
Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients



Abstract
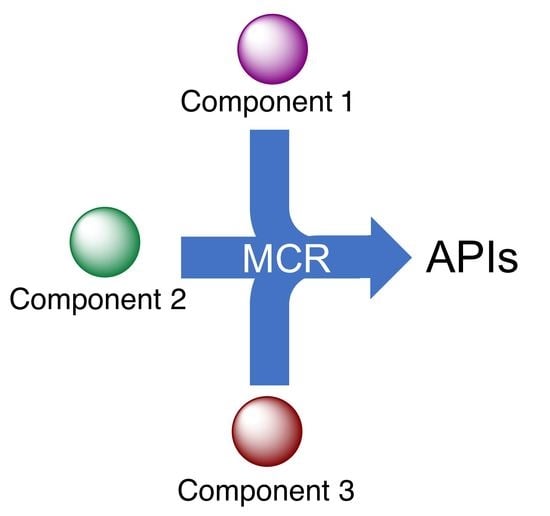
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Imine-Initiated Multicomponent Reactions
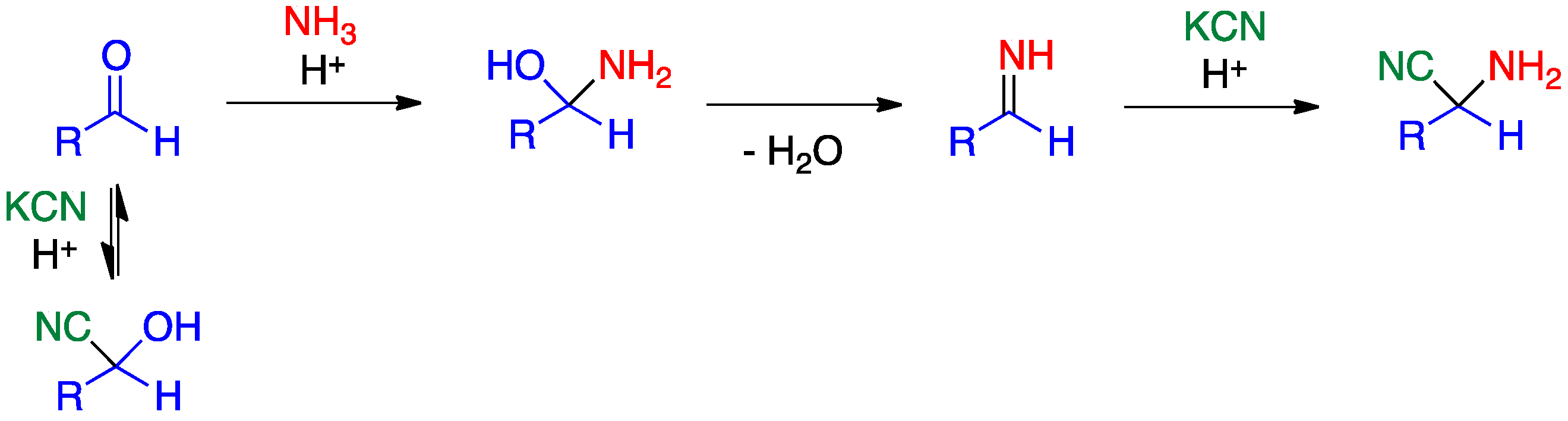
2.1. Strecker Reaction
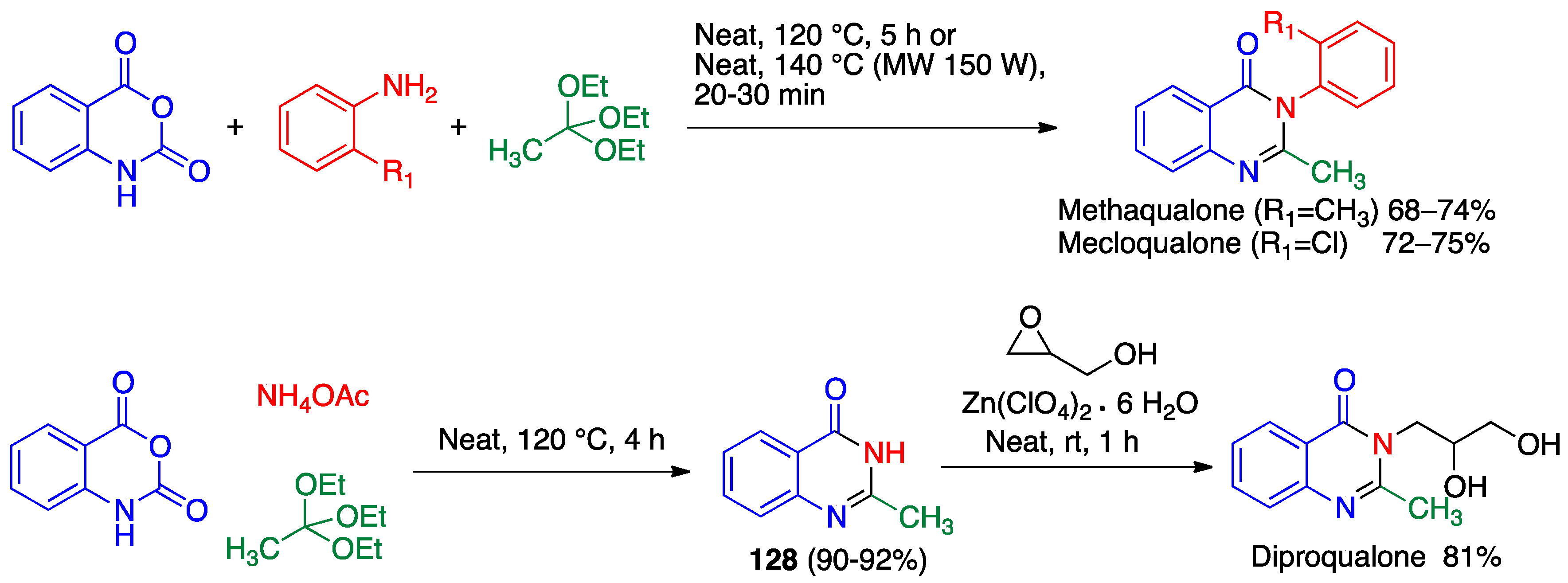
2.2. Bucherer-Bergs Reaction
2.3. Mannich Reaction
2.4. Petasis (Borono-Mannich) Reaction
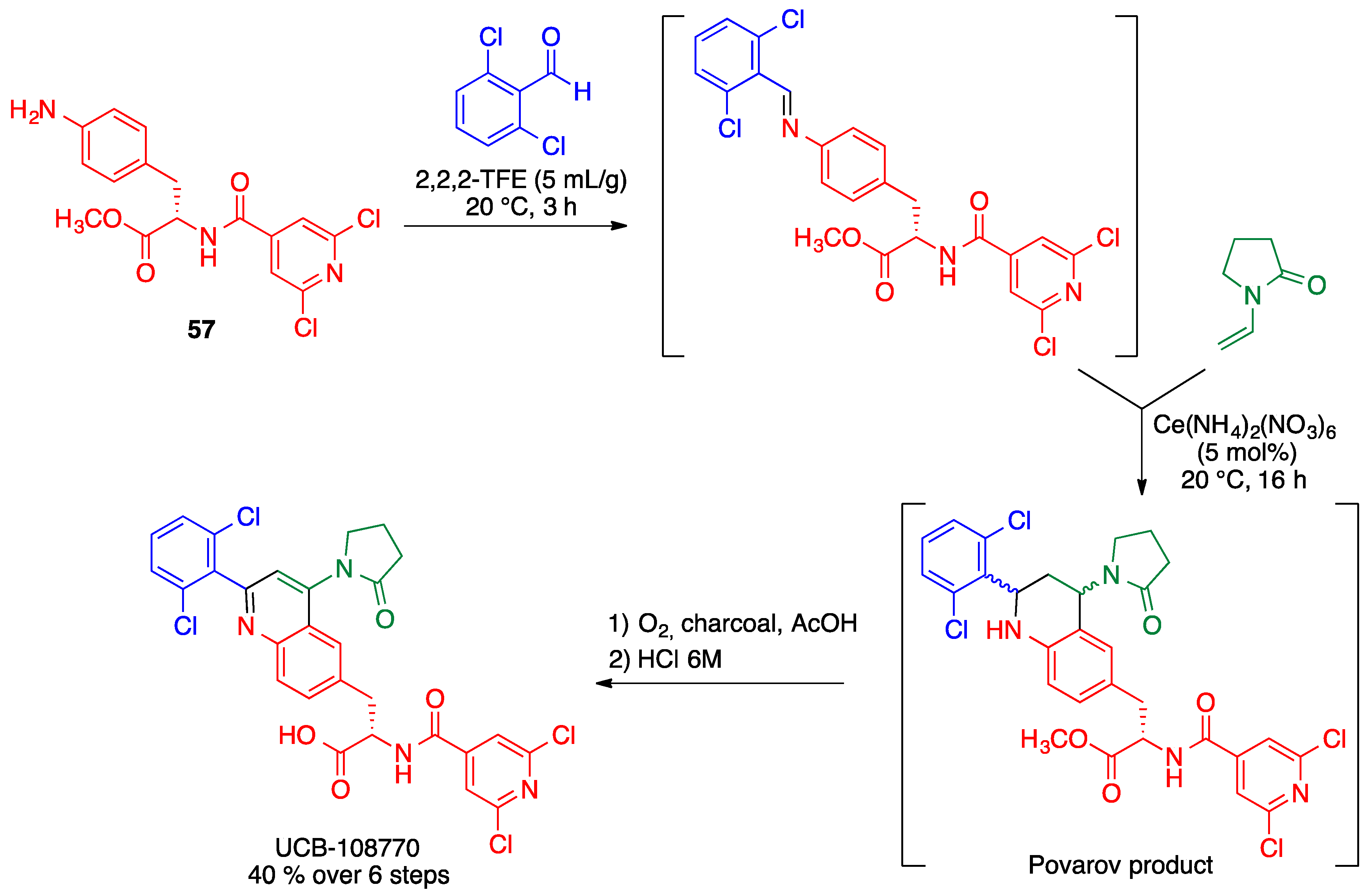
2.5. Povarov Reaction
2.6. Doebner Reaction
3. Enamine-Initiated Multicomponent Reactions
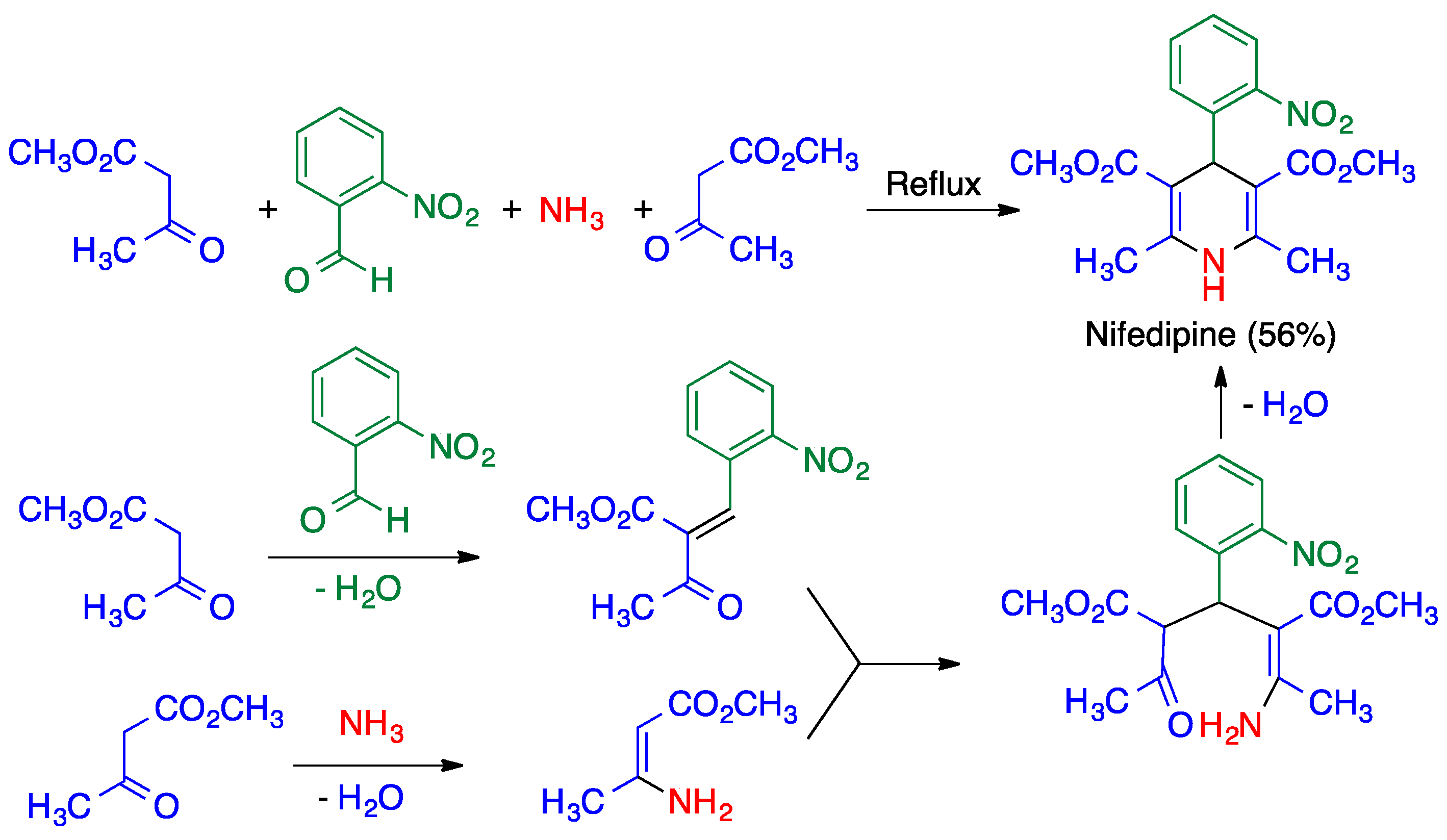
3.1. Hantzsch Dihydropyridine Synthesis
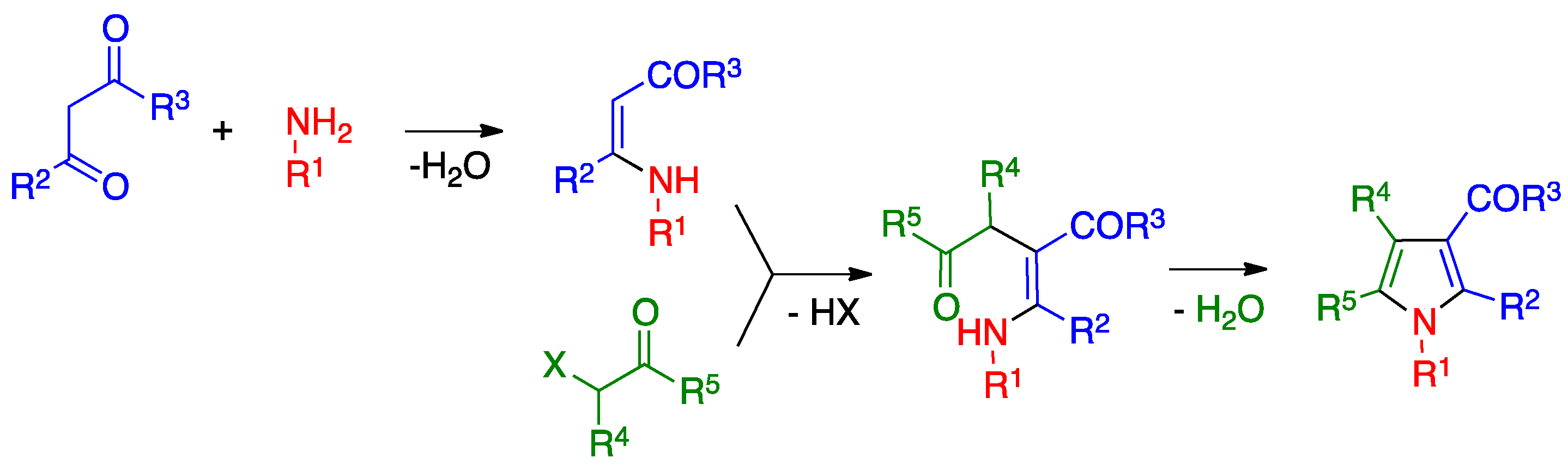
3.2. Hantzsch Pyrrole Synthesis
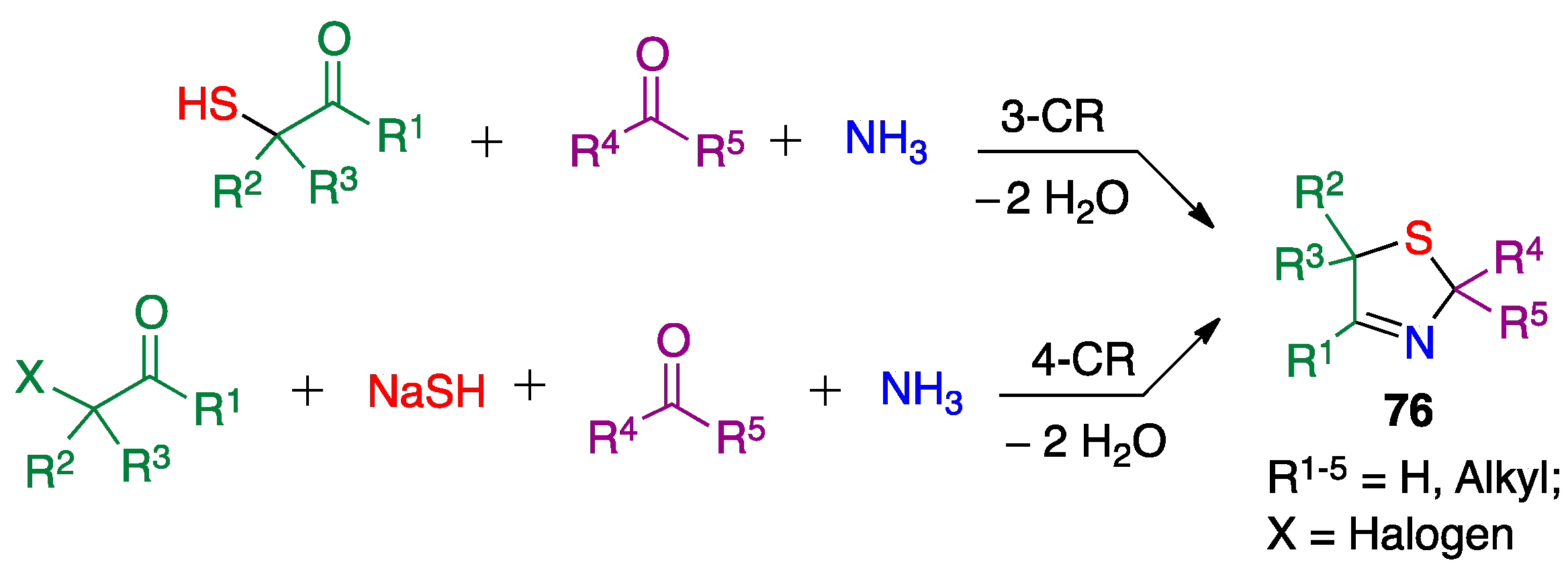
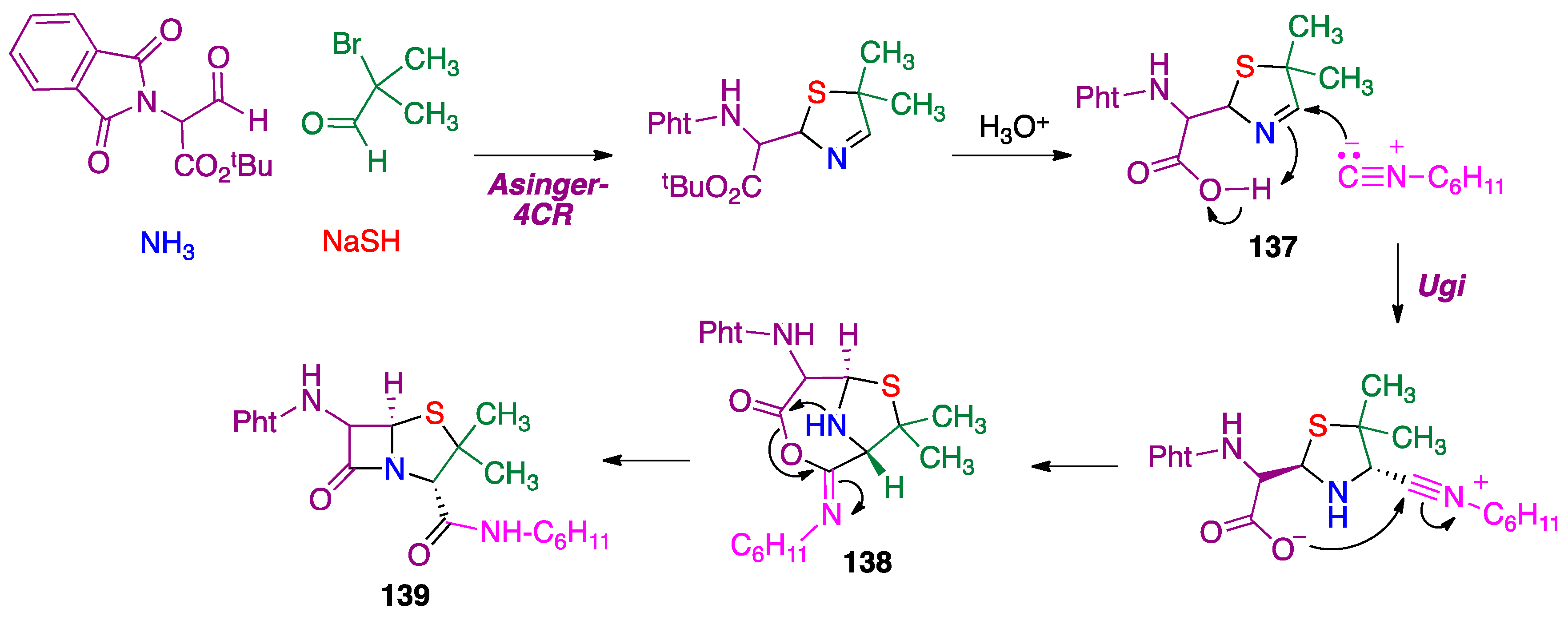
3.3. Asinger Reaction
4. Isonitrile-Based Multicomponent Reactions
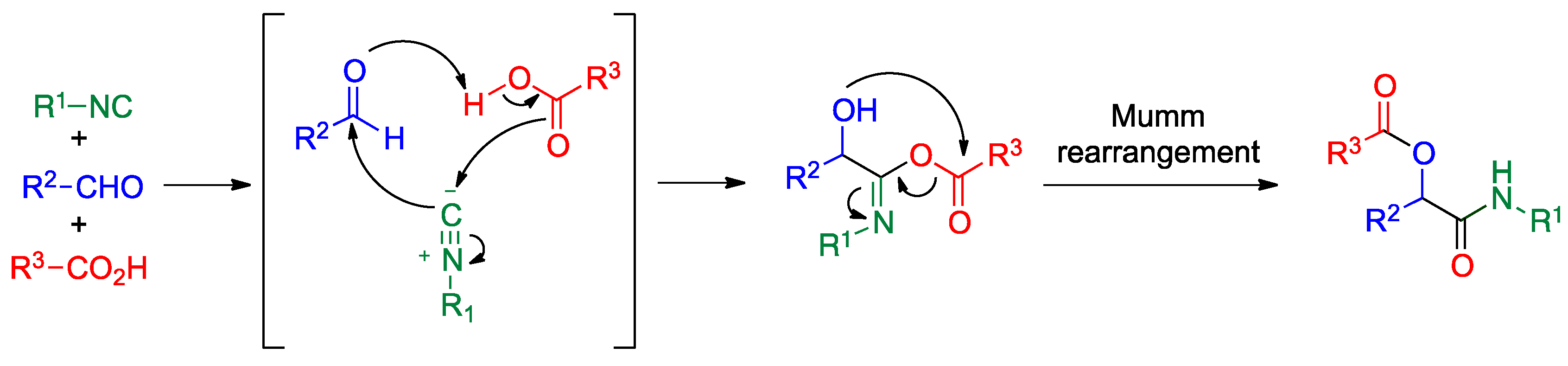
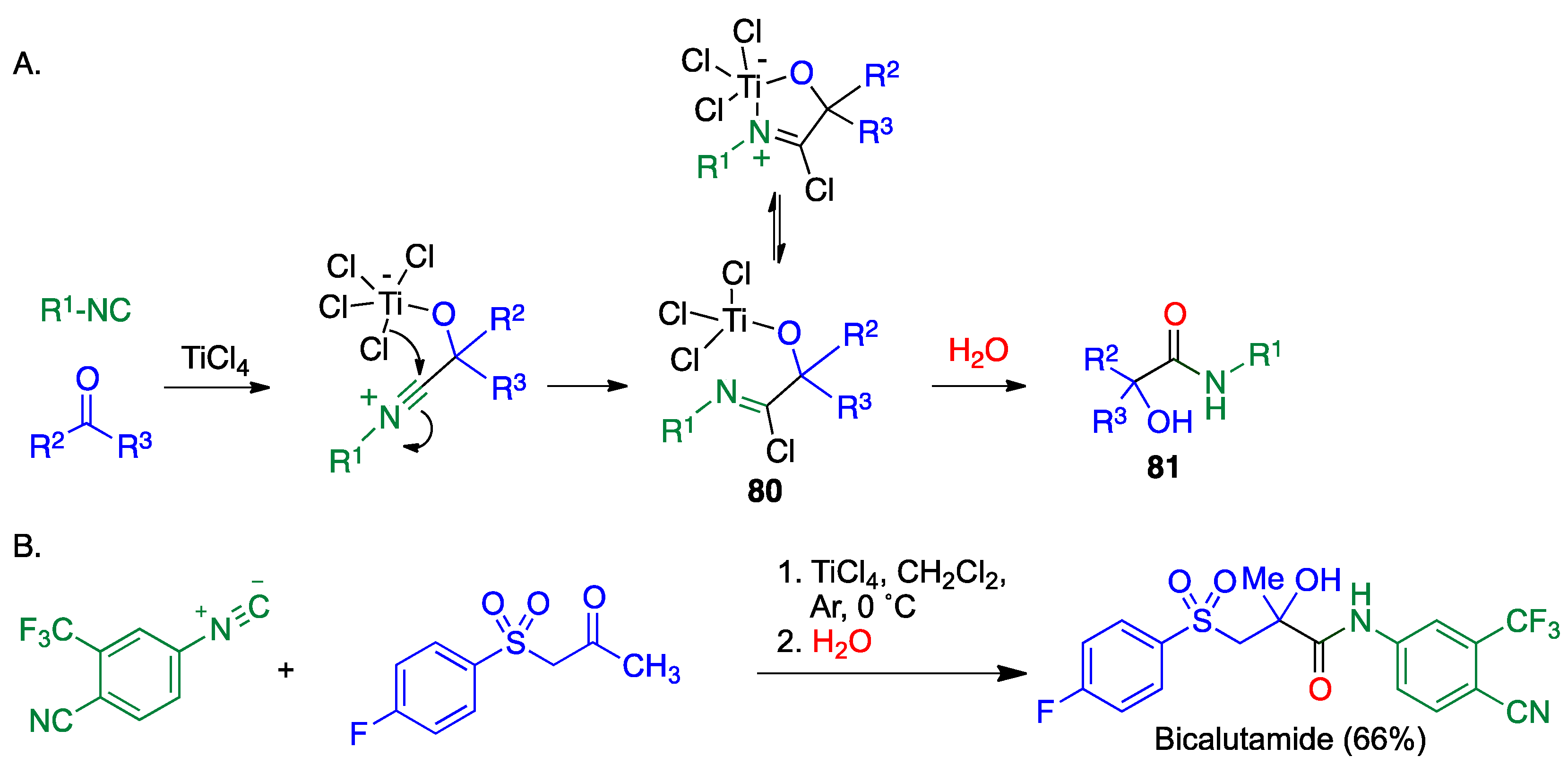
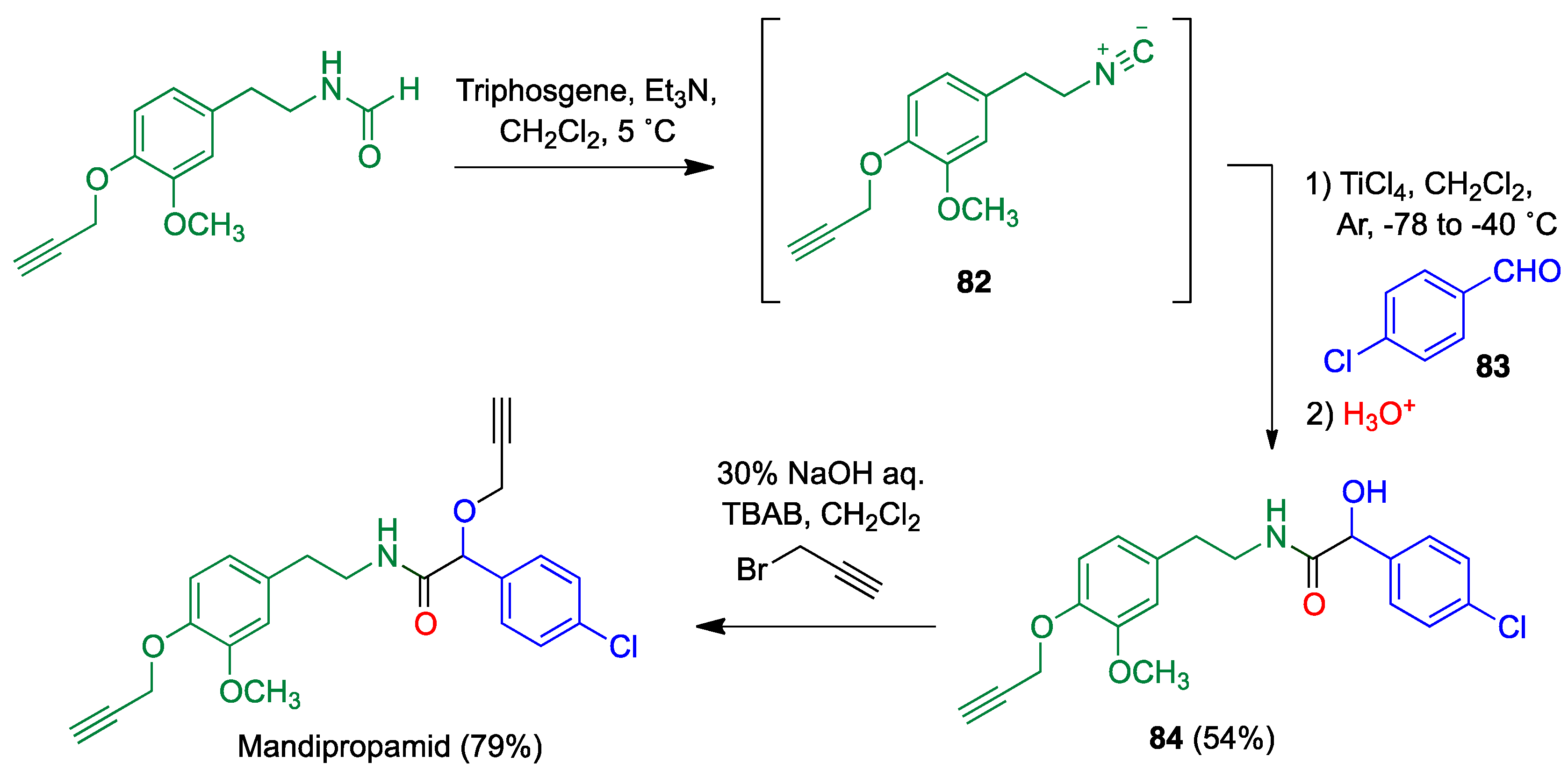
4.1. Passerini Reaction and Its Variants
4.2. Ugi Reaction
4.3. Van Leusen Reaction
5. Miscellaneous Named Multicomponent Reactions
5.1. Pauson–Khand Reaction
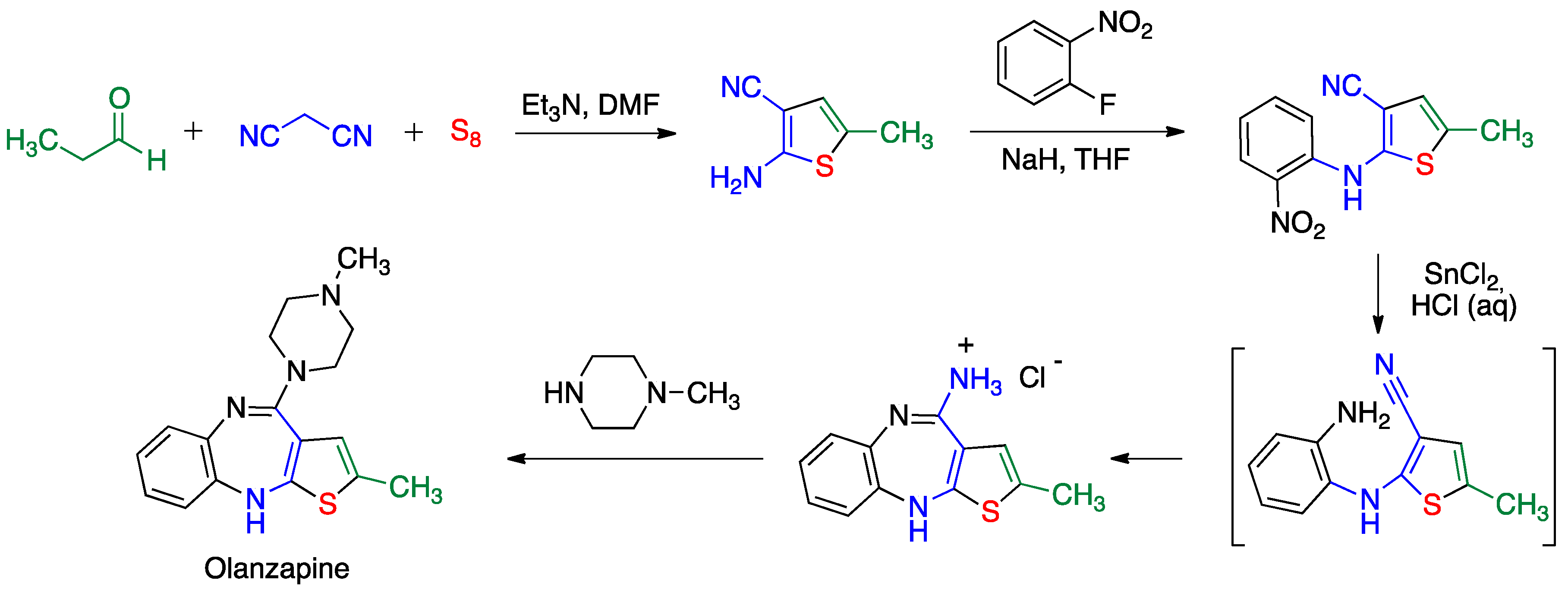
5.2. Gewald Reaction
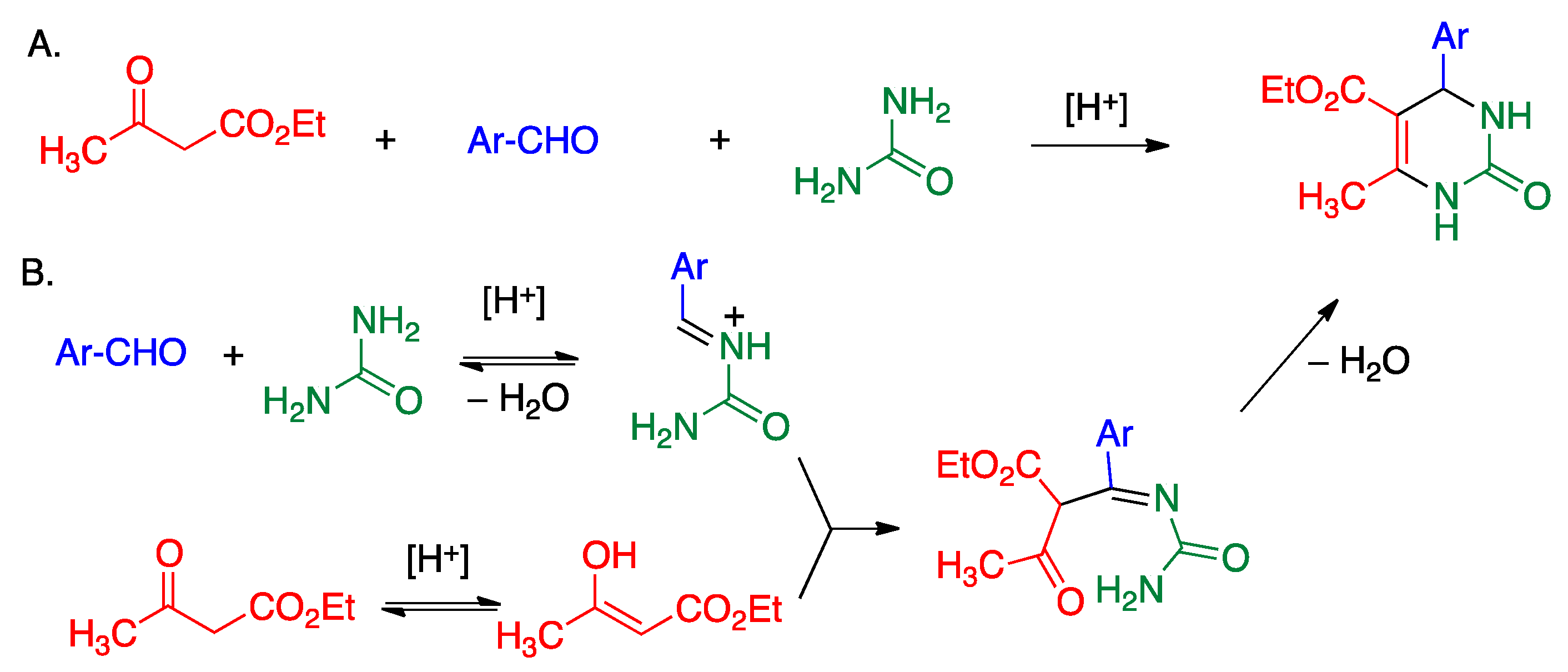
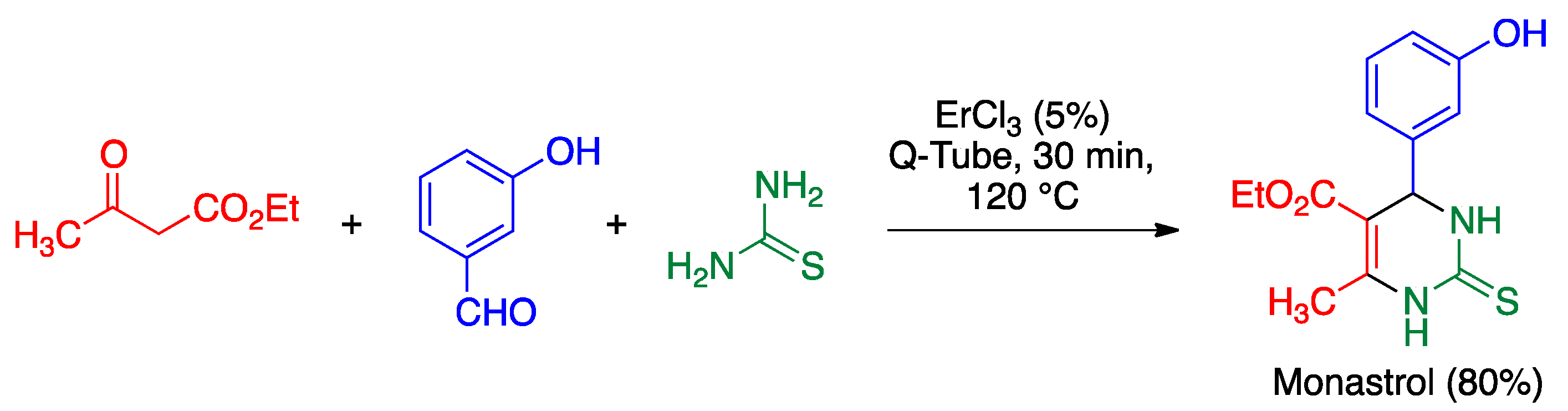
5.3. Biginelli Reaction
6. Miscellaneous Non-Named Multicomponent Reactions
7. Combination of Multicomponent Reactions
8. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheldon, R.A. The E factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Peters, D.S.; Pitts, C.R.; McClymont, K.S.; Stratton, T.P.; Bi, C.; Baran, P.S. Ideality in context: Motivations for total synthesis. Acc. Chem. Res. 2021, 54, 605–617. [Google Scholar] [CrossRef]
- Kalinski, C.; Umkehrer, M.; Weber, L.; Kolb, J.; Burdack, C.; Ross, G. On the industrial applications of MCRs: Molecular diversity in drug discovery and generic drug synthesis. Mol. Divers. 2010, 14, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.; Orru, R. Multicomponent reactions-Opportunities for the pharmaceutical industry. Drug Discov. Today Technol. 2013, 10, 15–20. [Google Scholar] [CrossRef]
- Radtke, L.; Marque, C.E.; Herrera, R.P. Multicomponent reactions in the synthesis of target molecules. In Multicomponent Reactions: Concepts and Applications for Design and Synthesis, 1st ed.; Marqués-López, E., Herrera, R.P., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2015; Chapter 6. [Google Scholar]
- Graebin, C.S.; Ribeiro, F.V.; Rogério, K.R.; Kummerle, A.E. Multicomponent reactions for the synthesis of bioactive compounds: A review. Curr. Org. Synth. 2019, 16, 855–899. [Google Scholar] [CrossRef]
- Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of biologically active molecules through multicomponent reactions. Molecules 2020, 25, 505. [Google Scholar] [CrossRef]
