google Scholar] [CrossRef]
- Campardelli, R.; Santo, I.E.; Albuquerque, E.C.; de Melo, S.V.; Della Porta, G.; Reverchon, E. Efficient encapsulation of proteins in submicro liposomes using a supercritical fluid assisted continuous process. J. Supercrit. Fluids 2016, 107, 163–169. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, Y.P.; Zhang, Y.T.; Dang, B.L.; Liu, Y.; Feng, N.P. Enhanced oral bioavailability of silymarin using liposomes containing a bile salt: Preparation by supercritical fluid technology and evaluation in vitro and in vivo. Int. J. Nanomed. 2015, 10, 6633. [Google Scholar] [CrossRef]
- Parmentier, J.; Hofhaus, G.; Thomas, S.; Cuesta, L.C.; Gropp, F.; Schroder, R.; Hartmann, K.; Fricker, G. Improved Oral Bioavailability of Human Growth Hormone by a Combination of Liposomes Containing Bio-Enhancers and Tetraether Lipids and Omeprazole. J. Pharm. Sci. 2014, 103, 3985–3993. [Google Scholar] [CrossRef]
- Costa, C.; Liu, Z.; Martins, J.P.; Correia, A.; Figueiredo, P.; Rahikkala, A.; Li, W.; Seitsonen, J.; Ruokolainen, J.; Hirvonen, S.P.; et al. All-in-one microfluidic assembly of insulin-loaded pH-responsive nano-in-microparticles for oral insulin delivery. Biomater. Sci. 2020, 8, 3270–3277. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Huang, H.H. A study on chitosan-coated liposomes as a carrier of bovine serum albumin as oral protein drug. J. Dispers. Sci. Technol. 2021, 42, 1494–1503. [Google Scholar] [CrossRef]
- Werle, M.; Takeuchi, H. Chitosan-aprotinin coated liposomes for oral peptide delivery: Development, characterisation and in vivo evaluation. Int. J. Pharm. 2009, 370, 26–32. [Google Scholar] [CrossRef]
- Suzuki, K.; Kim, K.S.; Bae, Y.H. Long-term oral administration of Exendin-4 to control type 2 diabetes in a rat model. J. Control. Release 2019, 294, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Qi, J.P.; Lu, Y.; He, W.; Li, X.Y.; Wu, W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 167–176. [Google Scholar] [CrossRef] [PubMed]
- He, H.S.; Lu, Y.; Qi, J.P.; Zhao, W.L.; Dong, X.C.; Wu, W. Biomimetic thiamine- and niacin-decorated liposomes for enhanced oral delivery of insulin. Acta Pharm. Sin. B 2018, 8, 97–105. [Google Scholar] [CrossRef]
- Wang, A.H.; Yang, T.T.; Fan, W.W.; Yang, Y.W.; Zhu, Q.L.; Guo, S.Y.; Zhu, C.L.; Yuan, Y.C.; Zhang, T.; Gan, Y. Protein Corona Liposomes Achieve Efficient Oral Insulin Delivery by Overcoming Mucus and Epithelial Barriers. Adv. Healthc. Mater. 2019, 8, 1801123. [Google Scholar] [CrossRef]
- Taipaleenmaki, E.; Christensen, G.; Brodszkij, E.; Mouritzen, S.A.; Gal, N.; Madsen, S.; Hedemann, M.S.; Knudsen, T.A.; Jensen, H.M.; Christiansen, S.L.; et al. Mucopenetrating polymer - Lipid hybrid nanovesicles as subunits in alginate beads as an oral formulation. J. Control. Release 2020, 322, 470–485. [Google Scholar] [CrossRef]
- Song, K.H.; Chung, S.J.; Shim, C.K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control. Release 2005, 106, 298–308. [Google Scholar] [CrossRef]
- Yu, Y.L.; Wu, Z.H.; Wu, J.W.; Shen, X.R.; Wu, R.N.; Zhou, M.L.; Li, L.; Huang, Y. Investigation of FcRn-Mediated Transepithelial Mechanisms for Oral Nanoparticle Delivery Systems. Adv. Ther. 2021, 4, 2100145. [Google Scholar] [CrossRef]
