
Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]pyridine Antileishmanial Pharmacophore


 ,
,  , , , and
, , , and 
Abstract
:
1. Introduction
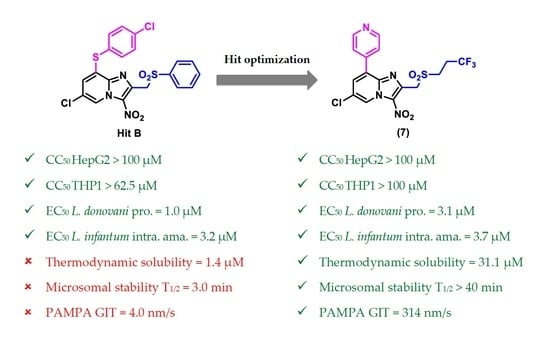
2. Results and Discussion
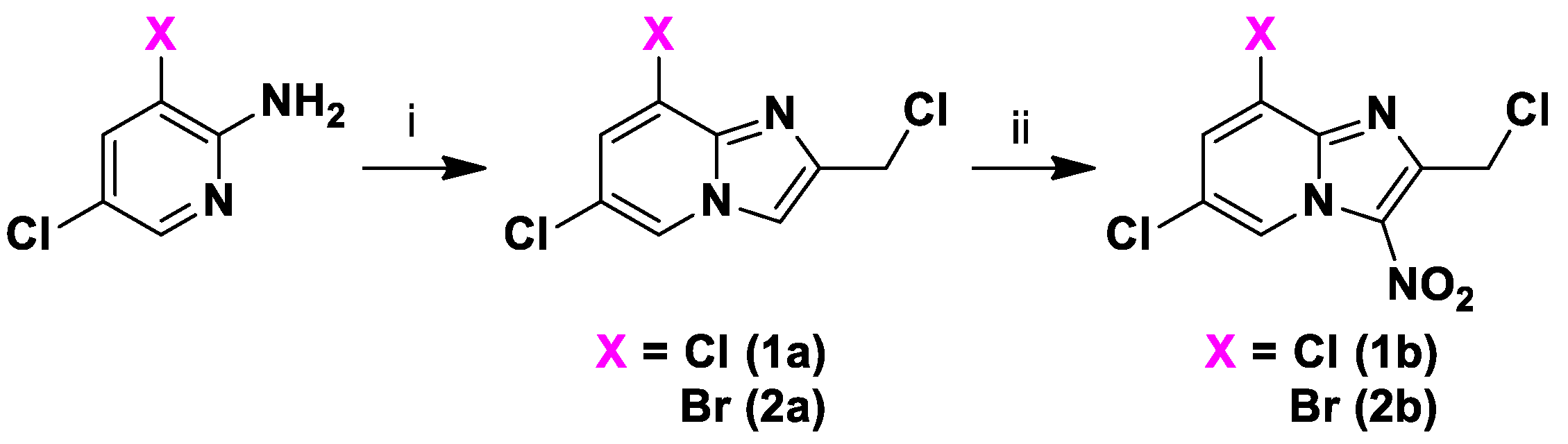
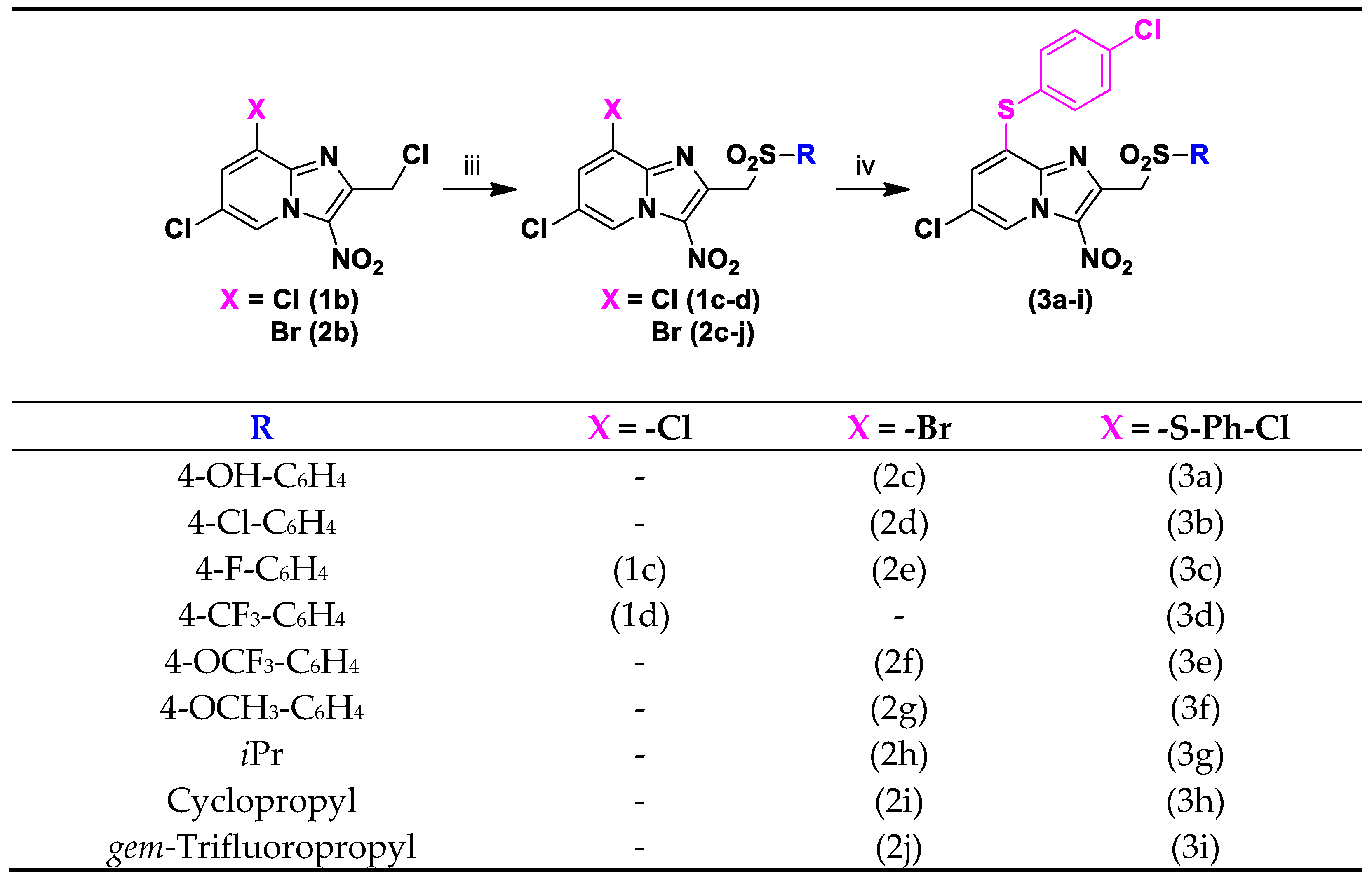
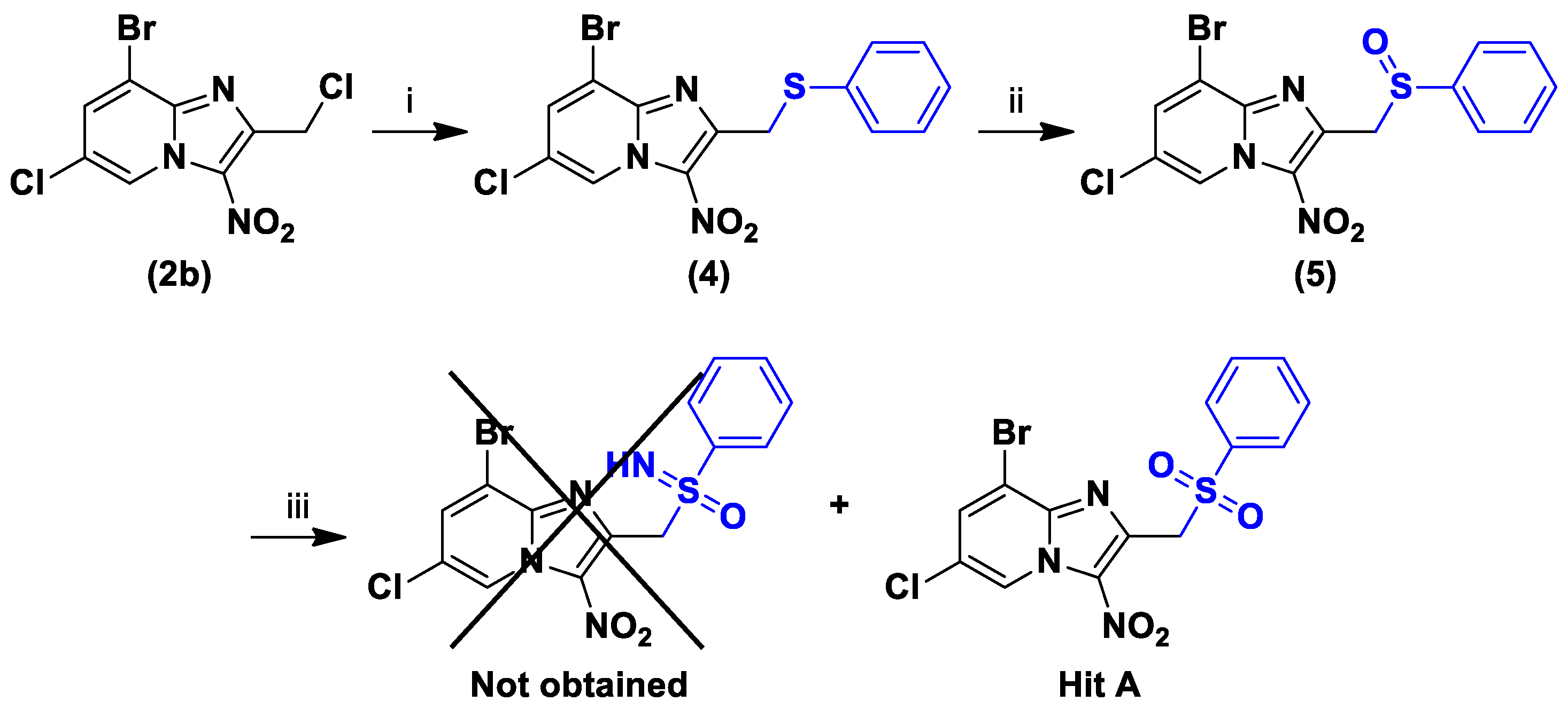
2.1. Synthesis
2.2. Biological Results
2.2.1. Antileishmanial Structure-Activity Relationships Study
2.2.2. Anti-Trypanosoma brucei Structure-Activity Relationship Study
2.2.3. In Vitro Physicochemical and Pharmacokinetic Study of Selected Key Compounds
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for the Preparation of Sodium Sulfinate
3.1.3. General Procedure for the Preparation of 8-Chloro-2-Sulfonylmethyl[1,2-a]pyridine derivatives (1c-1d)
6,8-Dichloro-2-[(4-fluorophenylsulfonyl)methyl]-3-nitroimidazo[1,2-a]pyridine (1c)
6,8-Dichloro-3-Nitro-2-[(4-Trifluoromethylphenylsulfonyl)methyl]imidazo[1,2-a]pyridine (1d)
3.1.4. General Procedure for the Preparation of 8-Bromo-2-Sulfonylmethyl[1,2-a]pyridine derivatives (2c-2j)
4-{[(8-Bromo-6-Chloro-3-Nitroimidazo[1,2-a]pyridin-2-yl)methyl]sulfonyl}phenol (2c)