- Younus, H.A.; al-Rashida, M.; Hameed, A.; Uroos, M.; Salar, U.; Rana, S.; Khan, K.M. Multicomponent reactions (MCR) in medicinal chemistry: A patent review (2010–2020). Expert Opin. Ther. Pat. 2021, 31, 267–289. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Galvis, C.E.P. Strecker reaction and α-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 2018, 74, 773–810. [Google Scholar] [CrossRef]
- Menéndez, J.C.; González-Trigo, G.; Söllhuber, M.M. The application of ultrasound to the Strecker synthesis on 9, 10-dimethoxy-1, 3, 4, 6, 7, 11b-hexahydrobenzo[a]quinolizin-2-one. Tetrahedron Lett. 1996, 27, 3285–3288. [Google Scholar] [CrossRef]
- Hernández, J.G.; Turberg, M.; Schiffers, I.; Bolm, C. Mechanochemical Strecker reaction: Access to α-aminonitriles and tetrahydroisoquinolines under ball-milling conditions. Chem. Eur. J. 2016, 22, 14513–14517. [Google Scholar] [CrossRef]
- Li, J.J. Name Reactions for Functional Group Transformations; Wiley: Hoboken, NJ, USA, 2007; pp. 477–478. [Google Scholar]
- Feldman, P.L.; Brackeen, M.F. A novel route to the 4-anilido-4-(methoxycarbonyl)piperidine class of analgetics. J. Org. Chem. 1990, 55, 4207–4209. [Google Scholar] [CrossRef]
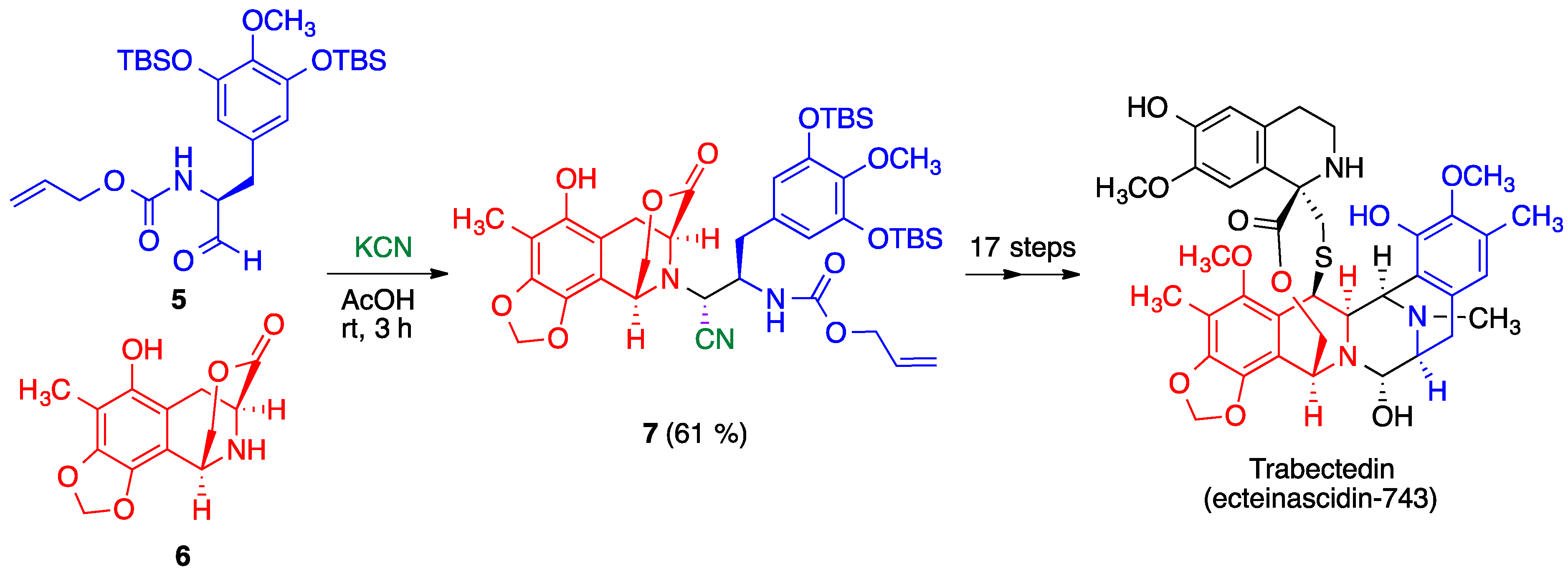
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
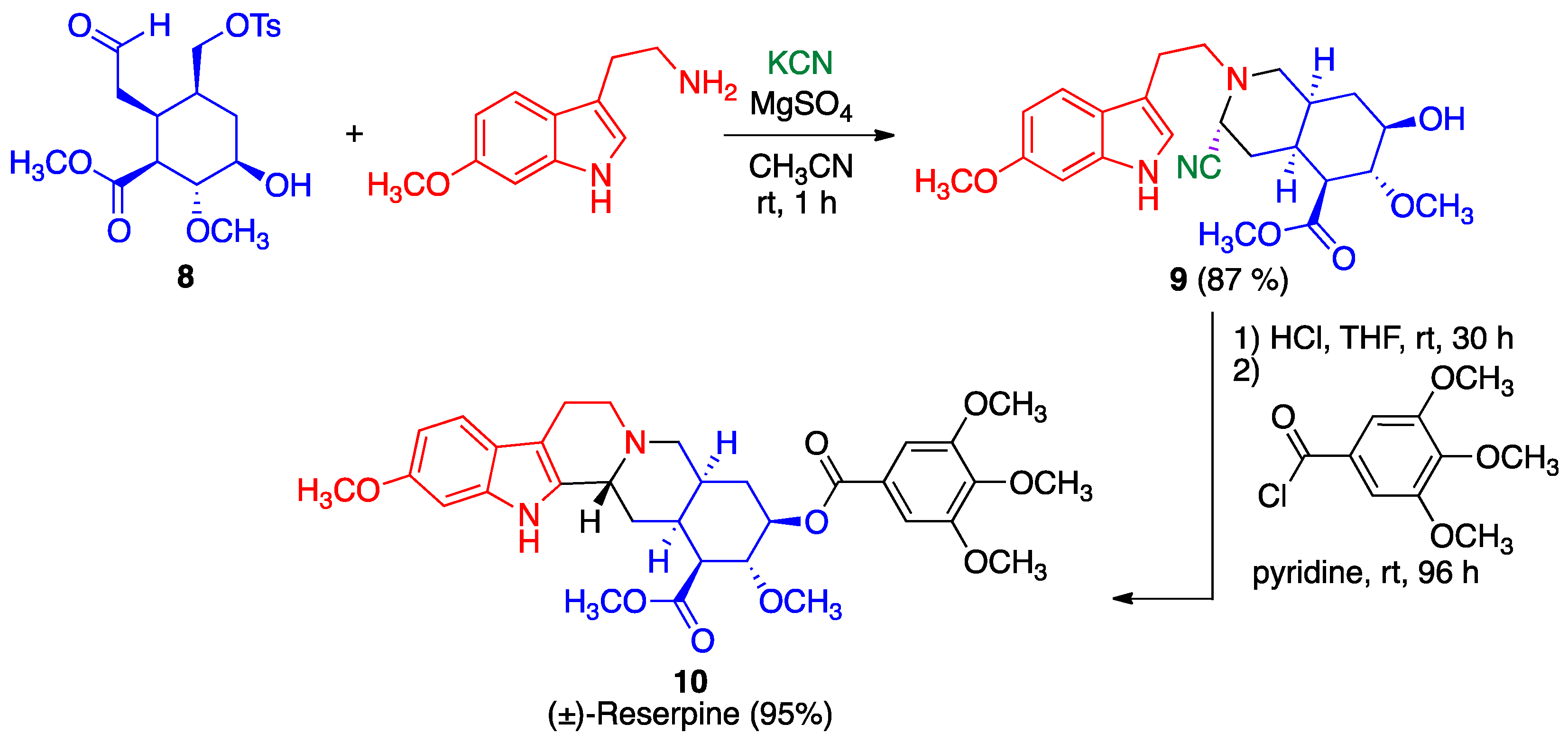
- Stork, G.; Tang, P.C.; Casey, M.; Goodman, B.; Toyota, M. Regiospecific and stereoselective syntheses of (±)-reserpine and (−)-reserpine. J. Am. Chem. Soc. 2005, 127, 16255–16262. [Google Scholar] [CrossRef] [PubMed]
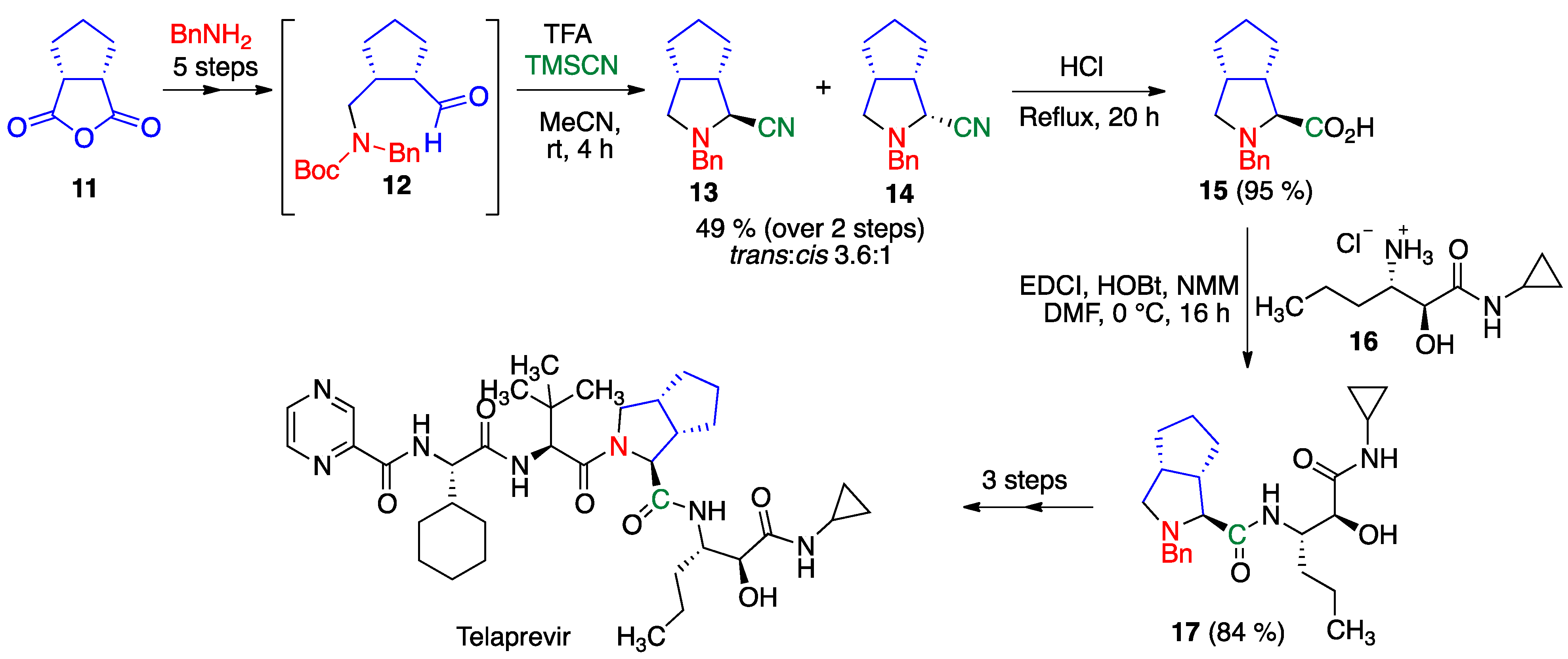
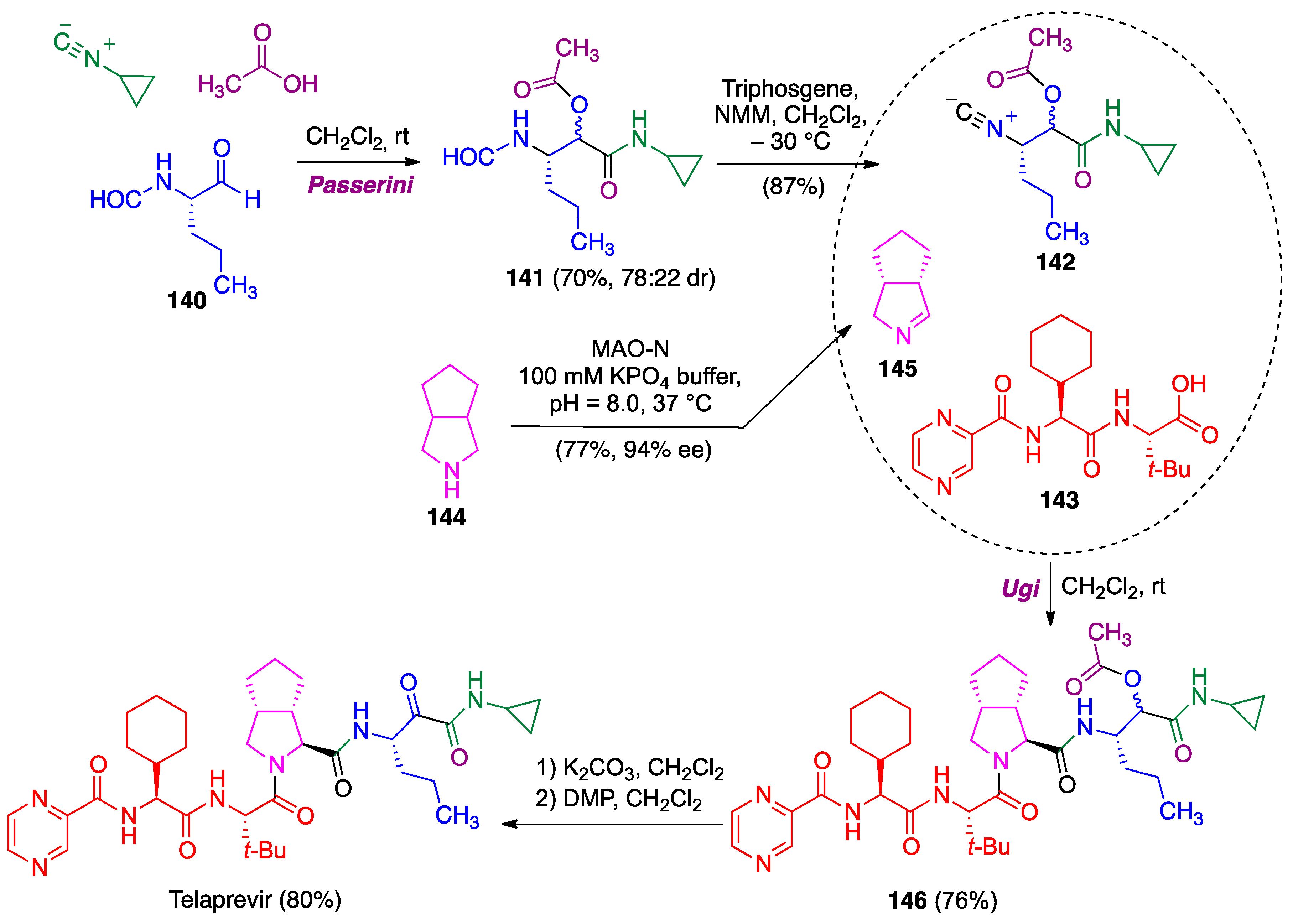
- Zhang, F.; Wen, X.; Xu, Q.L.; Sun, H. Asymmetric synthesis of 3, 4-disubstituted proline derivatives: Application in synthesis of hepatitis C virus protease inhibitor telaprevir. Eur. J. Org. Chem. 2014, 2014, 8101–8109. [Google Scholar] [CrossRef]
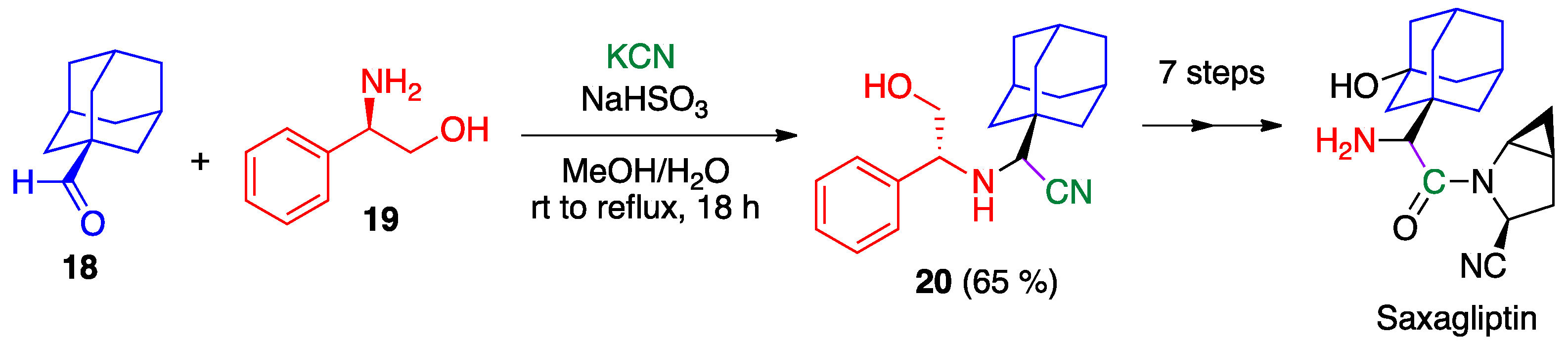
- Augeri, D.J.; Robl, J.A.; Betebenner, D.A.; Magnin, D.R.; Khanna, A.; Robertson, J.G.; Wang, A.; Simpkins, L.M.; Taunk, P.; Huang, Q.; et al. Discovery and preclinical profile of saxagliptin (BMS-477118): A highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 5025–5037. [Google Scholar] [CrossRef]
- Sadhukhan, A.; Saravanan, S.; Khan, N.U.H.; Kureshy, R.I.; Abdi, S.H.; Bajaj, H.C. Modified asymmetric Strecker reaction of aldehyde with secondary amine: A protocol for the synthesis of S-clopidogrel (an antiplatelet agent). J. Org. Chem. 2012, 77, 7076–7080. [Google Scholar] [CrossRef]
- Bousquet, A.; Calet, S.; Heymes, A. 2-Thienylglycidic Derivative, Process for Its Preparation and Its Use as Synthesis Intermediate. U.S. Patent 5,132,435, 21 July 1992. [Google Scholar]
- Bousquet, A.; Musolino, A. Hydroxyacetic ester Derivatives, Preparation Method and Use as Synthesis Intermediates. U.S. Patent 6,573,381B1, 3 June 2003. [Google Scholar]