- Foger, F.; Kopf, A.; Loretz, B.; Albrecht, K.; Bernkop-Schnurch, A. Correlation of in vitro and in vivo models for the oral absorption of peptide drugs. Amino Acids 2008, 35, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Kremsmayr, T.; Aljnabi, A.; Blanco-Canosa, J.B.; Tran, H.N.T.; Emidio, N.B.; Muttenthaler, M. On the Utility of Chemical Strategies to Improve Peptide Gut Stability. J. Med. Chem. 2022, 65, 6191–6206. [Google Scholar] [CrossRef] [PubMed]
- Buckton, L.K.; Rahimi, M.N.; McAlpine, S.R. Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development. Chem. A Eur. J. 2021, 27, 1487–1513. [Google Scholar] [CrossRef]
- Plaza-Oliver, M.; Santander-Ortega, M.J.; Lozano, M.V. Current approaches in lipid-based nanocarriers for oral drug delivery. Drug Deliv. Transl. Res. 2021, 11, 471–497. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A liposomal formulation for the oral application of the investigational hepatitis B drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef]
- Umbarkar, M.G. Niosome as a Novel Pharmaceutical Drug Delivery: A Brief Review Highlighting Formulation, Types, Composition and Application. Indian J. Pharm. Educ. Res. 2021, 55, S11–S28. [Google Scholar] [CrossRef]
- Muller, S.; Gruhle, K.; Meister, A.; Hause, G.; Drescher, S. Bolalipid-Doped Liposomes: Can Bolalipids Increase the Integrity of Liposomes Exposed to Gastrointestinal Fluids? Pharmaceutics 2019, 11, 646. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; McGuiggan, P.M. Forces between surfaces in liquids. Science 1988, 241, 795–800. [Google Scholar] [CrossRef]
- Jain, M.K.; Zakim, D. The spontaneous incorporation of proteins into preformed bilayers. Biochim. Et Biophys. Acta 1987, 906, 33–68. [Google Scholar] [CrossRef]
- Jones, M.N. The surface-properties of phospholipid liposome systems and their characterization. Adv. Colloid Interface Sci. 1995, 54, 93–128. [Google Scholar] [CrossRef]
- Colletier, J.-P.; Chaize, B.; Winterhalter, M.; Fournier, D. Protein encapsulation in liposomes: Efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.Y.; Kim, H.K.; Choo, J.; Seong, G.H.; Hien, T.B.D.; Lee, E.K. Effects of operating parameters on the efficiency of liposomal encapsulation of enzymes. Colloids Surf. B-Biointerfaces 2012, 94, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Zupancic, O.; Bernkop-Schnurch, A. Lipophilic peptide character - What oral barriers fear the most. J. Control. Release 2017, 255, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnurch, A. Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers. Adv. Drug Deliv. Rev. 2022, 182, 114097. [Google Scholar] [CrossRef]
- Forbes, N.; Hussain, M.T.; Briuglia, M.L.; Edwards, D.P.; ter Horst, J.H.; Szita, N.; Perrie, Y. Rapid and scale-independent microfluidic manufacture of liposomes entrapping protein incorporating in-line purification and at-line size monitoring. Int. J. Pharm. 2019, 556, 68–81. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. A versatile supercritical assisted process for the one-shot production of liposomes. J. Supercrit. Fluids 2019, 146, 136–143. [Google Scholar] [CrossRef]