8-Bromo-6-Chloro-2-[(4-Chlorophenylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (2d)
8-Bromo-6-Chloro-2-[(4-Fluorophenylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (2e)
8-Bromo-6-Chloro-3-Nitro-2-[(4-Trifluoromethoxyphenylsulfonyl)methyl]imidazo[1,2-a] pyridine (2f)
8-Bromo-6-Chloro-2-[(4-Methoxyphenylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (2g)
8-Bromo-6-Chloro-2-[(isopropylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (2h)
8-Bromo-6-Chloro-2-[(cyclopropylsulfonyl]methyl)-3-Nitroimidazo[1,2-a]pyridine (2i)
8-Bromo-6-Chloro-3-Nitro-2-[(3,3,3-Trifluoropropylsulfonyl)methyl]imidazo[1,2-a]pyridine (2j)
3.1.5. General Procedure for the Preparation of 8-Phenylthioimidazo[1,2-a]pyridine Derivatives (3a-3i)
4-{[(6-Chloro-8-[(4-Chlorophenyl)thio]-3-Nitroimidazo[1,2-a]pyridin-2-yl)methyl]sulfonyl}phenol (3a)
6-Chloro-2-[(4-Chlorophenylsulfonyl)methyl]-8-[(4-Chlorophenyl)thio]-3-Nitroimidazo[1,2-a]pyridine (3b)
6-Chloro-8-[(4-Chlorophenyl)thio]-2-[(4-Fluorophenylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (3c)
6-Chloro-8-[(4-Chlorophenyl)thio]-3-Nitro-2-[(4-Trifluoromethylphenylsulfonyl)methyl]imidazo[1,2-a]pyridine (3d)