- Wang, L.; Shen, J.; Tang, Y.; Chen, Y.; Wang, W.; Cai, Z.; Du, Z. Synthetic improvements in the preparation of clopidogrel. Org. Proc. Res. Devel. 2007, 11, 487–489. [Google Scholar] [CrossRef]
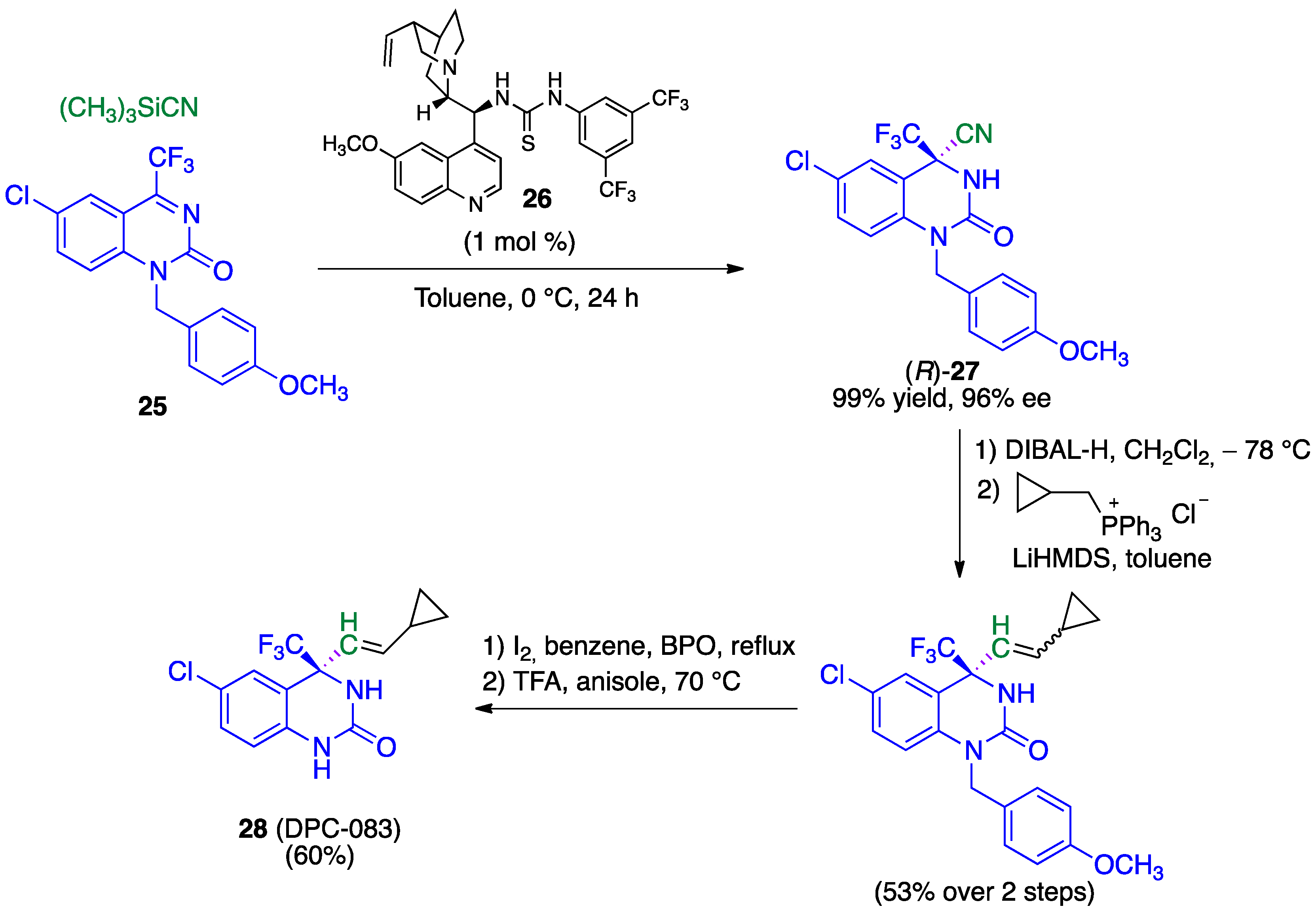
- Zhang, F.G.; Zhu, X.Y.; Li, S.; Nie, J.; Ma, J.A. Highly enantioselective organocatalytic Strecker reaction of cyclic N-acyl trifluoromethylketimines: Synthesis of anti-HIV drug DPC 083. Chem. Commun. 2012, 48, 11552–11554. [Google Scholar] [CrossRef]
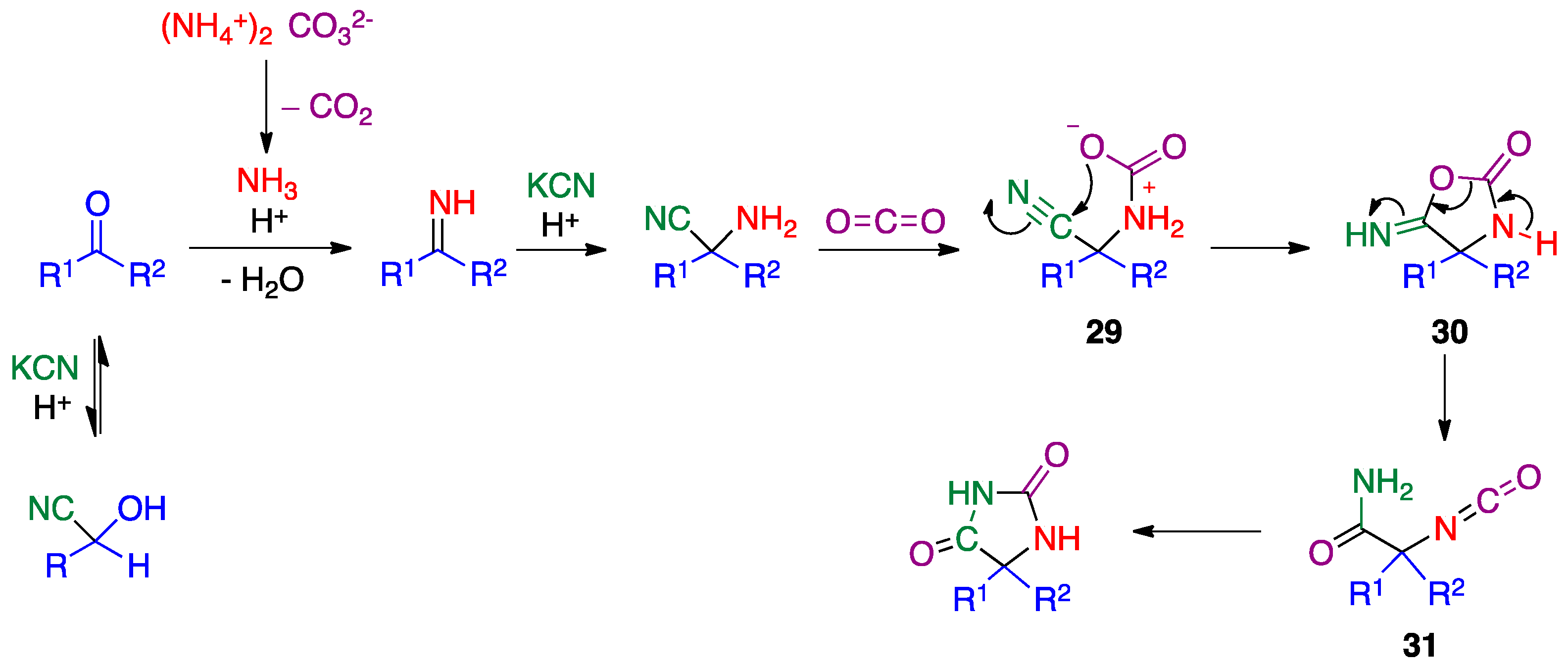
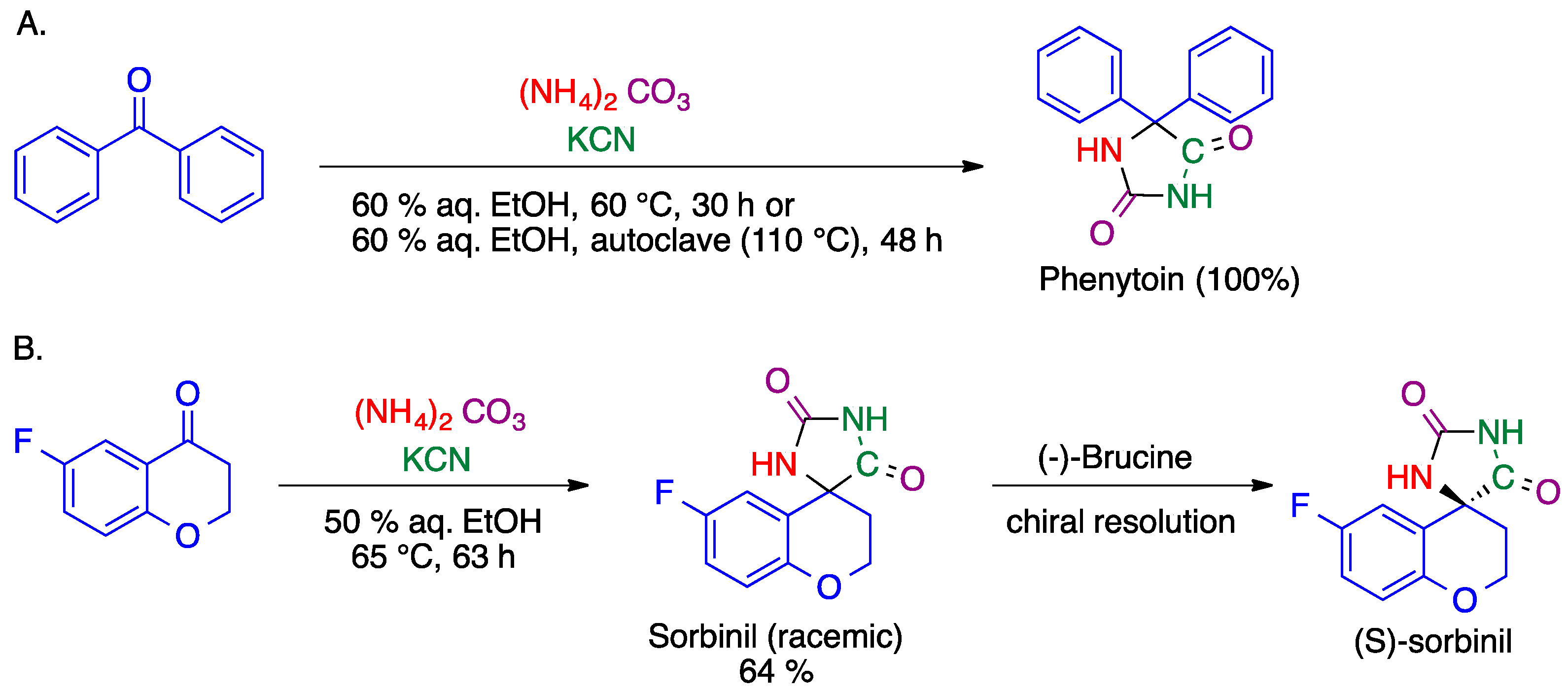
- Kalník, M.; Gabko, P.; Bella, M.; Koóš, M. The Bucherer–Bergs multicomponent synthesis of hydantoins—excellence in simplicity. Molecules 2021, 26, 4024. [Google Scholar] [CrossRef]
- Bergs, H. Verfahren zur Darstellung von Hydantoinen. German Patent DE566094, 14 December 1932. [Google Scholar]
- Bucherer, H.T.; Lieb, V.A. Syntheses of hydantoins. II. Formation of substituted hydantoins from aldehydes and ketones. J. Prakt. Chem. 1934, 141, 5–43. [Google Scholar] [CrossRef]
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1905, 38, 1671–1675. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Recent advances in the synthesis of hydantoins: The state of the art of a valuable scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar]
- Li, J.; Li, L.; Li, T.; Li, H.; Liu, J. An efficient and convenient procedure for the synthesis of 5,5-disubstituted hydantoins under ultrasound. Ultrason. Sonochem. 1996, 3, S141–S143. [Google Scholar] [CrossRef]
- Hoyer, H.L. Über das camphan-2-spiro-hydantoin. Chem. Ber. 1950, 83, 491–500. [Google Scholar] [CrossRef]
- Monteiro, J.L.; Pieber, B.; Corrêa, A.G.; Kappe, C.O. Continuous synthesis of hydantoins: Intensifying the Bucherer–Bergs reaction. Synlett 2016, 27, 83–87. [Google Scholar]
- Maddah, B. Highly efficient and rapid synthesis of diverse hydantoin derivatives using nano-ordered ZnO catalyst under mechanochemical ball milling. Iran. Chem. Commun. 2017, 5, 58–66. [Google Scholar]
- Henze, H.R. Method for Obtaining Hydantoins. U.S. Patent 2,409,754, 22 October 1946. [Google Scholar]
- Sarges, R. Hydantoin Therapeutic Agents. U.S. Patent 4,130,714, 19 December 1978. [Google Scholar]
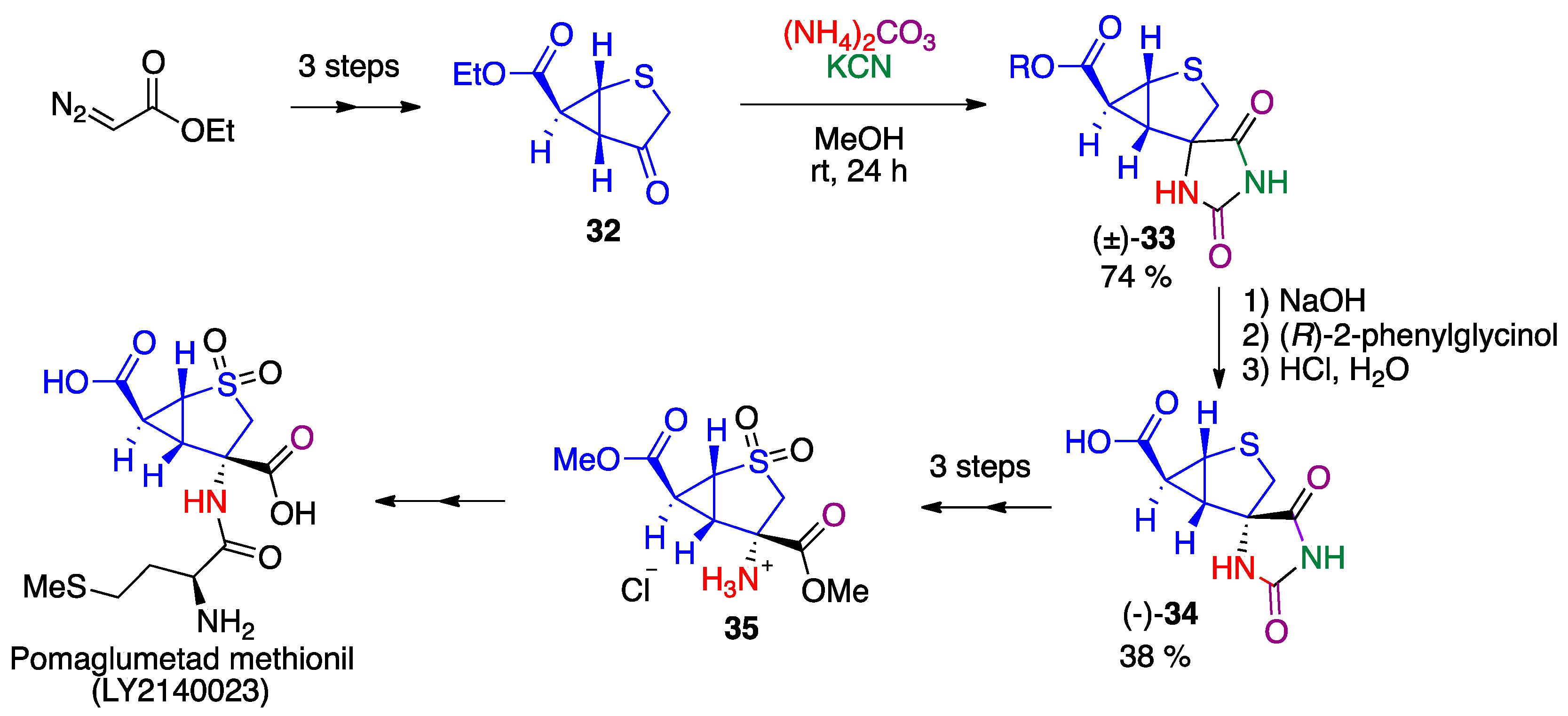
- Sonnenschein, S.F.; Grace, A.A. The mGluR2/3 agonist pomaglumetad methionil normalizes aberrant dopamine neuron activity via action in the ventral hippocampus. Neuropsychopharmacology 2020, 45, 2106–2113. [Google Scholar] [CrossRef]
- Waser, M.; Moher, E.D.; Borders, S.S.; Hansen, M.M.; Hoard, D.W.; Laurila, M.E.; LeTourneau, M.E.; Miller, R.D.; Phillips, M.L.; Sullivan, K.A.; et al. Process development for a key synthetic intermediate of LY2140023, a clinical candidate for the treatment of schizophrenia. Org. Process Res. Dev. 2011, 15, 1266–1274. [Google Scholar] [CrossRef]
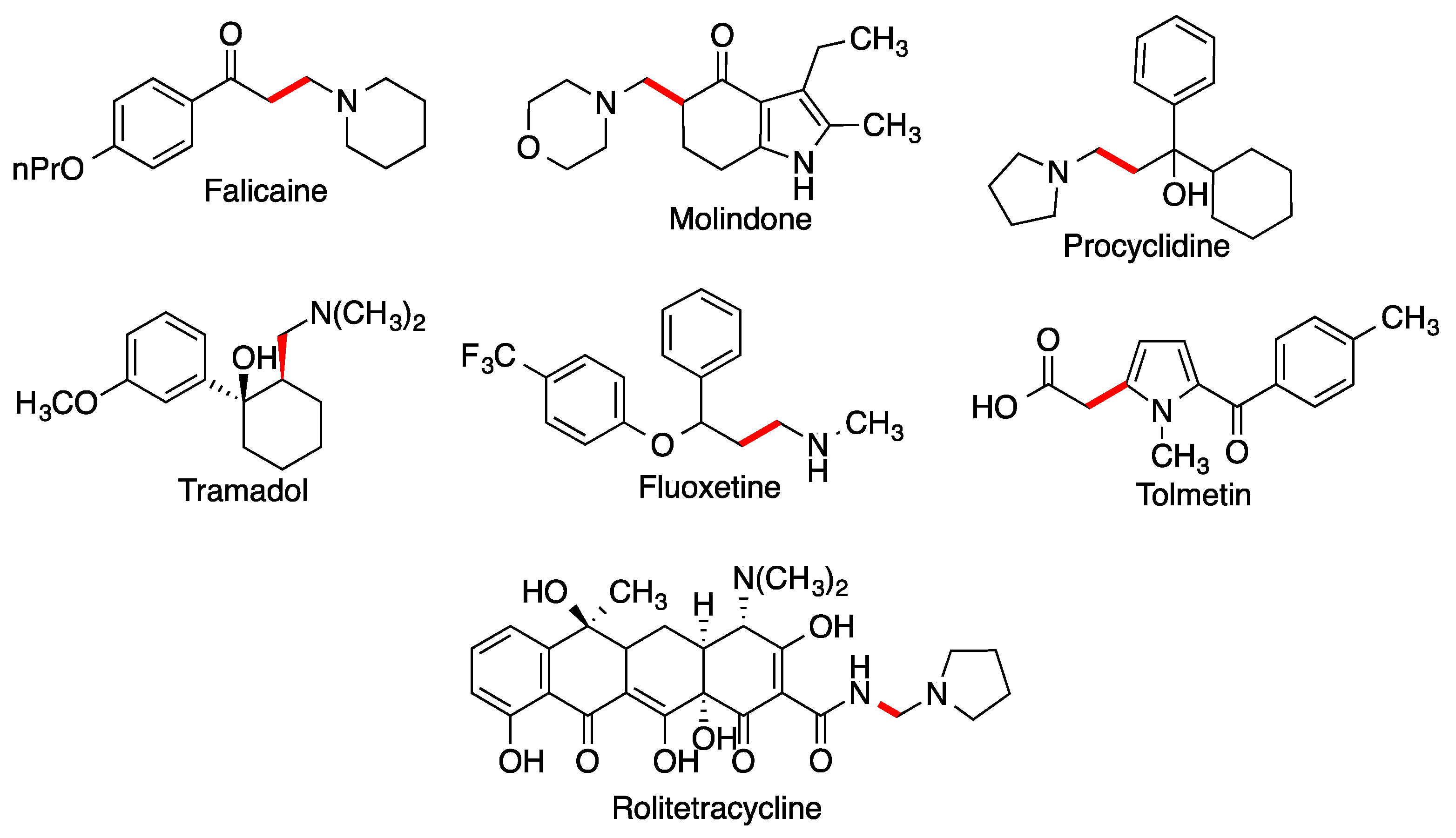