- Jash, A.; Ubeyitogullari, A.; Rizvi, S.S.H. Liposomes for oral delivery of protein and peptide-based therapeutics: Challenges, formulation strategies, and advances. J. Mater. Chem. B 2021, 9, 4773–4792. [Google Scholar] [CrossRef]
- Holme, M.N.; Rashid, M.H.; Thomas, M.R.; Barriga, H.M.G.; Herpoldt, K.L.; Heenan, R.K.; Dreiss, C.A.; Banuelos, J.L.; Xie, H.N.; Yarovsky, I.; et al. Fate of Liposomes in the Presence of Phospholipase C and D: From Atomic to Supramolecular Lipid Arrangement. Acs Cent. Sci. 2018, 4, 1023–1030. [Google Scholar] [CrossRef]
- Lai, W.F.; Wong, W.T.; Rogach, A.L. Molecular Design of Layer-by-Layer Functionalized Liposomes for Oral Drug Delivery. Acs Appl. Mater. Interfaces 2020, 12, 43341–43351. [Google Scholar] [CrossRef]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine Mapping of Pre-S Sequence Requirements for Hepatitis B Virus Large Envelope Protein-Mediated Receptor Interaction. J. Virol. 2010, 84, 1989–2000. [Google Scholar] [CrossRef]
- Cui, M.; Wu, W.; Hovgaard, L.; Lu, Y.; Chen, D.W.; Qi, J.P. Liposomes containing cholesterol analogues of botanical origin as drug delivery systems to enhance the oral absorption of insulin. Int. J. Pharm. 2015, 489, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.M.; Lu, Y.; Hovgaard, L.; Guan, P.P.; Tan, Y.A.; Lian, R.Y.; Qi, J.P.; Wu, W. Hypoglycemic activity and oral bioavailability of insulin-loaded liposomes containing bile salts in rats: The effect of cholate type, particle size and administered dose. Eur. J. Pharm. Biopharm. 2012, 81, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Ye, A.Q.; Han, F.F.; Han, J.Z. Advances and challenges in liposome digestion: Surface interaction, biological fate, and GIT modeling. Adv. Colloid Interface Sci. 2019, 263, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Gradauer, K.; Dunnhaupt, S.; Vonach, C.; Szollosi, H.; Pali-Scholl, I.; Mangge, H.; Jensen-Jarolim, E.; Bernkop-Schnurch, A.; Prassl, R. Thiomer-coated liposomes harbor permeation enhancing and efflux pump inhibitory properties. J. Control. Release 2013, 165, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, V.J.; Barnes, T.J.; Prestidge, C.A. Silica nanoparticle coated liposomes: A new type of hybrid nanocapsule for proteins. Int. J. Pharm. 2010, 392, 285–293. [Google Scholar] [CrossRef]
- Liu, X.; Chen, D.W.; Xie, L.P.; Zhang, R.Q. Oral colon-specific drug delivery for bee venom peptide: Development of a coated calcium alginate gel beads-entrapped liposome. J. Control. Release 2003, 93, 293–300. [Google Scholar] [CrossRef]
- Huang, A.W.; Su, Z.G.; Li, S.; Sun, M.J.; Xiao, Y.Y.; Ping, Q.N.; Deng, Y.P. Oral absorption enhancement of salmon calcitonin by using both N-trimethyl chitosan chloride and oligoarginines-modified liposomes as the carriers. Drug Deliv. 2014, 21, 388–396. [Google Scholar] [CrossRef]
- Yazdi, J.R.; Tafaghodi, M.; Sadri, K.; Mashreghi, M.; Nikpoor, A.R.; Nikoofal-Sahlabadi, S.; Chamani, J.; Vakili, R.; Moosavian, S.A.; Jaafari, M.R. Folate targeted PEGylated liposomes for the oral delivery of insulin: In vitro and in vivo studies. Colloids Surf. B-Biointerfaces 2020, 194, 111203. [Google Scholar] [CrossRef]
