4.1. Chemistry
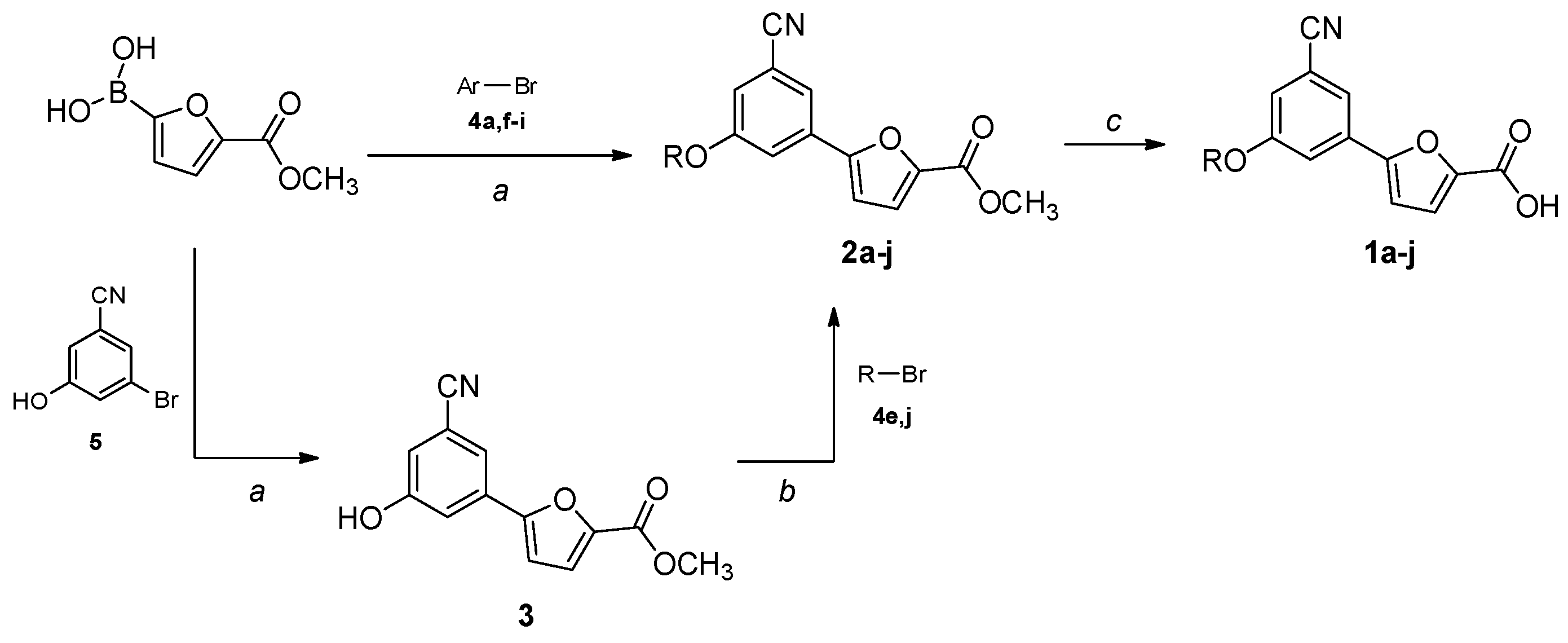
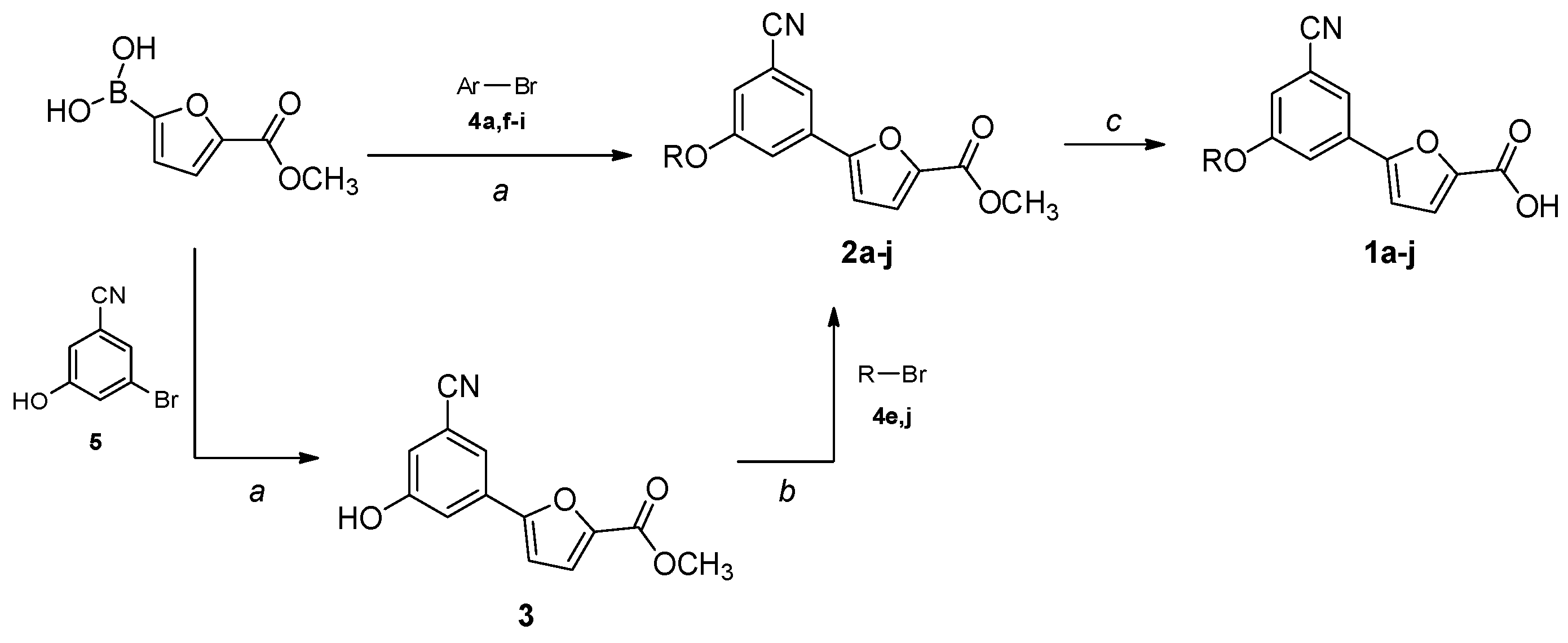
All starting reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, St. Louis, MI, USA; FluoroChem, Hadfield, UK) and used as received. Anhydrous solvents were utilized without further drying. The course of the reactions was followed by thin-layer chromatography (TLC) using aluminum-backed Silica Gel 60 plates (0.2 mm; Merck, Darmstadt, Germany). Microwave-assisted reactions were carried out with a Biotage® Initiator Classic (Biotage, Uppsala, Sweden). Crude products were purified by flash column chromatography on silica gel 60 (40–63 μm; Merck, Darmstadt, Germany) using the indicated solvent system. Melting points were determined in open capillary tubes with a Stuart SMP30 Melting Point Apparatus (Cole-Parmer Stuart, Stone, UK). All tested compounds were characterized by means of mono- and bi-dimensional NMR techniques, FT-IR, and ESI-MS. 1H and 13C NMR spectra were acquired at ambient temperature with a Varian Oxford 300 MHz instrument (Varian, Palo Alto, CA, USA) or a Bruker Avance 300 MHz instrument (Bruker, Billerica, MA, USA), operating at 300 MHz for 1H and 75 MHz for 13C. 2D NMR experiments were performed on a Bruker Avance Neo 400 MHz spectrometer. Chemical shifts are expressed in ppm (δ), and J-couplings are given in Hertz. The full decoupling mode was employed for 13C spectra when the relaxation times of the carbons did not allow for a sufficient resolution using the APT sequence. When necessary, the 2D-COSY and 2D-NOESY sequences were employed to unambiguously assign the hydrogen signals, while HSQC-DEPT experiments