- Allochio Filho, J.F.; Lemos, B.C.; de Souza, A.S.; Pinheiro, S.; Greco, S.J. Multicomponent Mannich reactions: General aspects, methodologies and applications. Tetrahedron 2017, 73, 6977–7004. [Google Scholar] [CrossRef]
- Córdova, A. The direct catalytic asymmetric Mannich reaction. Acc. Chem. Res. 2004, 37, 102–112. [Google Scholar] [CrossRef]
- Heravi, M.; Zadsirjan, V.; Bonzorgpour Savadjani, Z. Applications of Mannich reaction in total syntheses of natural products. Curr. Org. Chem. 2014, 28, 2857–2891. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Q.; Gao, S. Recent advances in the intramolecular Mannich reaction in natural products total synthesis. Org. Chem. Front. 2018, 5, 1049–1066. [Google Scholar] [CrossRef]
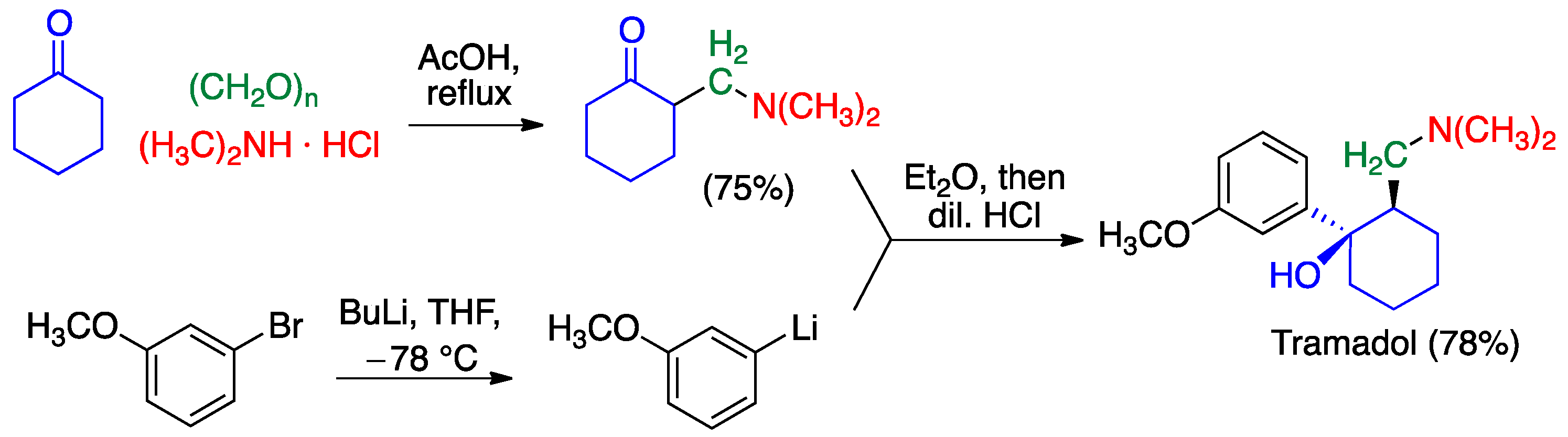
- Alvarado, C.; Guzmán, A.; Díaz, E.; Patiño, R. Synthesis of tramadol and analogs. J. Mex. Chem. Soc. 2005, 49, 324–327. [Google Scholar]
- Monos, T.M.; Jaworski, J.N.; Stephens, J.C.; Jamison, T.F. Continuous-flow synthesis of tramadol from cyclohexanone. Synlett 2020, 31, 1888–1893. [Google Scholar]
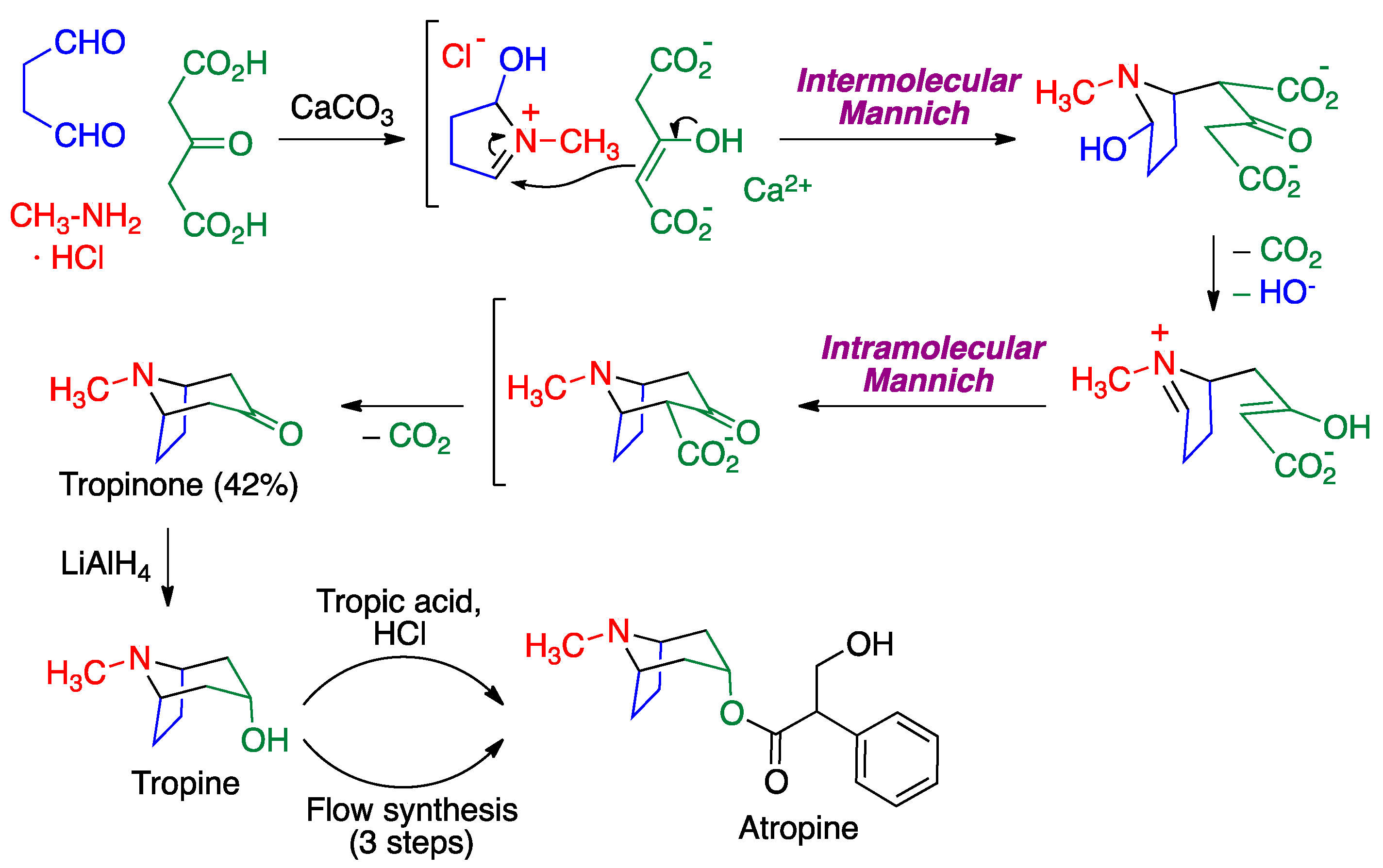
- Medley, J.W.; Movassaghi, M. Robinson’s landmark synthesis of tropinone. Chem. Commun. 2013, 49, 10775–10777. [Google Scholar] [CrossRef]
- Gore, V.; Joshi, R.; Tripathi, A.; Jadhav, M.; Bhandari, S. Crystalline Atropine Sulfate. WO 2014/102829 Al, 3 July 2020. [Google Scholar]
- Dai, C.; Snead, D.R.; Zhang, P.; Jamison, T.F. Continuous-flow synthesis and purification of atropine with sequential in-line separations of structurally similar impurities. J. Flow Chem. 2015, 5, 133–138. [Google Scholar] [CrossRef]
- Rosenblum, S.B.; Huynh, T.; Afonso, A.; Davis, H.R.; Yumibe, N.; Clader, J.W.; Burnett, D.A. Discovery of 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone (SCH 58235): A designed, potent, orally active inhibitor of cholesterol absorption. J. Med. Chem. 1998, 41, 973–980. [Google Scholar] [CrossRef]
- Minuto, F.; Lambruschini, C.; Basso, A. Ketene 3-component Staudinger reaction (K-3CSR) to β-lactams: A new entry in the class of photoinduced multicomponent reactions. Eur. J. Org. Chem. 2021, 2021, 3270–3273. [Google Scholar] [CrossRef]
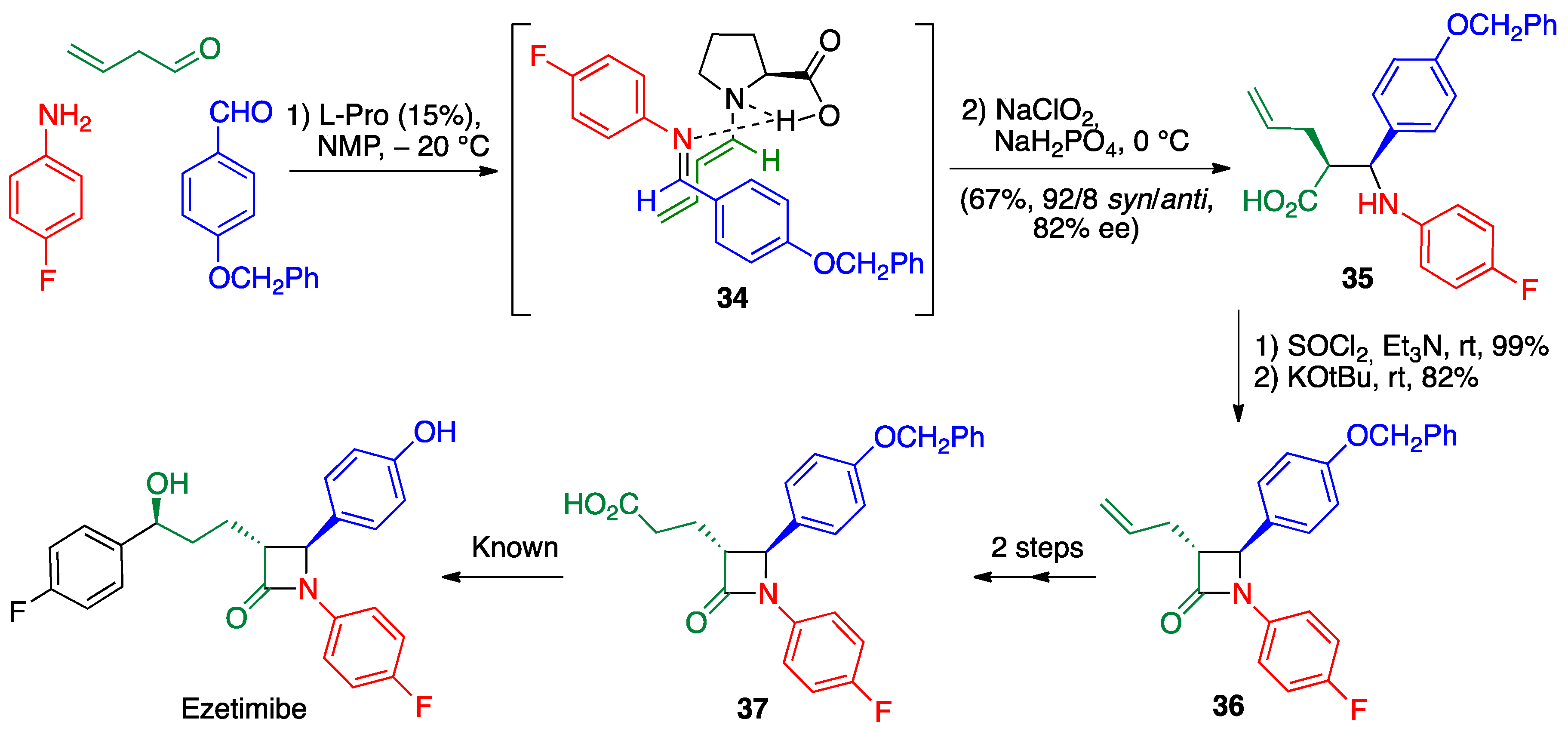
- Shimasaki, Y.; Koshino, S.; Hayashi, Y. Formal synthesis of ezetimibe using a proline-mediated, asymmetric, three-component Mannich reaction. Chem. Lett. 2016, 45, 30–32. [Google Scholar] [CrossRef]
- Bae, H.Y.; Kim, M.J.; Sim, J.H.; Song, C.E. Direct catalytic asymmetric reaction with dithiomalonates as excellent Mannich donors: Organocatalytic synthesis of (R)-sitagliptin. Angew. Chem. Int. Ed. 2016, 55, 10825–10829. [Google Scholar] [CrossRef]
- Wu, P.; Givskov, M.; Nielsen, T.E. Reactivity and synthetic applications of multicomponent Petasis reactions. Chem. Rev. 2019, 119, 11245–11290. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.Y.; Bataglion, G.A.; Ferreira, D.A.C.; Gatto, C.C.; Eberlin, M.N.; Neto, B.A.D. Insights on Petasis borono-Mannich reaction multicomponent reaction mechanism. RSC Adv. 2015, 5, 76337–76341. [Google Scholar] [CrossRef]