- Niu, M.M.; Tan, Y.N.; Guan, P.P.; Hovgaard, L.; Lu, Y.; Qi, J.P.; Lian, R.Y.; Li, X.Y.; Wu, W. Enhanced oral absorption of insulin-loaded liposomes containing bile salts: A mechanistic. Int. J. Pharm. 2014, 460, 119–130. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Pandya, P.; Giram, P.; Bhole, R.P.; Chang, H.I.; Raut, S.Y. Nanocarriers based oral lymphatic drug targeting: Strategic bioavailability enhancement approaches. J. Drug Deliv. Sci. Technol. 2021, 64, 102585. [Google Scholar] [CrossRef]
- Shukla, A.; Katare, O.P.; Singh, B.; Vyas, S.P. M-cell targeted delivery of recombinant hepatitis B surface antigen using cholera toxin B subunit conjugated bilosomes. Int. J. Pharm. 2010, 385, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.N.; Vyas, S.P. Investigation of lectinized liposomes as M-cell targeted carrier-adjuvant for mucosal immunization. Colloids Surf. B-Biointerfaces 2011, 82, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Elz, A.S.; Trevaskis, N.L.; Porter, C.J.H.; Bowen, J.M.; Prestidge, C.A. Smart design approaches for orally administered lipophilic prodrugs to promote lymphatic transport. J. Control. Release 2022, 341, 676–701. [Google Scholar] [CrossRef]
- Zhang, Z.C.; Lu, Y.; Qi, J.P.; Wu, W. An update on oral drug delivery via intestinal lymphatic transport. Acta Pharm. Sin. B 2021, 11, 2449–2468. [Google Scholar] [CrossRef]
- Caliph, S.M.; Charman, W.N.; Porter, C.J.H. Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J. Pharm. Sci. 2000, 89, 1073–1084. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Verma, A.; Saharan, V.A. Lipid Drug Carriers for Cancer Therapeutics: An Insight into Lymphatic Targeting, P-gp, CYP3A4 Modulation and Bioavailability Enhancement. Adv. Pharm. Bull. 2020, 10, 524–541. [Google Scholar] [CrossRef]
- Romana, B.; Hassan, M.M.; Sonvico, F.; Pereira, G.G.; Mason, A.F.; Thordarson, P.; Bremmell, K.E.; Barnes, T.J.; Prestidge, C.A. A liposome-micelle-hybrid (LMH) oral delivery system for poorly water- soluble drugs: Enhancing solubilisation and intestinal transport. Eur. J. Pharm. Biopharm. 2020, 154, 338–347. [Google Scholar] [CrossRef]
- Rege, B.D.; Kao, J.P.Y.; Polli, J.E. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2002, 16, 237–246. [Google Scholar] [CrossRef]
- Rathod, S.; Desai, H.; Patil, R.; Sarolia, J. Non-ionic Surfactants as a P-Glycoprotein(P-gp) Efflux Inhibitor for Optimal Drug Delivery-A Concise Outlook. Aaps Pharmscitech 2022, 23, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Dong, K.; Wang, Z.G.; Miao, R.M.; Lu, W.J.; Wu, X.Y. Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine. Pharmaceutics 2021, 13, 1261. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, L.; Lynch, C.; Haidar, S.; Polli, J.; Wang, H.B. Effects of Commonly Used Excipients on the Expression of CYP3A4 in Colon and Liver Cells. Pharm. Res. 2010, 27, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Yoshioka, Y.; Morishita, Y.; Yoshida, T.; Uji, M.; Nagano, K.; Mukai, Y.; Kamada, H.; Tsunoda, S.; Higashisaka, K.; et al. Size and surface modification of amorphous silica particles determine their effects on the activity of human CYP3A4 in vitro. Nanoscale Res. Lett. 2014, 9, 651. [Google Scholar] [CrossRef] [Green Version]