were performed to aid the assignment of 13C NMR signals. ATR-FT-IR spectra were acquired with a Perkin Elmer Spectrum One FT-IR (Perkin Elmer, Waltham, MA, USA), equipped with a Perkin Elmer Universal ATR sampling accessory consisting of a diamond crystal. Analyses were performed in a spectral region between 4000 and 650 cm−1 and analyzed by transmittance technique with 28 scansions and 4 cm−1 resolutions. MS analyses were carried out with a Thermo Fisher (Waltham, MA, USA) LCQ Fleet system, equipped with an ESI electrospray ionization source and an Ion Trap mass analyzer; ionization: ESI positive or ESI negative; capillary temperature: 250 °C; source voltage: 5.50 kV; source current: 4.00 μA; multipole 1 and 2 offset: −5.50 V and −7.50 V, respectively; intermultipole lens voltage: −16.00 V; trap DC offset voltage: −10.00 V. The purity (≥95%) of the tested compounds was assessed by means of reversed-phase HPLC (Waters, Milford, MA, USA) on a Phenomenex Luna® 3 µm C18(2) 100 Å, 100 mm × 4.6 mm column (Phenomenex, Torrance, CA, USA) using the following operative conditions: mobile phase: 80:20 methanol/water + 0.05% TFA (isocratic mode); flow rate: 1 mL/min; detector λ: 254 nm; time: 20 min; temperature: 23 °C. The synthesis of the final compounds (1a–j) and ester intermediates (2a–j) is described in the following paragraphs. Proton and carbon assignments are based on 1D and 2D NMR data and are given for all final compounds; atom numbering is detailed in the SM. The preparation of the remaining synthetic precursors (3, 4a,e–j, 5) is reported in the SM. Compounds 2b–d were obtained from commercially available bromo-derivatives.