- Kalinski, C.; Lemoine, H.; Schmidt, J.; Burdack, C.; Kolb, J.; Umkehrer, M.; Ross, G. Multicomponent reactions as a powerful tool for generic drug synthesis. Synthesis 2008, 24, 4007–4011. [Google Scholar] [CrossRef]
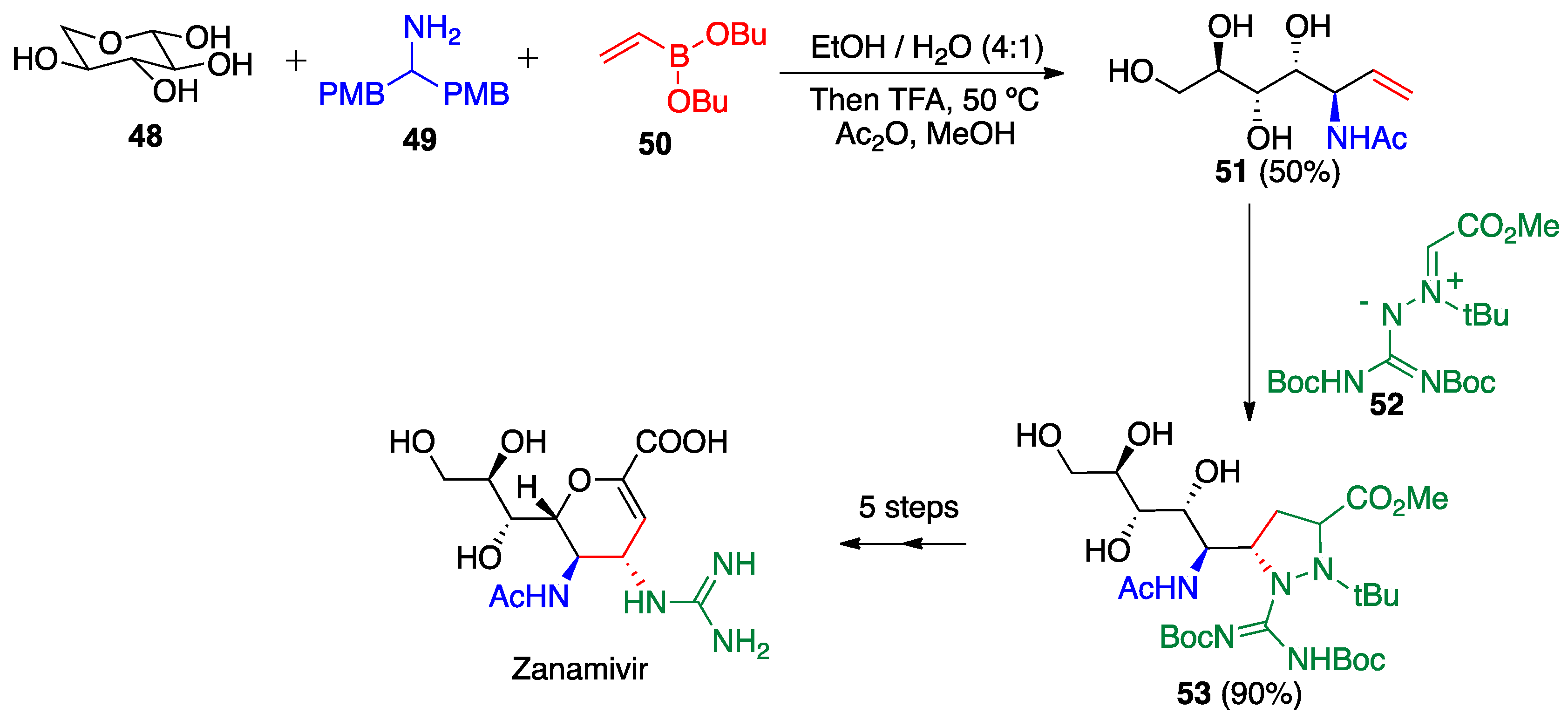
- Zhangyong, H.; Zeming, C.; Kui, Y.; Jin, W. Novel Method for Synthesis of Anti-Influenza Medicament Zanamivir. Chinese Patent CN104744415A, 26 December 2013. [Google Scholar]
- Lemos, B.C.; Filho, E.V.; Fiorot, R.G.; Medici, F.; Greco, S.J.; Benaglia, M. Enantioselective Povarov reactions: An update of a powerful catalytic synthetic methodology. Eur. J. Org. Chem. 2022, 2022, e202101171. [Google Scholar] [CrossRef]
- Clerigué, J.; Ramos, M.T.; Menéndez, J.C. Enantioselective catalytic Povarov reactions. Org. Biomol. Chem. 2022, 20, 1550–1581. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Sáez, J.A.; Mekelleche, S.M. Understanding the mechanism of the Povarov reaction. A DFT study. RSC Adv. 2014, 4, 25268–25278. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Layeb, H.; Domingo, L.R. A DFT study of the mechanism of Brønsted acid catalysed Povarov reactions. Tetrahedron 2015, 71, 9339–9345. [Google Scholar] [CrossRef]
- Mahi, M.A.; Mekelleche, S.M.; Benchouk, W.; Aurell, M.J.; Domingo, L.R. Theoretical study of the regio-and stereoselectivity of the intramolecular Povarov reactions yielding 5H-chromeno [2,3-c] acridine derivatives. RSC Adv. 2016, 6, 15759–15769. [Google Scholar] [CrossRef]
- Isambert, N.; Cruz, M.; Arévalo, M.J.; Gómez, E.; Lavilla, R. Enol esters: Versatile substrates for Mannich-type multicomponent reactions. Org. Lett. 2007, 9, 4199–4202. [Google Scholar] [CrossRef]
- Sridharan, V.; Avendaño, C.; Menéndez, J.C. New findings on the cerium(IV) ammonium nitrate catalyzed Povarov reaction: Stereoselective synthesis of 4-alkoxy-2-aryl-1,2,3,4-tetrahydroquinoline derivatives. Synthesis 2008, 2008, 1039–1044. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; Arrieta, A.; Cossío, F.P.; Ezpeleta, J.M.; Fuertes, M.; Rubiales, G. Lewis acid activated aza-Diels–Alder reaction of N-(3-pyridyl) aldimines: An experimental and computational study. Eur. J. Org. Chem. 2010, 11, 2091–2099. [Google Scholar] [CrossRef]
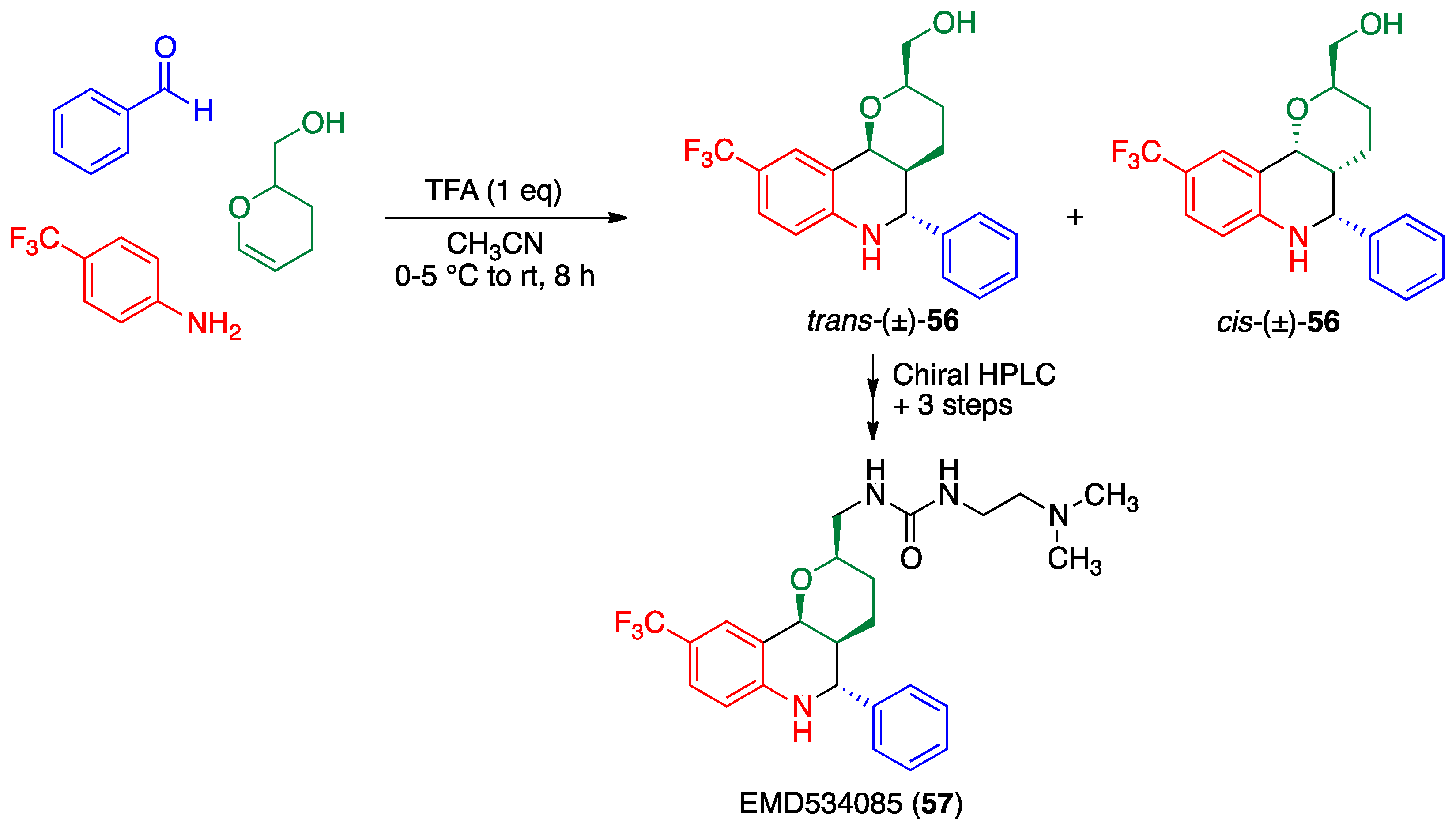
- Schiemann, K.; Finsinger, D.; Zenke, F.; Amendt, C.; Knöchel, T.; Bruge, D.; Buchstaller, H.P.; Emde, U.; Stähle, W.; Anzali, S. The discovery and optimization of hexahydro-2H-pyrano [3,2-c] quinolines (HHPQs) as potent and selective inhibitors of the mitotic kinesin-5. Bioorg. Med. Chem. Lett. 2010, 20, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Cerfontaine, P.; Driessens, F.; Deltent, M.-F.; Petit, S. Efficient multikilogram synthesis of a VLA-4 antagonist via a Povarov reaction. Org. Process Res. Dev. 2020, 24, 807–816. [Google Scholar] [CrossRef]