| Agent | Phospholipid | Formulation | Property | Modification | EE (%) ± SD | Zeta (mv) ± SD | MD (nm) ± SD | Gain | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Insulin | DMPG (PSC) | Stirring ultrasonic | Anionic phospholipid | - | 70.9 ± 2.0 | 6.2 ± 0.5 | 29.8 ± 2.3 | Degradation of insulin was reduced | [6] |
| Salmon calcitonin | DPPC DPPE-MCC | TFH + FTC | Amphoteric phospholipid | CS–TGA and CS–TGA–MNA modification | 69 ± 12.0 | 27.9 ± 1.1 | 604.8 ± 29.6 | Reduces blood calcium by 35% | [7] |
| BSA | SPC | Supercritical assisted process | Amphoteric phospholipid | - | 95 ± 3.0 | 25 ± 5.0 | 250 ± 58.0 | Up to 90% encapsulation rate | [8] |
| Silymarin (SM) | SPC | Supercritical assisted process | Amphoteric phospholipid | SGC modification | 91.4 | −62.3 | 160.50 | SM-Lip-SEDS Cmax, AUC increases | [9] |
| HGH | EPC (GCTE) | DAC | Amphoteric phospholipid | GCTE | 31.2 ± 0.5 | 41.0 ± 1.2 | 229.7 ± 12.8 | 3.4% oral bioavailability | [10] |
| RhIns | PC, DSPE-PEG, Chol | MHF | Amphoteric phospholipid | Chitosan coated with double emulsion carrier | 91 ± 4 | 23 ± 1.0 | 363 ± 54 | The stability and permeability of rhIns increase | [11] |
| BSA | SPC | RPE | Cationic phospholipid | chitosan coated | 44.2 ± 0.3 | 33.1 ± 0.6 | 173.7 ± 5.6 | More stable | [12] |
| Calcitonin | DSPC DCP Chol | TFH | Amphoteric phospholipid | Protease inhibitor modified chitosan | >75 | 39.9 ± 1.6 | 4460.0 | Increases the AAC | [13] |
| Exendin-4 | DOPC DOTAP | RPE | Anionic phospholipid | GCA modified chitosan coating | 74.2 ± 2.5 | −31 ± 0.2 | 229 ± 4.0 | 19% oral bioavailability | [14] |
| Insulin | SPC | RPE | Amphoteric phospholipid | Biotin-DSPE promotes absorption | - | 38.5 ± 3.5 | 150.0 | 12% oral bioavailability | [15] |
| Insulin | SPC | RPE | Amphoteric phospholipid | Thiamine and nicotinic acid Decoration | 30.6 ± 2.4 | - | 125.6 ± 2.9 | 2.5% oral bioavailability | [16] |
| Insulin | EPC: Chol DOTAP | TFH | Cationic phospholipid | Protein adsorption | 28.7 ± 5.1 | −23.1 ± 0.5 | 164.7 ± 5.3 | 12% oral bioavailability | [17] |
| Cy5-amine | POPC POPS, | TFH | Anionic phospholipid | Alginate microcapsule | - | −12.0± 1.0 | 124 ± 13.0 | Longer residence time in the intestine | [18] |
| Salmon calcitonin | PC | TFH | Amphoteric phospholipid | Bile salt modification | 54.9 ± 4.1 | - | 741 ± 76.9 | 7.1-times higher bioavailability of sCT | [19] |
| Insulin | SPC Chol | TFH | Amphoteric phospholipid | FcBP receptor modification | 70.9 ± 2.0 | 6.2 ± 0.5 | 29.8 ± 2.3 | Blood sugar decreased by 47.87% | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Wen, Z.; Zhang, W.; Wu, W. Recent Advances in Oral Peptide or Protein-Based Drug Liposomes. Pharmaceuticals 2022, 15, 1072. https://doi.org/10.3390/ph15091072
Cui J, Wen Z, Zhang W, Wu W. Recent Advances in Oral Peptide or Protein-Based Drug Liposomes. Pharmaceuticals. 2022; 15(9):1072. https://doi.org/10.3390/ph15091072
Chicago/Turabian StyleCui, Jian, Zhiwei Wen, Wei Zhang, and Wei Wu. 2022. "Recent Advances in Oral Peptide or Protein-Based Drug Liposomes" Pharmaceuticals 15, no. 9: 1072. https://doi.org/10.3390/ph15091072
APA StyleCui, J., Wen, Z., Zhang, W., & Wu, W. (2022). Recent Advances in Oral Peptide or Protein-Based Drug Liposomes. Pharmaceuticals, 15(9), 1072. https://doi.org/10.3390/ph15091072