6-Chloro-8-[(4-Chlorophenyl)thio]-3-Nitro-2-[(4-Trifluoromethoxyphenylsulfonyl)methyl]imidazo[1,2-a]pyridine (3e)
6-Chloro-8-[(4-Chlorophenyl)thio]-2-[(4-Methoxyphenylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (3f)
6-Chloro-8-[(4-Chlorophenyl)thio]-2-[(isopropylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (3g)
6-Chloro-8-[(4-Chlorophenyl)thio]-2-[(cyclopropylsulfonyl)methyl]-3-Nitroimidazo[1,2-a]pyridine (3h)
6-Chloro-8-[(4-Chlorophenyl)thio]-3-Nitro-2-[(3,3,3-Trifluoropropylsulfonyl)methyl]imidazo[1,2-a]pyridine (3i)
3.1.6. 8-Bromo-6-Chloro-3-Nitro-2-[(phenylsulfinyl)methyl]imidazo[1,2-a]pyridine (5)
3.1.7. 6-Chloro-8-[(4-Chlorophenyl)sulfinyl]-3-Nitro-2-[(3,3,3-Trifluoropropylsulfonyl)methyl] imidazo[1,2-a]pyridine (6a)
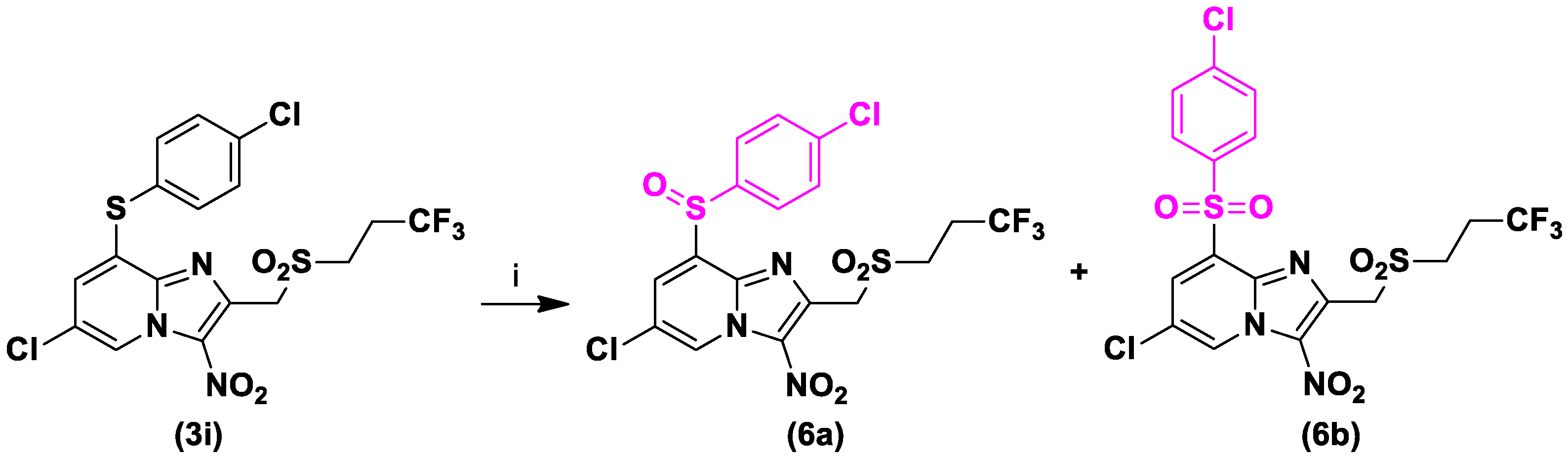
3.1.8. 6-Chloro-8-[(4-Chlorophenyl)sulfonyl]-3-Nitro-2-[(3,3,3-Trifluoropropylsulfonyl)methyl] imidazo[1,2-a]pyridine (6b)
3.1.9. 6-Chloro-3-Nitro-8-(Pyridin-4-yl)-2-[(3,3,3-Trifluoropropylsulfonyl)methyl]imidazo[1,2-a]pyridine (7)
3.2. Electrochemistry
3.3. Biology
3.3.1. Antileishmanial Activity against L. donovani Promastigotes
3.3.2. Antileishmanial Activity against L. infantum Promastigotes
3.3.3. Antileishmanial Activity on L. infantum Axenic Amastigotes
- Luciferase activity and determination of EC50 were performed as above.
3.3.4. Antileishmanial Activity on L. infantum Intracellular Amastigotes
- Luciferase activity and determination of EC50 were performed as above.
3.3.5. Antitrypanosomal Evaluation on T. b. brucei BSF Trypomastigotes
3.3.6. Cytotoxicity Evaluation on HepG2 Cell Line
3.3.7. Cytotoxicity on THP-1 Cell Line
3.3.8. Plasma Protein Binding
3.3.9. Microsomal Stability
3.3.10. Parallel Artificial Membrane Permeability Assay (PAMPA)
3.3.11. Thermodynamic Solubility at pH 7.4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Working to Overcome the Global Impact of Neglected Tropical Diseases: First WHO Report on Neglected Tropical Diseases. Available online: https://apps.who.int/iris/handle/10665/44440 (accessed on 21 March 2022).
- World Health Organization (WHO). Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 21 March 2022).
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Research Priorities for Chagas Disease, Human African Trypanosomiasis and Leishmaniasis. Available online: https://apps.who.int/iris/handle/10665/77472 (accessed on 21 March 2022).