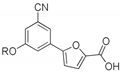
5-(3-Cyano-5-phenoxyphenyl)furan-2-carboxylic acid (1a). General Procedure A. The appropriate ester (43 mg, 0.135 mmol) was dissolved in a solution of THF/H2O 2:1 (1.0 mL). LiOH·H2O (17 mg, 0.407 mmol) was added while the solution was kept in an ice bath. The reaction mixture was stirred at r.t. for a variable time (1–6 h). THF was evaporated under reduced pressure, and the aqueous phase was brought to an acidic pH with 3 M HCl; the precipitated solid was filtered in vacuo and washed with small portions of a cyclohexane/EtOAc 6:4 mixture. Further purification strategies were adopted, when needed to afford the desired product. Starting compound: methyl 5-(3-benzyl-5-cyanophenyl)furan-2-carboxylate (2a). Time: 1 h. Yield: 80%. Aspect: white solid. Mp: 228 °C (dec.). TLC (DCM–MeOH 8:2): Rf = 0.41. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.55–12.93 (br s exch. D2O, 1H, COOH), 8.05 (d, J = 1.5 Hz, 1H, H7), 7.64 (t, J = 1.9 Hz, 1H, H11), 7.47 (t, J = 1.7 Hz, 1H, H9), 7.43 (dd, J = 7.5, 1.7 Hz, 2H, H14, H14′), 7.36 (d, J = 3.7 Hz, 1H, H3), 7.32 (d, J = 3.7 Hz, 1 H, H4), 7.23 (t, J = 7.4 Hz, 1H, H15), 7.13 (dd, J = 7.5, 1.7 Hz, 2H, H13, H13′).13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.47 C10, 158.32 COOH, 155.87 C5, 153.63 C12, 145.75 C10, 132.69 C6, 130.87 C14, C14′, 125.13 C15, 123.45 C7, 121.87 C3, 120.07 C9, 119.73 C13, C13′, 118.51 C11, 118.11 CN, 114.29 C8, 110.94 C4. FTIR (ATR): ν = 3116, 3086, 3063, 2956, 2924, 2919, 2852, 2233, 1732, 1693, 1592, 1569, 1519, 1438, 1417, 1363, 1298, 1216, 1030, 803 cm−1. ESI-MS (m/z) calcd. for C18H11NO4 305.07, found 305.02 [M–H]−, 609.57 = [2M–H]−. HPLC purity: 96.7%.
5-(3-(Benzyloxy)-5-cyanophenyl)furan-2-carboxylic acid (1b). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-(benzyloxy)-5-cyanophenyl)furan-2-carboxylate (2b). Time: 90 min. Purification: trituration in cold DCM. Yield: 80%. Aspect: yellowish solid. Mp: 187 °C. TLC (DCM–MeOH 9:1): Rf = 0.20. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.50–12.90 (br s exch. D2O, 1H, COOH), 7.84 (t, J = 1.4 Hz, 1H, H7), 7.71 (dd, J = 2.5, 1.4 Hz, 1H, H11), 7.55 (dd, J = 2.5, 1.4 Hz, 1H, H9), 7.50–7.45 (m, 2H, H13, H13′), 7.43–7.36 (m, 3H, H14, H14′, H15), 7.35 (d, J = 3.6 Hz, 1H, H3), 7.32 (d, J = 3.6 Hz, 1H, H4), 5.20 (s, 1H, CH2). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.62 C10, 159.51 COOH, 154.00 C5, 145.81 C2, 136.58 C6, 132.19 C12, 128.98 C14, C14′, 128.61 C15, 128.45 C13, C13′, 120.88 C7, 119.96 C3, 118.61 CN, 118.29 C9, 115.97 C11, 113.71C8, 110.56 C4, 70.47 CH2. FTIR (ATR): ν = 3489, 3405, 3121, 3087, 2968, 2935, 2882, 2232, 1679, 1594, 1517, 1436, 1366, 1329, 1302, 1288, 1216, 1167, 1048, 1025, 963, 761 cm−1. ESI-MS (m/z) calcd. for C19H13NO4 319.08, found 318.4 [M-H]−. HPLC purity: 97.7%.