- Liu, H.; Dagousset, G.; Masson, G.; Retailleau, P.; Zhu, J. Chiral Brønsted acid-catalyzed enantioselective three-component Povarov reaction. J. Am. Chem. Soc. 2009, 131, 4598–4599. [Google Scholar] [CrossRef] [PubMed]
- Dagousset, G.; Zhu, J.; Masson, G. Chiral phosphoric acid-catalyzed enantioselective three-component Povarov reaction using enecarbamates as dienophiles: Highly diastereo- and enantioselective synthesis of substituted 4-aminotetrahydroquinolines. J. Am. Chem. Soc. 2011, 133, 14804–14813. [Google Scholar] [CrossRef]
- Döbner, O. Ueber α-alkylcinchoninsäuren und α-alkylchinoline. Justus Liebigs Ann. Chem. 1887, 242, 265–289. [Google Scholar] [CrossRef]
- Wang, D.Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; Volume 1, pp. 921–923. [Google Scholar]
- Domling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Bossert, F.; Elberfeld, W.; Water, W. 4-Aryl-1,4-Dihydropyridines. U.S. Patent US 3,485,847, 23 December 1969. [Google Scholar]
- Yiu, S.-H.; Knaus, E.E. An improved synthesis of high-purity felodipine. Org. Prep. Proc. Int. 1996, 28, 91–95. [Google Scholar] [CrossRef]
- Leonardi, M.; Estévez, V.; Villacampa, M.; Menéndez, J.C. The Hantzsch pyrrole synthesis: Non-conventional variations and applications of a neglected classical reaction. Synthesis 2019, 51, 816–828. [Google Scholar]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Three-component access to pyrroles promoted by the CAN–silver nitrate system under high-speed vibration milling conditions: A generalization of the Hantzsch pyrrole synthesis. Chem. Commun. 2013, 49, 591–593. [Google Scholar] [CrossRef]
- Estévez, V.; Sridharan, V.; Sabaté, S.; Villacampa, M.; Menéndez, J.C. Three-component synthesis of pyrrole-related nitrogen heterocycles by a Hantzsch-type process: Comparison between conventional and high-speed vibration milling conditions. Asian J. Org. Chem. 2016, 5, 652–662. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Concise synthesis of atorvastatin lactone under high-speed vibration milling conditions. Org. Chem. Front. 2014, 1, 458–463. [Google Scholar] [CrossRef]
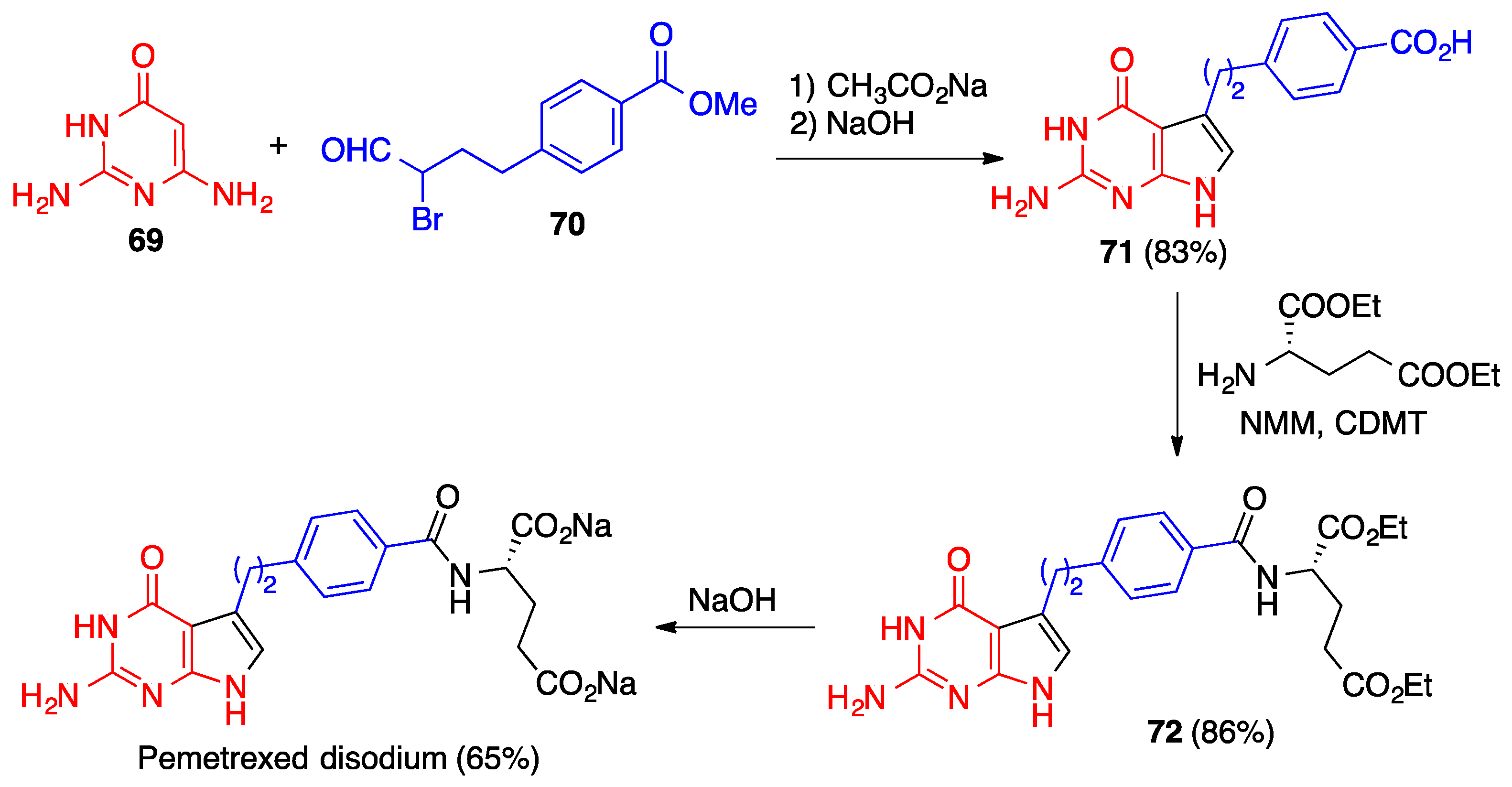
- Qi, H.; Wen, J.; Li, L.; Bai, R.; Chen, L.; Wang, D. An efficient synthesis of pemetrexed disodium. J. Heterocycl. Chem. 2015, 52, 1565–1569. [Google Scholar] [CrossRef]
- Griboura, N.; Gatzonas, K.; Neochoritis, C.G. Still relevant today: The Asinger multicomponent reaction. ChemMedChem 2021, 16, 1997–2020. [Google Scholar] [CrossRef]
- Delangle, P.; Mintz, E. Chelation therapy in Wilson’s disease: From D-penicillamine to the design of selective bioinspired intracellular Cu(I) chelators. Dalton Trans. 2012, 41, 6359–6370. [Google Scholar] [CrossRef]