- Musa, A.; Khalil, E.; Hailu, A.; Olobo, J.; Balasegaram, M.; Omollo, R.; Edwards, T.; Rashid, J.; Mbui, J.; Musa, B.; et al. Sodium Stibogluconate (SSG) & Paromomycin Combination Compared to SSG for Visceral Leishmaniasis in East Africa: A Randomised Controlled Trial. PLoS Negl. Trop. Dis. 2012, 6, e1674. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. An Update on Pharmacotherapy for Leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; More, D.K.; Singh, M.K.; Singh, V.P.; Sharma, S.; Makharia, A.; Kumar, P.C.K.; Murray, H.W. Failure of Pentavalent Antimony in Visceral Leishmaniasis in India: Report from the Center of the Indian Epidemic. Clin. Infect. Dis. 2000, 31, 1104–1107. [Google Scholar] [CrossRef]
- Deray, G. Amphotericin B Nephrotoxicity. J. Antimicrob. Chemother. 2002, 49, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.R.H.; Bicanic, T.; Salim, R.; Hope, W. Liposomal Amphotericin B (AmBisome®): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef]
- Dorlo, T.P.C.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A Review of Its Pharmacology and Therapeutic Efficacy in the Treatment of Leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef] [PubMed]
- Drug for Neglected Diseases Initiative (DNDi), Research & Development Portfolio. Available online: https://dndi.org/research-development/portfolio/ (accessed on 21 March 2022).
- Ferreira, H.D. Clinico-therapeutic trial with benzonidazole in Chagas’ disease. Rev. Inst. Med. Trop. São Paulo 1976, 8, 357–364. [Google Scholar]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Fairlamb, A.H. Fexinidazole for the treatment of human African trypanosomiasis. Drugs Today 2019, 55, 705–712. [Google Scholar] [CrossRef]
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 13088–13095. [Google Scholar] [CrossRef]
- Hall, B.S.; Wilkinson, S.R. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 2012, 56, 115–123. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Castera-Ducros, C.; Paloque, L.; Verhaeghe, P.; Casanova, M.; Cantelli, C.; Hutter, S.; Tanguy, F.; Laget, M.; Remusat, V.; Cohen, A.; et al. Targeting the human parasite Leishmania donovani: Discovery of a new promising anti-infectious pharmacophore in 3-nitroimidazo[1,2-a]pyridine series. Bioorg. Med. Chem. 2013, 21, 7155–7164. [Google Scholar] [CrossRef]
- Fersing, C.; Basmaciyan, L.; Boudot, C.; Pedron, J.; Hutter, S.; Cohen, A.; Castera-Ducros, C.; Primas, N.; Laget, M.; Casanova, M.; et al. Nongenotoxic 3-nitroimidazo[1,2-a]pyridines are NTR1 substrates that display potent in vitro antileishmanial activity. ACS Med. Chem. Lett. 2019, 10, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Mäder, P.; Kattner, L. Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem. 2020, 63, 14243–14275. [Google Scholar] [CrossRef]
- Lohier, J.F.; Glachet, T.; Marzag, H.; Gaumont, A.C.; Reboul, V. Mechanistic investigation of the NH-sulfoximination of sulfide. Evidence for λ6-sulfanenitrile intermediates. Chem. Commun. 2017, 53, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Miao, K.; Yun, H.; Shen, H.C.; Zhao, W.; Liang, C. Eaton’s reagent-mediated metal-free and efficient synthesis of NH-sulfoximines. Tetrahedron Lett. 2017, 58, 333–337. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Pedron, J.; Hutter, S.; Primas, N.; Castera-Ducros, C.; Bourgeade-Delmas, S.; Sournia-Saquet, A.; Moreau, A.; Cohen, A.; et al. 8-Aryl-6-chloro-3-nitro-2-(phenylsulfonylmethyl)imidazo[1,2-a]pyridines as potent antitrypanosomatid molecules bioactivated by type 1 nitroreductases. Eur. J. Med. Chem. 2018, 157, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Boudot, C.; Paoli-Lombardo, R.; Primas, N.; Pinault, E.; Hutter, S.; Castera-Ducros, C.; Kabri, Y.; Pedron, J.; Bourgeade-Delmas, S.; et al. Antikinetoplastid SAR Study in 3-Nitroimidazopyridine Series: Identification of a Novel Non-Genotoxic and Potent Anti-T. b. Brucei Hit-Compound with Improved Pharmacokinetic Properties. Eur. J. Med. Chem. 2020, 206, 112668. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial Pharmacology and Therapeutics of Atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug. Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue® Assay to Determine Drug Sensitivity of African Trypanosomes (T.b. Rhodesiense and T.b. Gambiense) in Vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Baltz, T.; Baltz, D.; Giroud, C.; Crockett, J. Cultivation in a Semi-Defined Medium of Animal Infective Forms of Trypanosoma Brucei, T. Equiperdum, T. Evansi, T. Rhodesiense and T. Gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar] [CrossRef]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Fréon, A.; et al. Design of Donecopride, a Dual Serotonin Subtype 4 Receptor Agonist/Acetylcholinesterase Inhibitor with Potential Interest for Alzheimer’s Disease Treatment. Proc. Natl. Acad. Sci. USA 2014, 111, e3825–e3830. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | |
|---|---|---|
| Hit A | Hit B | |
| EC50 L. donovani promastigotes (µM) | 5.3 ± 1.1 | 1.0 ± 0.3 |
| EC50 L. infantum promastigotes (µM) | 8.7 ± 0.7 | 0.6 ± 0.1 |
| EC50 L. infantum axenic amastigotes (µM) | 16.4 ± 1.3 | 1.7 ± 0.3 |
| EC50 L. infantum amastigotes intramacro. (µM) | 2.1 ± 0.1 | 3.2 ± 0.1 |
| CC50 HepG2 (µM) | >15.6 | >100 |
| CC50 THP1 (µM) | >100 | >62.5 |
| Microsomal stability: half-life (min) | 11 | 3 |
| Thermodynamic solubility at pH 7.4 (µM) | 6.9 | 1.4 |
 | Cell Viability | Activity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R2 | R8 | CC50 HepG2 (µM) | CC50 THP1 (µM) | EC50 L. donovani Pro. (µM) | EC50 L. infantum Pro. (µM) | EC50 L. infantum Axenic Ama. (µM) | EC50 L. infantum Intra. Ama. (µM) | SI HepG2/THP1 L. infantum Intra. Ama. | E0 (V) | ClogP |
| 2c | -CH2-SO2-Ph-4-OH | -Br | >3.9 a | >100 c | >3.1 a | >100 c | >5.0 c | - | - | - | 2.4 |
| 2d | -CH2-SO2-Ph-4-Cl | -Br | >12.5 a | >31.2 a | 3.0 ± 0.4 | 3.5 ± 0.4 | 2.1 ± 0.3 | - | - | −0.62 | 3.28 |
| 1c | -CH2-SO2-Ph-4-F | -Cl | >7.8 a | 22.5 ± 1.1 | 5.8 ± 1.9 | >100 c | >1.6 a | - | - | −0.6 | 2.98 |
| 2e | -CH2-SO2-Ph-4-F | -Br | >7.8 a | 33.3 ± 1.4 | 2.8 ± 0.5 | >100 c | 3.3 ± 0.1 | - | - | −0.64 | 3.06 |
| 1d | -CH2-SO2-Ph-4-CF3 | -Cl | >15.6 a | 23.2 ± 1.9 | 4.1 ± 0.4 | >100 c | 2.2 ± 0.1 | - | - | −0.6 | 3.72 |
| 2f | -CH2-SO2-Ph-4-OCF3 | -Br | >25 a | >100 c | 4.4 ± 0.1 | 6.7 ± 0.8 | 2.7 ± 0.4 | 4.2 ± 0.3 | >6.0/>23.8 | −0.62 | 3.59 |
| 2g | -CH2-SO2-Ph-4-OCH3 | -Br | 3.9 ± 1.3 | >100 c | 2.8 ± 0.9 | 9.3 ± 1.2 | 2.3 ± 0.3 | 2.2 ± 0.1 | 1.8/>45.5 | −0.64 | 2.75 |
| 2h | -CH2-SO2-iPr | -Br | >62.5 | >100 c | >25 c | 61.2 ± 3.4 | >50 c | - | - | −0.65 | 2.15 |
| 2i | -CH2-SO2-Cyclopropyl | -Br | >6.2 a | >50 | >25 c | >50 c | >50 c | - | - | −0.63 | 1.97 |