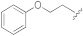
5-(3-Cyano-5-phenethoxyphenyl)furan-2-carboxylic acid (1c). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-phenethoxyphenyl)furan-2-carboxylate (2c). Time: 4 h. Purification: recrystallization from DCM/hexane. Yield: 85%. Aspect: white solid. Mp: 202 °C. TLC (DCM–MeOH 9:1): Rf = 0.23. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.95–12.64 (br s exch. D2O, 1H, COOH), 7.80 (t, J = 1.4 Hz, 1H, H7), 7.58 (dd, J = 2.5, 1.5 Hz, 1H, H11), 7.46 (dd, J = 2.5, 1.5 Hz, 1H, H9), 7.38–7.26 (m, 6H, H14, H14′, H13, H13′, H4, H3), 7.25–7.17 (m, 1H, H15), 4.33 (t, J = 6.8 Hz, 2H, CH2O), 3.06 (t, J = 6.8 Hz, 2H, CH2). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.65 C10, 159.60 COOH, 153.98 C5, 145.80 C2, 138.48 C12, 132.14 C6, 129.45 C14, C14′, 128.80 C13, C13′, 126.82 C15, 120.69 C7, 119.89 C3, 118.60 C9, 115.55 C11, 118.60 CN, 113.73 C8, 110.54 C4, 69.40 CH2O, 35.14 CH2. FTIR (ATR): ν = 3087, 3059, 3026, 2976, 2947, 2932, 2841, 2235, 1696, 1679, 1607, 1593, 1522, 1436, 1365, 1334, 1304, 1272, 1219, 1163, 1062, 1030, 959, 754 cm−1. ESI-MS (m/z) calcd. for C20H15NO4 333.10, found 333.01 [M-H]−, 665.66 [2M-H]−. HPLC purity: 98.5%.
5-(3-Cyano-5-(3-phenylpropoxy)phenyl)furan-2-carboxylic acid (1d). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-(3-phenylpropoxy)phenyl)furan-2-carboxylate (2d). Time: 2 h. Purification: flash column chromatography (DCM–MeOH 9:1). Yield: 80%. Aspect: white solid. Mp: 186 °C. TLC (DCM–MeOH 9:1): Rf = 0.20. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.70–12.70 (br s exch. D2O, 1H, COOH), 7.81 (t, J = 1.4 Hz, H7), 7.60 (dd, J = 2.5, 1.4 Hz, 1H, H11), 7.44 (dd, J = 2.5, 1.4 Hz, 1H, H9,), 7.34 (d, J = 3.6 Hz, 1H, H3), 7.30 (d, J = 3.6 Hz, 1H, H4), 7.30–7.12 (m, 5H, H16, H16′, H17, H17′, H18), 4.09 (t, J = 6.2, 2H, H12), 2.74 (dd, J = 8.6, 6.5 Hz, 2H, H14), 2.02 (m, 2H, H13). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.79 C10, 159.68 COOH, 153.97 C5, 145.89 C2, 141.69 C15, 132.17 C6, 128.81 C17, C17′ C16, C16′, 126.33 C18, 120.68 C7, 119.84 C3, 118.64 CN, 117.79 C9, 115.63 C11, 113.74 C8, 110.51 C4, 68.12 C12, 31.81 C14, 30.62 C13. FTIR (ATR): ν = 3403, 3120, 3084, 3026, 2951, 2884, 2228, 1680, 1592, 1517, 1429, 1363, 1333, 1215, 1164, 1039, 958, 869, 797, 699 cm−1. ESI-MS (m/z) calcd. for C21H17NO4 347.12, found 346.61 [M-H]−, 693.37 [2M-H]−. HPLC purity: 95.1%.
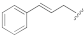
(E)-5-(3-(Cinnamyloxy)-5-cyanophenyl)furan-2-carboxylic acid (1e). The compound was obtained according to General Procedure A. Starting compound: (E)-methyl 5-(3-(cinnamyloxy)-5-cyanophenyl)furan-2-carboxylate (2e). Time: 1 h. Yield: 82%. Aspect: white solid. Mp: 170 °C. TLC (DCM–MeOH 9:1): Rf = 0.14. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.88–12.65 (br s exch. D2O, 1H, COOH), 7.83 (t, J = 1.4 Hz, H7), 7.67 (dd, J = 2.1, 1.5 Hz, 1H, H11), 7.55–7.44 (m, 3H, H9, H16, H16′), 7.41–7.16 (m, 5H, H3, H4, H17, H17′, H18), 6.81 (d, J = 16.0 Hz, 1H, H14), 6.51 (dt, J = 16.0, 5.8 Hz, 1H, H13), 4.86 (dd, J = 5.9, 1.4 Hz, 2H, H12). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.16 C10, 158.89 COOH, 153.31 C5, 145.70 C2, 135.96 C15, 133.05 C14, 131.76 C6, 128.60 C17, C17′, 127.93 C18, 126.46 C16, C16′, 123.92 C13, 120.28 C7, 119.08 C3, 118.09 CN, 117.73 C9, 115.34 C11, 113.21 C8, 109.95 C4, 68.80 C12. FTIR (ATR): ν = 3391, 3019, 3080, 3027, 2929, 2667, 2576, 2232, 1678, 1593, 1569, 1519, 1436, 1364, 1331, 1269, 1168, 1146, 1030, 961, 862, 806, 692 cm−1. ESI-MS (m/z) calcd. for C21H15NO4 345.35, found 344.83 [M-H]−, 689.40 [2M-H]−. HPLC purity: 95.6%.