- Wiegert, W.M.; Offermans, H.; Scherberich, P. D-Penicilamine–production and properties. Angew. Chem. Int. Ed. 1975, 14, 330–336. [Google Scholar] [CrossRef]
- Reguera, L.; Méndez, Y.; Humpierre, A.R.; Valdés, O.; Rivera, D.G. Multicomponent reactions in ligation and bioconjugation chemistry. Acc. Chem. Res. 2018, 51, 1475–1486. [Google Scholar] [CrossRef]
- Sornay, C.; Hessmann, S.; Erb, S.; Dovgan, I.; Ehkirch, A.; Botzanowski, T.; Cianférani, S.; Wagner, A.; Chaubet, G. Investigating Ugi/Passerini multicomponent reactions for the site-selective conjugation of native trastuzumab. Chem. Eur. J. 2020, 26, 13797–13805. [Google Scholar] [CrossRef]
- Javanbakht, S.; Shaabani, A. Multicomponent reactions-based modified/functionalized materials in the biomedical platforms. ACS Appl. Bio Mater. 2020, 3, 156–174. [Google Scholar] [CrossRef]
- Banfi, L.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R. The 100 facets of the Passerini reaction. Chem. Sci. 2021, 12, 15445–15472. [Google Scholar] [CrossRef]
- Seebach, D.; Adam, G.; Gees, T.; Schiess, M.; Weigand, W. Scope and limitations of the TiCl4-mediated additions of isocyanides to aldehydes and ketones with formation of α-hydroxycarboxylic acid amides. Chem. Ber. 1988, 121, 507–517. [Google Scholar] [CrossRef]
- Schiess, M.; Seebach, D. N-Methyl-C-(trichlortitanio)formimidoylchlorid. Ein effizientes reagenz zur homologisierung von aldehyden und ketonen zu α-hydroxy-carbonsäureamiden. Helv. Chim. Acta 1983, 66, 1618–1623. [Google Scholar] [CrossRef]
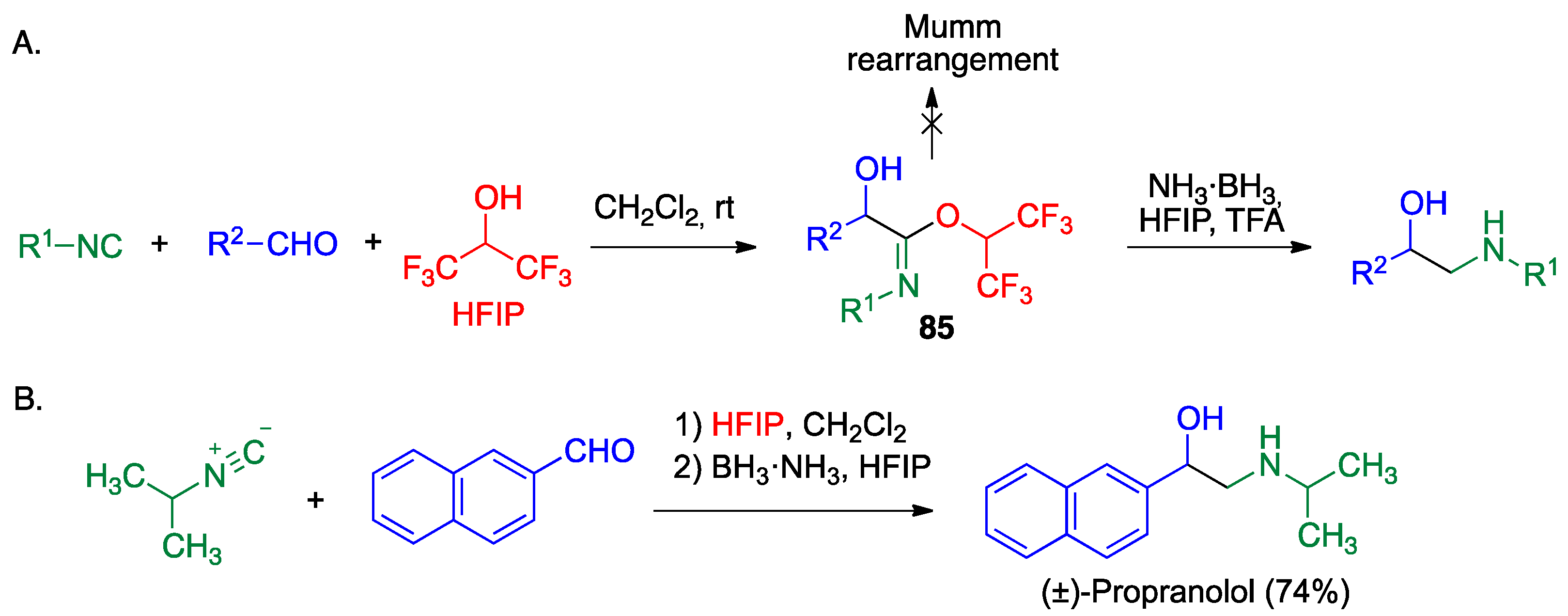
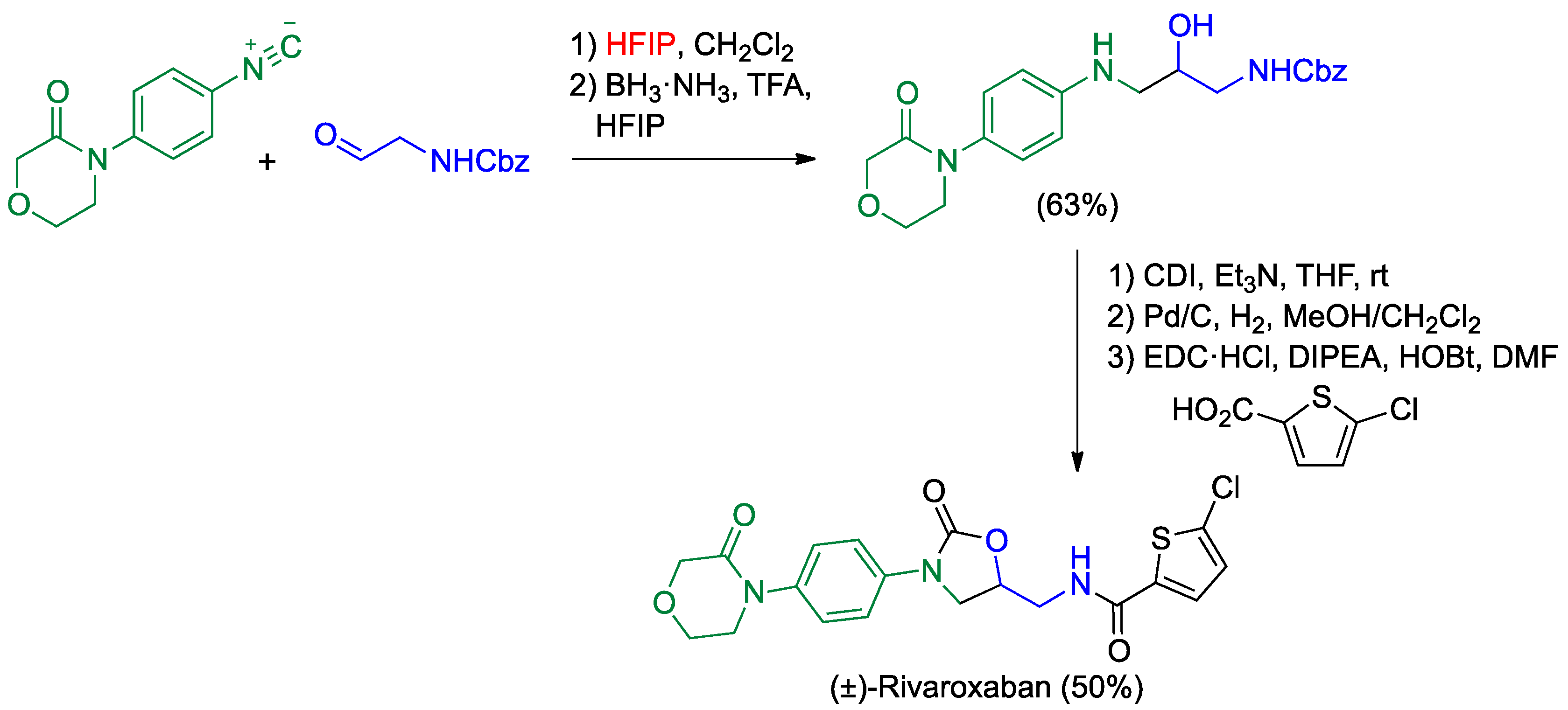
- Saya, J.M.; Berabez, R.; Broersen, P.; Schuringa, I.; Kruithof, A.; Orru, R.V.A.; Ruijter, E. Hexafluoroisopropanol as the acid component in the Passerini reaction: One-pot access to β-amino alcohols. Org. Lett. 2018, 20, 3988–3991. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Fan, Y. The first catalytic, asymmetric α-additions of isocyanides. Lewis-base-catalyzed, enantioselective Passerini-type reactions. J. Am. Chem. Soc. 2003, 125, 7825–7827. [Google Scholar] [CrossRef] [PubMed]
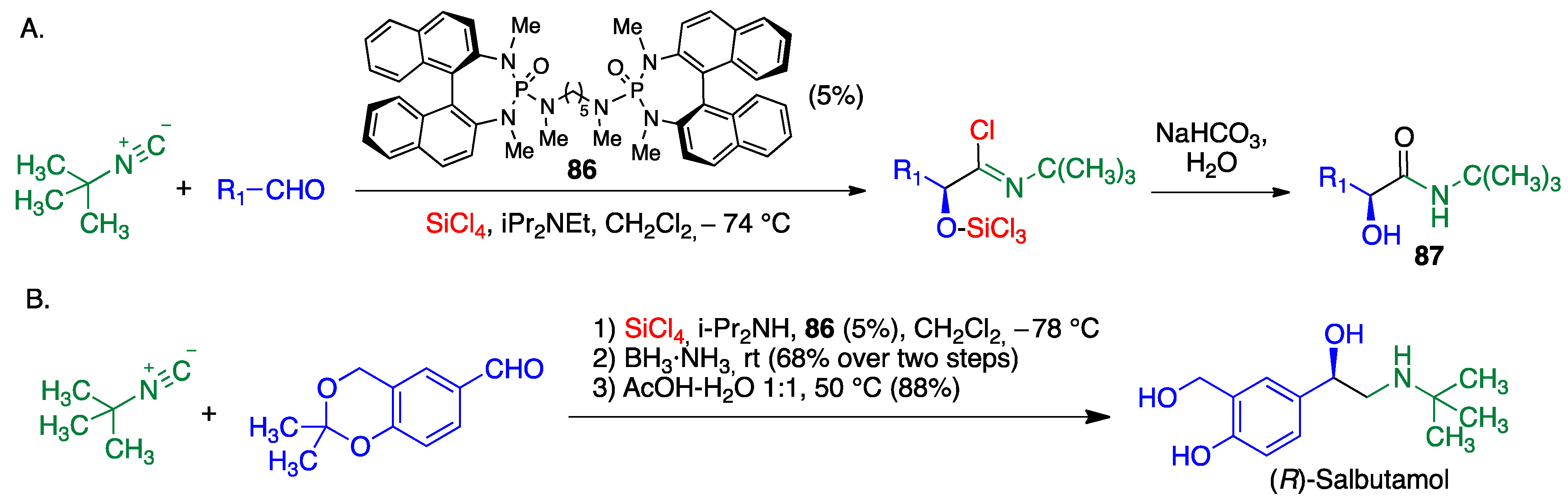
- Gernet, A.; Ratovelomanana-Vidal, V.; Pirat, J.L.; Virieux, D.; Ayad, T. Efficient synthesis of 2-amino-1-arylethanols through a Lewis base-catalyzed SiCl4-mediated asymmetric Passerini-type reaction. Eur. J. Org. Chem. 2020, 2020, 6497–6500. [Google Scholar] [CrossRef]
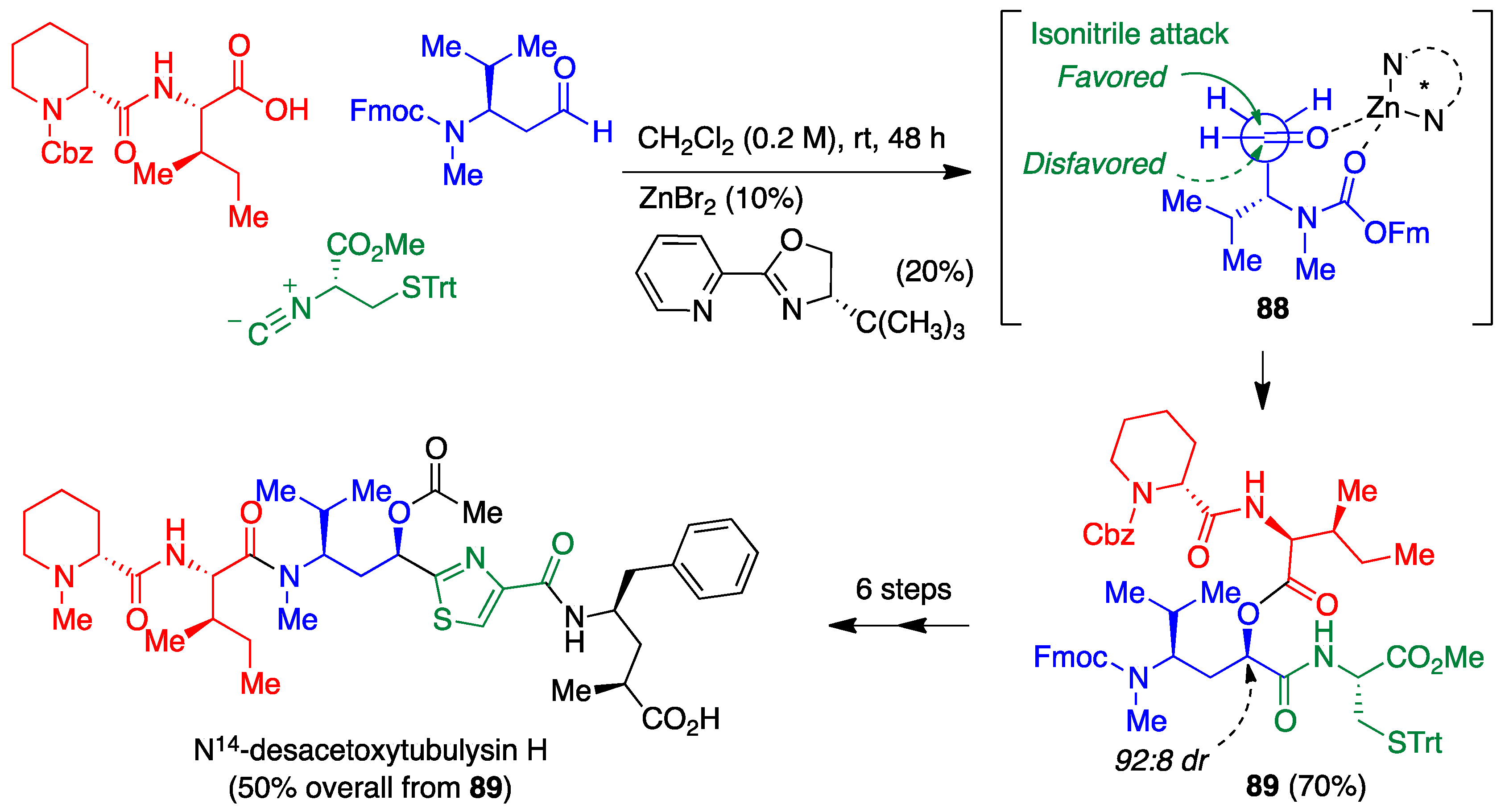
- Vishwanatha, T.M.; Giepmans, B.; Goda, S.K.; Dömling, A. Tubulysin synthesis featuring stereoselective catalysis and highly convergent multicomponent assembly. Org. Lett. 2020, 22, 5396–5400. [Google Scholar] [CrossRef]
- Hoffmann, J.; Gorges, J.; Junk, L.; and Kazmaier, U. Synthesis of pretubulysin-derivatives via the TubUgi-approach. Org. Biomol. Chem. 2015, 13, 6010–6020. [Google Scholar] [CrossRef]