| 2j | -CH2-SO2-gem-Trifluoropropyl | -Br | 42.9 ± 12.3 | 26.0 ± 1.2 | 14.4 ± 1.6 | 1.9 ± 0.4 | 20.1 ± 5.6 | - | - | −0.6 | 2.91 |
| 3a | -CH2-SO2-Ph-4-OH | 4-Cl-Ph-S- | 22.3 ± 2.5 | 28.2 ± 0.1 | 0.8 ± 0.2 | 5.6 ± 0.9 | 1.4 ± 0.3 | 2.7 ± 0.5 | 8.3/10.4 | - | 3.96 |
| 3b | -CH2-SO2-Ph-4-Cl | 4-Cl-Ph-S- | 8.9 ± 1.3 | >15.6 a | 1.4 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 0.7 ± 0.1 | 12.7/>22.3 | −0.68 | 4.83 |
| 3c | -CH2-SO2-Ph-4-F | 4-Cl-Ph-S- | >15.6 a | >100 c | 0.9 ± 0.1 | 1.5 ± 0.1 | 0.2 ± 0.1 | 1.6 ± 0.1 | >9.8/>62.5 | −0.61 | 4.67 |
| 3d | -CH2-SO2-Ph-4-CF3 | 4-Cl-Ph-S- | >3.9 a | >50 | 1.8 ± 0.2 | 3.3 ± 0.4 | 0.8 ± 0.1 | 4.4 ± 0.2 | >0.9/>11.4 | −0.56 | 5.28 |
| 3e | -CH2-SO2-Ph-4-OCF3 | 4-Cl-Ph-S- | >12.5 a | >100 c | 2.6 ± 0.2 | 3.1 ± 0.2 | 0.8 ± 0.2 | 1.6 ± 0.1 | >7.8/>62.5 | −0.66 | 5.21 |
| 3f | -CH2-SO2-Ph-4-OCH3 | 4-Cl-Ph-S- | >3.9 a | >50 | 1.2 ± 0.1 | 1.2 ± 0.1 | 0.5 ± 0.1 | 0.9 ± 0.1 | >4.3/>55.6 | −0.66 | 4.38 |
| 3g | -CH2-SO2-iPr | 4-Cl-Ph-S- | 22.2 ± 5.5 | >31.2 a | 2.2 ± 0.3 | 2.4 ± 0.6 | 3.2 ± 0.1 | - | - | −0.63 | 3.73 |
| 3h | -CH2-SO2-Cyclopropyl | 4-Cl-Ph-S- | 22.3 ± 2.5 | >62.5 | 2.2 ± 0.3 | 2.2 ± 0.3 | 2.4 ± 0.1 | 3.7 ± 0.2 | 6.0/>16.9 | −0.65 | 3.63 |
| 3i | -CH2-SO2-gem-Trifluoropropyl | 4-Cl-Ph-S- | >6.2 a | >50 | 1.4 ± 0.2 | 1.4 ± 0.1 | 0.8 ± 0.3 | 1.2 ± 0.1 | >5.2/>41.7 | −0.64 | 4.4 |
| 6a | -CH2-SO2-gem-Trifluoropropyl | 4-Cl-Ph-SO- | >5 | >100 | 1.9 ± 0.3 | 7.4 ± 0.4 | 5.0 ± 0.1 | 3.3 ± 0.2 | >1.5/>30.3 | - | 3.79 |
| 6b | -CH2-SO2-gem-Trifluoropropyl | 4-Cl-Ph-SO2- | >10 | >100 | 1.3 ± 0.2 | 13.7 ± 0.9 | 2.3 ± 0.3 | 8.3 ± 0.8 | >1.2/>12.0 | - | 3.63 |
| 7 | -CH2-SO2-gem-Trifluoropropyl | 4-pyridinyl | >100 | >100 | 3.1 ± 0.6 | 14.0 ± 0.4 | 14.0 ± 0.9 | 3.7 ± 0.4 | >27.0/>27.0 | −0.61 | 2.93 |
| Ref. [1] | Hit A | >15.6 a | >100 c | 5.3 ± 1.1 | 8.7 ± 0.7 | 16.4 ± 1.3 | 2.1 ± 0.1 | >7.4/>47.6 | −0.65 | 2.81 | |
| Ref. [2] | Hit B | >100 | >62.5 | 1.0 ± 0.3 | 0.6 ± 0.1 | 1.7 ± 0.3 | 3.2 ± 0.1 | >31.3/>19.5 | −0.63 | 4.37 | |
| Ref. [3] | Doxorubicin b | 0.20 ± 0.02 | 0.8 ± 0.1 | - | - | - | - | - | - | 0.52 | |
| Ref. [4] | Amphotericin B d | 8.8 ± 0.3 | 12.1 ± 1.3 | 0.07 ± 0.01 | 0.04 ± 0.01 | 0.08 ± 0.02 | 0.06 ± 0.01 | 146.7/201.7 | - | −0.26 | |
| Ref. [5] | Miltefosine d | 85.0 ± 8.8 | 28.1 ± 3.4 | 3.1 ± 0.2 | 18.2 ± 0.5 | 0.30 ± 0.02 | 0.40 ± 0.14 | 212.5/70.3 | - | 3.83 | |
| Ref. [6] | Fexinidazole d | >100 | >100 | 1.2 ± 0.2 | 1.0 ± 0.1 | 7.8 ± 2.3 | 15.9 ± 3.2 | >6.3/>6.3 | −0.83 | 1.61 | |
| Ref. [7] | Fexinidazole sulfone d | - | >100 | 1.2 ± 0.2 | 1.1 ± 0.1 | 4.3 ± 0.1 | 17.8 ± 1.4 | -/>5.6 | - | 0.66 | |
| CC50 HepG2 (µM) | EC50 T. b. brucei BSF (µM) | SI T. b. brucei c | |
|---|---|---|---|
| 2c | >3.9 a | 0.40 ± 0.03 | >9.8 |
| 3a | 22.3 ± 2.5 | 0.78 ± 0.05 | 28.6 |
| 3c | >15.6 a | 0.57 ± 0.02 | >27.4 |
| 3d | >3.9 a | 0.89 ± 0.07 | >4.4 |
| Hit A | >15.6 a | 1.4 ± 0.4 | >11.1 |
| Hit B | >100 | 0.95 ± 0.05 | >105.3 |
| Suraminb | >100 | 0.08 ± 0.04 | >1250 |
| Fexinidazoleb | >200 | 1.4 ± 0.5 | >142.9 |
| Thermodynamic Solubility at pH 7.4 in PBS (µM) | Mouse Microsomal Stability T1/2 (min) | PAMPA GIT at pH 7.4 Pe (nm/s) | Albumin a Binding (%) | |
|---|---|---|---|---|
| 2f | 8.6 ± 2.3 | - | - | - |
| 2g | 5.7 ± 1.3 | - | - | - |
| 3b | <2 | 4.5 | - | - |
| 3c | <2 | 5.7 | - | - |
| 3d | 2.5 ± 0.7 | - | - | - |