5-(3-Cyano-5-(2-phenoxyethoxy)phenyl)furan-2-carboxylic acid (1f). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-(2-phenoxyethoxy)phenyl)furan-2-carboxylate (2f). Time: 3 h. Yield: 92%. Aspect: white solid. Mp: 195 °C. TLC (DCM–MeOH 9:1): Rf = 0.18. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.07–12.92 (br s exch. D2O, 1H, COOH), 7.83 (t, J = 1.4 Hz, 1H, H7), 7.69 (dd, J = 2.5, 1.5 Hz, 1H, H11), 7.52 (dd, J = 2.5, 1.5 Hz, 1H, H9), 7.35 (d, J = 3.6 Hz, 1H, H3), 7.32 (d, J = 3.6 Hz, 1H, H4), 7.30–7.24 (m, 2H, H16, 16′), 7.02 − 6.89 (m, 3H, H15, H15′, H17), 4.46 (m, 2H, H12 or H13), 4.32 (m, 2H, H13 or H12). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.56 C10, 159.54 C14, 158.66 COOH, 154.07 C5, 145.65 C2, 132.19 C6, 129.97 C16, C16′, 121.28 C7, 120.89 C3, 119.99 C9, 118.55 CN, 118.18 C11, 115.80 C17, 114.98 C15, C15′, 113.77 C8, 110.54 C4, 67.77 CH2, 66.50 CH2. FTIR (ATR): ν = 3115, 3082, 2973, 2941, 2878, 2695, 2571, 2521, 2234, 1690, 1675, 1609, 1594, 1520, 1488, 1447, 1417, 1339, 1300, 1209, 1167, 1055, 862, 750 cm−1. ESI-MS (m/z) calcd. for C20H15NO5 349.34, found 348.40 [M-H]−, 697.22 [2M-H]−. HPLC purity: 98.4%.
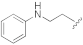
5-(3-Cyano-5-(2-(phenylamino)ethoxy)phenyl)furan-2-carboxylic acid (1g). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-(2- (phenylamino)ethoxy)phenyl)furan-2-carboxylate (2g). Time: 4 h. Yield: 81%. Aspect: white solid. Mp: 140 °C. TLC (DCM–MeOH 9:1): Rf = 0.44. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.80 (t, J = 1.4 Hz, 1H, H7), 7.62 (t, J = 2.0 Hz, 1H, H11), 7.43 (dd, J = 2.0, 1.4 Hz 1H, H9), 7.28 (d, 1H, J = 3.6 Hz, H3), 7.18 (d, J = 3.6 Hz, 1H, H4), 7.11–7.01 (m, 2H, H16, H16′), 6.62 (m, 2H, H15, H15′) 6.53 (t, J = 7.2 Hz, 1H, H17), 5.78 (br s exch. D2O, 1H, NH), 4.24 (t, J = 5.4 Hz, 2H, H12), 3.43 (t, partially overlapped with DMSO water peak, J = 5.4 Hz, 2H, H13). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 160.07 C10, 159.70 COOH, 158.97 C14, 153.14 C5, 148.90 C2, 132.44 C6, 129.38 C16, C16′, 120.60 C7, 118.65 CN, 118.61 C3, 117.68 C9, 116.33 C11, 115.44 C17, 113.62 C8, 112.53 C15, C15′, 110.37 C4, 67.71 C12, 42.46 C13. FTIR (ATR): ν = 3385, 3115, 3055, 3027, 2962, 2928, 2853, 2647, 2554, 2231, 1643, 1594, 1517, 1435, 1367, 1326, 1259, 1154, 1092, 1056, 1023, 962, 868, 798, 692 cm−1. ESI-MS (m/z) calcd. for C20H16N2O4 348.11, found 347.98 [M-H]−, 695.67 [2M-H]−. HPLC purity: 97.6%.
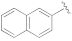
5-(3-Cyano-5-(naphthalen-2-yloxy)phenyl)furan-2-carboxylic acid (1h). The compound was obtained according to General Procedure A. Starting compound: methyl 4-(3-cyano-5-(naphthalen-2-yloxy)phenyl)cyclopenta-1,3-dienecarboxylate (2h). Time: 1 h. Yield: 79%. Aspect: white solid. Mp: 226 °C. TLC (DCM–MeOH 9:1): Rf = 0.16. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.30 (s exch. D2O, 1H, COOH), 8.08–8.07 (m, 1H, H7), 8.02 (d, J = 8.9, 1H, H18), 7.99–7.91 (m, 1H, H15), 7.90–7.84 (m, 1H, H15), 7.75 (t, J = 2.0 Hz, 1H, H11), 7.58 (t, J = 1.9 Hz, 1H, H9), 7.56 (d, J = 2.5 Hz, 1H, H19), 7.55–7.45 (m, 2H, H16, H17), 7.38–7.34 (m, 2H, H13, H3), 7.30 (d, J = 3.5 Hz, 1H, H4). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.95 C10, 158.62 COOH, 154.22 C12, 153.94 C5, 146.41 C2, 134.78 C20, 133.21 C6, 131.41 C14, 131.16 C21, 128.63 C18, 128.22 C5, 127.72 C16, 126.23 C17, 124.07 C7, 122.68 C9, 120.51 C13, 120.34 C3, 119.38 C11, 118.56 CN, 115.70 C19, 114.77 C8, 111.40 C4. FTIR (ATR): ν = 3117, 3057, 2962, 2919, 2232, 1697, 1591, 1570, 1520, 1418, 1362, 1296, 1214, 1029, 806 cm−1. ESI-MS (m/z) calcd. for C22H13NO4 355.35, found 354.92 [M-H]−, 709.41 [2M-H]−. HPLC purity: 97.9%.
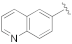
5-(3-Cyano-5-(quinolin-7-yloxy)phenyl)furan-2-carboxylic acid (1i). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-(quinolin-7-yloxy)phenyl)furan-2-carboxylate (2i). Time: 3 h. Yield: 89%. Aspect: yellow solid. Mp: 263 °C (dec.). TLC (DCM–MeOH 9:1): Rf = 0.12. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.26 (s exch. D2O, 1H, COOH), 8.87 (dd, J = 4.3, 1.7 Hz, 1H, H17), 8.39 (dd, J = 8.3, 1.8 Hz, 1H, H15), 8.12 (d, J = 1.5 Hz, 1H, H7), 8.08 (d, J = 8.7 Hz, 1H, H14), 7.83 (t, J = 1.9 Hz, 1H, H11), 7.69 (t, J = 1.8 Hz, 1H, H9), 7.53–7.43 (m, 3H, H13, H16, H19), 7.39 (d, J = 3.7 Hz, 1H, H3), 7.32 (d, J = 3.7 Hz, 1H, H4). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.45 C10, 157.41 COOH, 157,16 C12, 153.59 C5, 151.80 C17, 149.18 C20, 145.76 C2, 136.44 C15, 132.87 C6, 131.05 C14, 125.48 C21, 124.27 C19, 123.05 C13, 121.19 C3, 120.41 C7, 120.10 C9, 119.79 C16, 118. 04 CN, 115.57 C11, 114. 51 C8, 111.06 C4. FTIR (ATR): ν = 3268, 3118, 3078, 2919, 2852, 2232, 1733, 1692, 1657, 1593, 1571, 1520, 1505, 1441, 1416, 1363, 1294, 1257, 1213, 1164, 1093, 1015, 798 cm−1. ESI-MS (m/z) calcd. for C21H12N2O4 356.08, found 357.37 [M+H]+. HPLC purity: 96.5%.
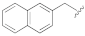
5-(3-Cyano-5-(naphthalen-2-ylmethoxy)phenyl)furan-2-carboxylic acid (1j). The compound was obtained according to General Procedure A. Starting compound: methyl 5-(3-cyano-5-(naphthalen-2-ylmethoxy)phenyl)furan-2-carboxylate (2j). Time: 6 h. Yield: 85%. Aspect: white solid. Mp: 209 °C (dec.). TLC (DCM–MeOH 7:3): Rf = 0.26. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.08–8.01 (m, 1H, H19), 8.00–7.89 (m, 3H, H14, H15, H18), 7.85 (t, J = 1.4 Hz, 1H, H7), 7.79 (dd, J = 2.4, 1.4, Hz, 1H, H11), 7.62 (dd, J = 8.4, 1.7 Hz, 1H, H13), 7.59 (dd, J = 2.5, 1.3 Hz, 1H, H9), 7.58–7.51 (m, 2H, H17, H16), 7.36 (d, J = 3.6 Hz, 1H, H4), 7.28 (d, J = 3.5 Hz, 1H, H3). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 159.82 C10, 159.53 COOH, 153.56 C5, 146.87 C2, 134.22 C20, 133.22 C6, 133.10 C12, 132.40 C21, 128.61 C14, 128.30 C15/18, 128.07 C18/15, 127.07 C19, 126.84 C16/17, 126.72 C17/16, 126.22 C13, 120.89 C7, 119.24 C3, 118.62 CN, 118.20 C9, 116.01 C11, 113.70 C8, 111.50 C4, 70.60 CH2. FTIR (ATR): ν = 3383, 3262, 3120, 3060, 2923, 2232, 1692, 1658, 1593, 1572, 1520, 1441, 1363, 1296, 1214, 1029, 803 cm−1. ESI-MS (m/z) calcd. for C23H15NO4 369.37, found 368.65 [M-H]−, 737.06 [2M-H]−. HPLC purity: 96.1%.