| 3e | 64.7 ± 7.3 | - | - | - |
| 3f | <2 | - | - | - |
| 3h | 3.0 ± 0.4 | - | - | - |
| 3i | 12.4 ± 3.0 | 9.1 | 6.3 ± 0.8 | 99.76 |
| 6b | - | 7.9 | - | - |
| 7 | 31.1 ± 3.7 | >40 | 314.0 ± 87.2 | 88.65 |
| Hit A | 6.9 ± 1.5 | 11.0 | 173.2 ± 38.8 | 96.50 |
| Hit B | 1.4 ± 0.5 | 3.0 | 4.0 ± 0.6 | 99.95 |
| Key Hit Selection Criteria | Compound 7 |
|---|---|
| Demonstrate an EC50 < 10 µM against the intracellular amastigote form of L. donovani or L. infantum | EC50 L. infantum intra. ama. = 3.7 µM |
| Chemical structure should be confirmed by identification, re-synthesis or re-purification | ✓ |
| Primary screening data should be validated on a selection of hit compounds (>90% pure) | Purity > 90% for Hit A, Hit B and all synthesized compounds (checked by LC/MS analyses) |
| Acceptable in vitro response | ✓ |
| Preliminary knowledge of the structure-activity relationship is desirable | ✓ |
| No highly reactive or unstable moieties in the pharmacophore, be amenable to structural variation by chemical or biochemical synthesis, pass basic drug-like filters such as pan-assay interference filters (PAINS), to eliminate promiscuous hits that lack target specificity, and the conformity to the “rule of five” is preferred | PAINS = 0 a Rule of five = 0 violation a |
| Demonstrate a greater than 10-fold selectivity window for cytotoxicity using a mammalian cell line | SI HepG2 > 27 SI THP1 > 27 |
| Demonstrate adequate selectivity in a biochemical counter-assay | Only relevant for target-based screens, not applicable in this case |
| No serious intellectual property conflicts should exist | ✓ |
| No major synthesis or formulation issues should be anticipated (compounds should ideally be synthesized in ≤5 steps with an acceptable yield and acceptable solubility) | Synthesized in 4 steps |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoli-Lombardo, R.; Primas, N.; Bourgeade-Delmas, S.; Hutter, S.; Sournia-Saquet, A.; Boudot, C.; Brenot, E.; Castera-Ducros, C.; Corvaisier, S.; Since, M.; et al. Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]pyridine Antileishmanial Pharmacophore. Pharmaceuticals 2022, 15, 998. https://doi.org/10.3390/ph15080998
Paoli-Lombardo R, Primas N, Bourgeade-Delmas S, Hutter S, Sournia-Saquet A, Boudot C, Brenot E, Castera-Ducros C, Corvaisier S, Since M, et al. Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]pyridine Antileishmanial Pharmacophore. Pharmaceuticals. 2022; 15(8):998. https://doi.org/10.3390/ph15080998
Chicago/Turabian StylePaoli-Lombardo, Romain, Nicolas Primas, Sandra Bourgeade-Delmas, Sébastien Hutter, Alix Sournia-Saquet, Clotilde Boudot, Emilie Brenot, Caroline Castera-Ducros, Sophie Corvaisier, Marc Since, and et al. 2022. "Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]pyridine Antileishmanial Pharmacophore" Pharmaceuticals 15, no. 8: 998. https://doi.org/10.3390/ph15080998
APA StylePaoli-Lombardo, R., Primas, N., Bourgeade-Delmas, S., Hutter, S., Sournia-Saquet, A., Boudot, C., Brenot, E., Castera-Ducros, C., Corvaisier, S., Since, M., Malzert-Fréon, A., Courtioux, B., Valentin, A., Verhaeghe, P., Azas, N., Rathelot, P., & Vanelle, P. (2022). Improving Aqueous Solubility and In Vitro Pharmacokinetic Properties of the 3-Nitroimidazo[1,2-a]pyridine Antileishmanial Pharmacophore. Pharmaceuticals, 15(8), 998. https://doi.org/10.3390/ph15080998