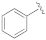
Methyl 5-(3-benzyl-5-cyanophenyl)furan-2-carboxylate (2a).General Procedure B. The appropriate bromo-derivative (70 mg, 0.255 mmol), (5-(methoxycarbonyl)furan-2-yl)boronic acid (56 mg, 0.330 mmol) and bis(triphenylphosphine)palladium dichloride (9 mg, 0.013 mmol) were dissolved in dry 1,4-dioxane (1.5 mL) under N2 atmosphere. A 2M Na2CO3 solution (0.26 mL, 0.255 mmol) was added, and the resulting mixture was stirred in a microwave synthesizer at 60 °C for 80 min. The reaction mixture was filtered on a pad of celite, diluted with water (2 mL), and extracted with EtOAc (3 × 3 mL). The organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (cyclohexane–EtOAc 8:2) to afford pure 2a as a white solid. Starting compound: 3-bromo-5-phenoxybenzonitrile (4a). Yield: 55%. Mp: 131 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.39. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.78–7.74 (m, 1H, HAr), 7.65–7.60 (m, 1H, HAr), 7.47–7.37 (m, 3H, HAr), 7.28–7.19 (m, partially overlapped with solvent peak, 4H, HAr), 7.14–7.11 (m, 1H, HAr), 7.09–7.02 (m, 2H, HAr), 6.79 (d, J = 3.7 Hz, 1H, HAr), 3.92 (s, 3H, OCH3).
Methyl 5-(3-(benzyloxy)-5-cyanophenyl)furan-2-carboxylate (2b). General Procedure C. Methyl 5-(3-cyano-5-hydroxyphenyl)furan-2-carboxylate (3, 150 mg, 0.620 mmol) was suspended in dry acetone (3.05 mL) under a nitrogen atmosphere. Oven-dried K2CO3 (343 mg, 2.48 mmol) was added, and the mixture was stirred for 10 min. Then, a solution of the suitable bromo-derivative (192 mg, 1.12 mmol) in dry acetone (0.76 mL) was added, and the reaction mixture was heated at reflux for a variable time for each substrate (3–72 h). Then, the suspension was filtered in vacuo to remove K2CO3, and acetone was evaporated under reduced pressure. The crude product was purified to afford the desired product. Starting compound: (bromomethyl)-benzene. Time: 3 h. Purification: crystallization from DCM/hexane. Yield: 75%. Aspect: off-white solid. Mp: 144 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.41. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.64 (t, J = 1.4 Hz, 1H, HAr), 7.60 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 7.49–7.32 (m, 5H, HAr), 7.29–7.23 (m, partially overlapped with solvent peak, 1H, HAr), 7.18 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 6.79 (d, J = 3.6 Hz, 1H, HAr), 5.14 (s, 2H, OCH2), 3.93 (s, 3H, OCH3).
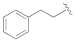
Methyl 5-(3-cyano-5-phenethoxyphenyl)furan-2-carboxylate (2c). The compound was obtained according to General Procedure C. Starting compound: (2-bromoethyl)-benzene. Time: 72 h. Purification: flash column chromatography (cyclohexane–EtOAc 8:2). Yield: 35%. Aspect: pearl-white solid. Mp: 150 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.40. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.60 (t, J = 1.4 Hz, 1H, HAr), 7.49 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 7.39–7.27 (m, partially overlapped with solvent peak, 5H, HAr), 7.25 (d, J = 3.7 Hz, 1H, HAr), 7.09 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 6.79 (d, J = 3.7 Hz, 1H, HAr), 4.25 (t, J = 6.9 Hz, 2H, OCH2), 3.93 (s, 3H, OCH3), 3.14 (t, J = 6.9 Hz, 2H, CH2).
Methyl 5-(3-cyano-5-(3-phenylpropoxy)phenyl)furan-2-carboxylate (2d). The compound was obtained according to General Procedure C. Starting compound: (3-propyl)benzene. Time: 16 h. Purification: flash column chromatography (cyclohexane–EtOAc 8:2). Yield: 35%. Aspect: yellowish solid. Mp: 90 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.39. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.61 (t, J = 1.4 Hz, 1H, HAr), 7.50 (dd, J = 2.4, 1.4 Hz, 1H, HAr), 7.38–7.16 (m, partially overlapped with solvent peak, 6H, HAr), 7.08 (dd, J = 2.4, 1.4 Hz, 1H, HAr), 6.79 (d, J = 3.6 Hz, 1H, HAr), 4.03 (t, J = 6.2 Hz, 2H, OCH2), 3.93 (s, 3H, OCH3), 2.83 (t, J = 7.5 Hz, 2H, CH2), 2.23–2.07 (m, 2H, CH2).
(E)-Methyl 5-(3-(cinnamyloxy)-5-cyanophenyl)furan-2-carboxylate (2e). The compound was obtained according to General Procedure C. Starting compound: (E)-(3-bromoprop-1-en-1-yl)benzene (4e). Time: 4 h. Purification: trituration in cold Et2O. Yield: 40%. Aspect: white solid. Mp: 129 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.33. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.64 (t, J = 1.4 Hz, 1H, HAr), 7.58 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 7.47–7.39 (m, 2H, HAr), 7.40–7.22 (m, partially overlapped with solvent peak, 4H, HAr), 7.17 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 6.83–6.73 (m, 2H, HAr), 6.39 (dt, J = 16.0, 5.8 Hz, 1H, CH), 4.78 (dd, J = 5.8, 1.5 Hz, 2H, CH2), 3.93 (s, 3H, OCH3).
Methyl 5-(3-cyano-5-(2-phenoxyethoxy)phenyl)furan-2-carboxylate (2f). The compound was obtained according to General Procedure B. Starting compound: 3-bromo-5-(2-phenoxyethoxy)benzonitrile (4f). Purification: flash column chromatography (cyclohexane–EtOAc 8:2). Yield: 80%. Aspect: white solid. Mp: 134 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.37. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.66 (t, J = 1.5 Hz, 1H, HAr), 7.59 (dd, J = 2.5, 1.5 Hz, 1H, HAr), 7.38–7.23 (m, partially overlapped with solvent peak, 3H, HAr), 7.18 (dd, J = 2.5, 1.5 Hz, 1H, HAr), 7.07–6.92 (m, 3H, HAr), 6.81 (d, J = 3.6 Hz, 1H, HAr), 4.44–4.31 (m, 4H, CH2), 3.93 (s, 3H, OCH3).
Methyl 5-(3-cyano-5-(2-(phenylamino)ethoxy)phenyl)furan-2-carboxylate (2g). The compound was obtained according to General Procedure B. Starting compound: 3-bromo-5-(2-(phenylamino)ethoxy)benzonitrile (4g). Purification: flash column chromatography (cyclohexane–EtOAc 7:3), recrystallization from DCM/hexane. Yield: 40%. Aspect: brownish solid. Mp: 160 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.20. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.64 (t, J = 1.4 Hz, 1H, HAr), 7.53 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 7.32–7.16 (m, partially overlapped with solvent peak, 3H, HAr), 7.12 (dd, J = 2.5, 1.4 Hz, 1H, HAr), 6.84–6.63 (m, 4H, HAr), 4.24 (t, J = 5.2 Hz, 2H, CH2), 4.07 (br s exch. D2O, 1H, NH), 3.93 (s, 3H, OCH3), 3.67–3.52 (m, 2H, CH2).
Methyl 5-(3-cyano-5-(naphthalen-2-yloxy)phenyl)furan-2-carboxylate (2h). The compound was obtained according to General Procedure B. Starting compound: 3-bromo-5-(naphthalen-2-yloxy)benzonitrile (4h). Purification: flash column chromatography (cyclohexane–EtOAc 8:2). Yield: 35%. Aspect: white solid. Mp: 155 °C. TLC (cyclohexane–EtOAc 8:2): Rf = 0.35. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.96–7.83 (m, 2H, HAr), 7.83–7.74 (m, 2H, HAr), 7.71–7.65 (m, 1H, HAr), 7.58–7.40 (m, 3H, HAr), 7.30–7.17 (m, partially overlapped with solvent peak, 3H, HAr), 6.79 (d, J = 3.6 Hz, 1H, HAr), 3.91 (s, 3H, OCH3).
Methyl 5-(3-cyano-5-(quinolin-7-yloxy)phenyl)furan-2-carboxylate (2i). The compound was obtained according to General Procedure B. Starting compound: 3-bromo-5-(quinolin-7-yloxy)benzonitrile (4i). Purification: flash column chromatography (cyclohexane–EtOAc 6:4). Yield: 40%. Aspect: white solid. Mp: 142 °C. TLC (cyclohexane–EtOAc 6:4): Rf = 0.30. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.92 (dd, J = 4.5, 1.7 Hz, 1H, HAr), 8.33 (d, J = 8.2 Hz, 1H, HAr), 7.95 (d, J = 8.9 Hz, 1H, HAr), 7.87 (t, J = 1.4 Hz, 1H, HAr), 7.80–7.70 (m, 2H, HAr), 7.50 (dd, J = 8.2, 4.5 Hz, 1H, HAr), 7.42 (dd, J = 8.9, 2.3 Hz, 1H, HAr), 7.30 (dd, J = 2.3, 1.4 Hz, 1H, HAr), 7.28–7.22 (m, partially overlapped with solvent peak, 1H, HAr), 6.84 (d, J = 3.6 Hz, 1H, HAr), 3.91 (s, 3H, OCH3).
Methyl 5-(3-cyano-5-(naphthalen-2-ylmethoxy)phenyl)furan-2-carboxylate (2j). The compound was obtained according to General Procedure C. Starting compound: 2-(bromomethyl)naphthalene (4j). Time: 5 h. Purification: trituration in cold Et2O. Yield: 66%. Aspect: white solid. Mp: 195 °C (dec.). TLC (cyclohexane–EtOAc 8:2): Rf = 0.28. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.97–7.80 (m, 4H, HAr), 7.70–7.60 (m, 2H, HAr), 7.57–7.48 (m, 3H, HAr), 7.28–7.24 (m, partially overlapped with solvent peak, 1H, HAr), 7.22 (dd, J = 2.4, 1.4 Hz, 1H, HAr), 6.80 (d, J = 3.6 Hz, 1H, HAr), 5.31 (s, 2H, OCH2), 3.93 (s, 3H, OCH3).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}