- Chandgude, A.L.; Dömling, A. An efficient Passerini tetrazole reaction (PT-3CR). Green Chem. 2016, 18, 3718–3721. [Google Scholar] [CrossRef]
- Fouad, M.A.; Abdel-Hamid, H.; Ayoup, M.S. Two decades of recent advances of Ugi reactions: Synthetic and pharmaceutical applications. RSC Adv. 2020, 10, 42644–42681. [Google Scholar] [CrossRef]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- Flores-Reyes, J.C.; Islas-Jácome, A.; González-Zamora, E. The Ugi three-component reaction and its variants. Org. Chem. Front. 2021, 8, 5460–5515. [Google Scholar] [CrossRef]
- Enders, D. Industrial applications of multicomponent reactions (MCRS). In Stereoselective Multiple Bond-Forming Transformations in Organic Synthesis; Rodriguez, J., Bonne, D., Eds.; Wiley: Hoboken, NJ, USA, 2015; Chapter 6. [Google Scholar]
- Ugi, I.; Werner, B.; Dömling, A. The chemistry of isocyanides, their multicomponent reactions and their libraries. Molecules 2003, 8, 53–66. [Google Scholar] [CrossRef]
- Ugi, I.; Sternbrücker, C. Process for Preparing Amino-Carboxylic Acid Amides. U.S. Patent 3,247,200, 10 April 1966. [Google Scholar]
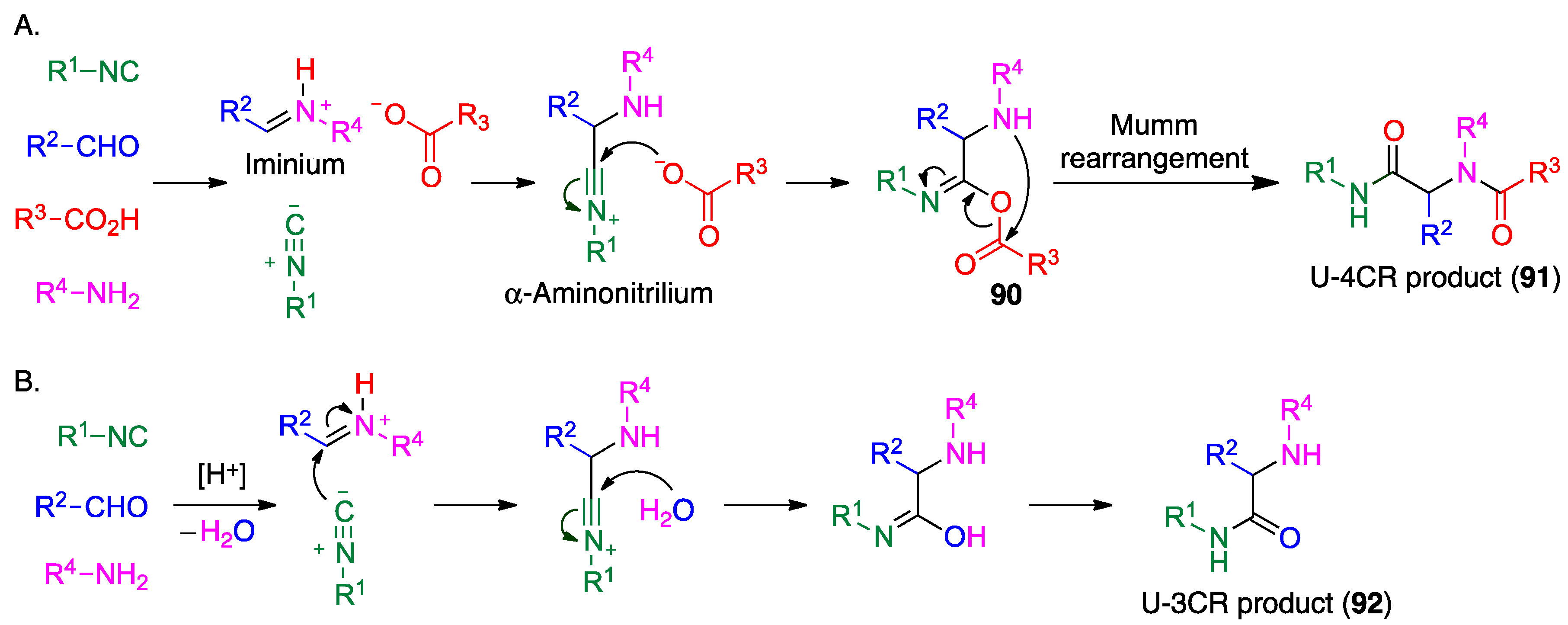
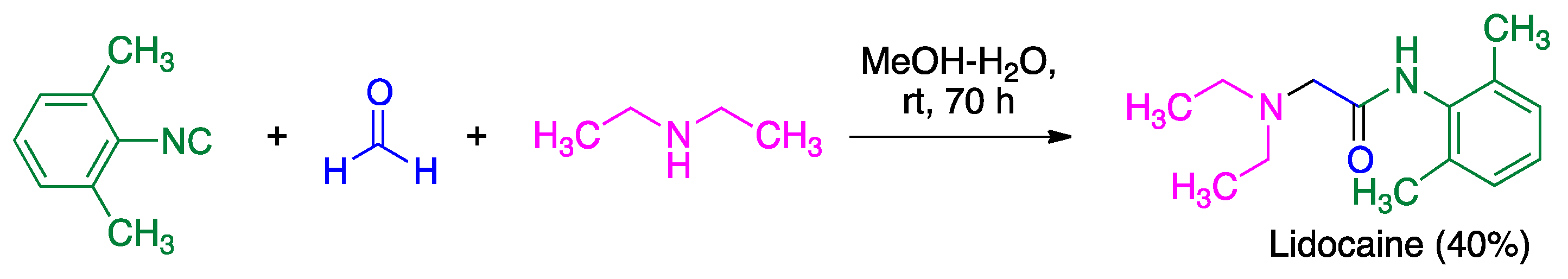
- Thomaidi, M.; Vagiaki, L.-E.; Tripolitsiotis, N.P.; Angeli, G.K.; Zarganes-Tzitzikas, T.; Sidiropoulou, K.; Neochoritis, C.G. Local anesthetics via multicomponent reactions. ChemMedChem 2022, 17, e202200246. [Google Scholar] [CrossRef] [PubMed]
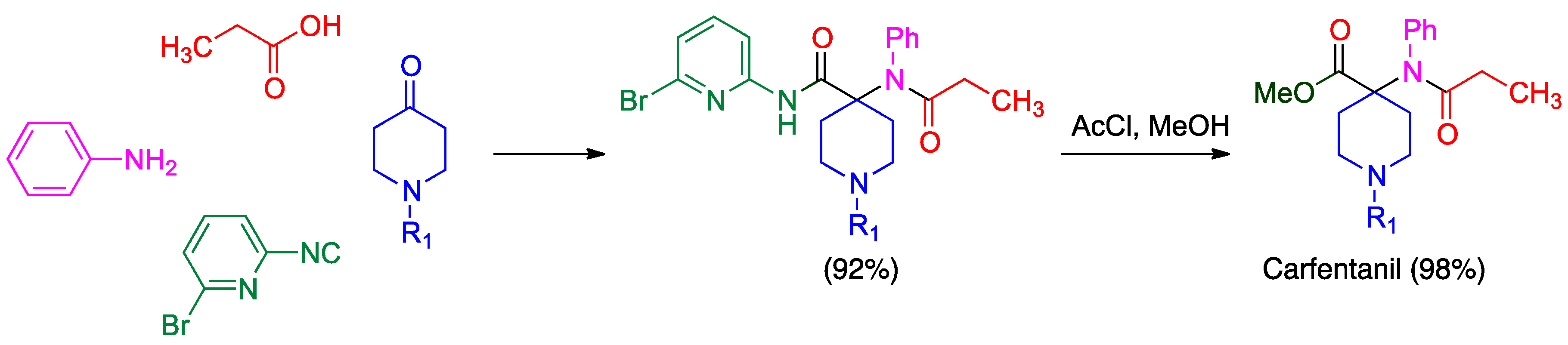
- Van der Heijden, G.; Jong, J.A.W.; Ruijter, E.; Orru, R.V.A. 2-Bromo-6-isocyanopyridine as a universal convertible isocyanide for multicomponent chemistry. Org. Lett. 2016, 18, 984–987. [Google Scholar] [CrossRef]
- Váradi, A.; Palmer, T.C.; Haselton, N.; Afonin, D.; Subrath, J.J.; Le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Marrone, G.F.; Borics, A.; et al. Synthesis of carfentanil amide opioids using the Ugi multicomponent reaction. ACS Chem. Neurosci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
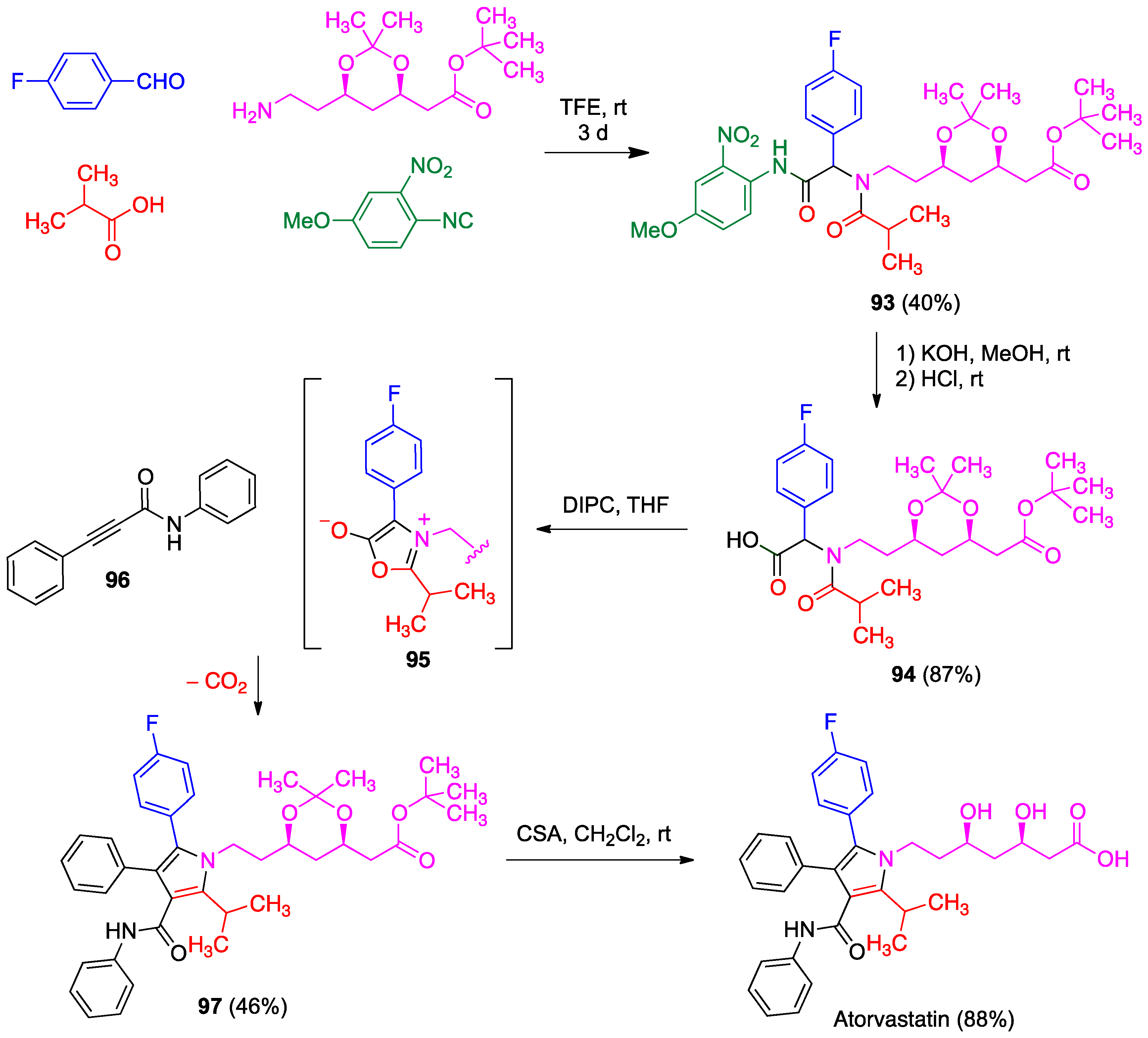
- Zarganes-Tzitzikas, T.; Neochoritis, C.G.; Dömling, A. Atorvastatin (Lipitor) by MCR. ACS Med. Chem. Lett. 2019, 10, 389–392. [Google Scholar] [CrossRef]
- Lopchuk, J.M.; Gribble, G. Total synthesis of atorvastatin via a late-stage, regioselective 1,3-dipolar münchnone cycloaddition. Tetrahedron Lett. 2015, 56, 3208–3211. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Levetiracetam: A review of its use in epilepsy. Drugs 2011, 71, 489–514. [Google Scholar]
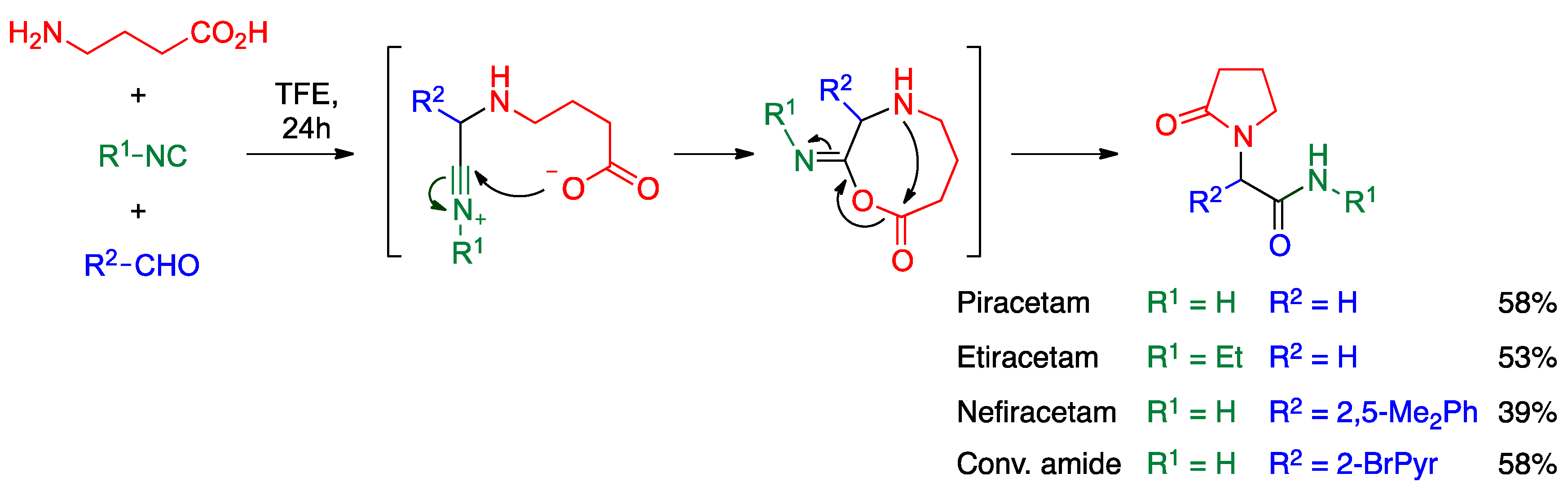
- Cioc, R.C.; van Riempst, L.S.; Schuckman, P.; Ruijter, E.; Orru, R.V. Ugi four-center three-component reaction as a direct approach to racetams. Synthesis 2017, 49, 1664–1674. [Google Scholar]
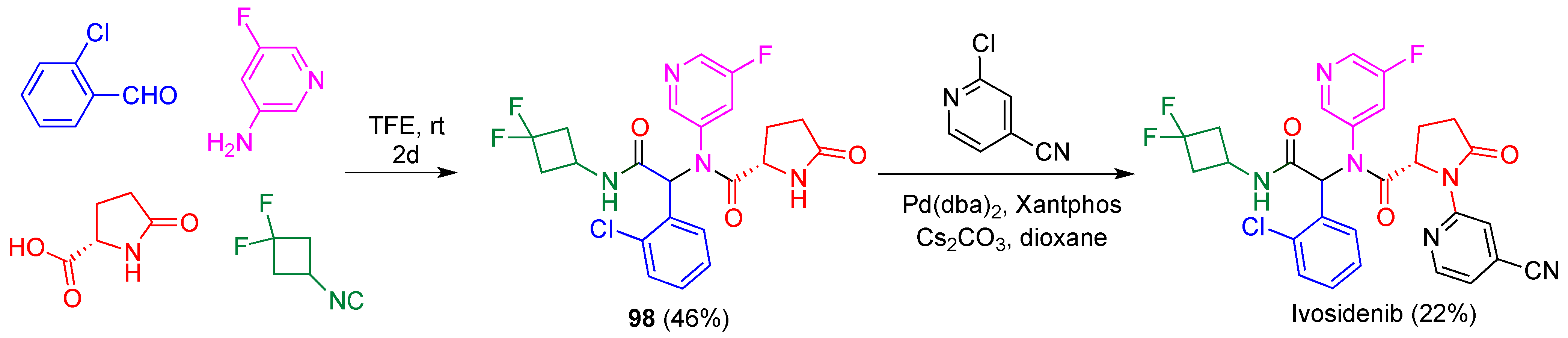
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Let. 2018, 9, 300–305. [Google Scholar] [CrossRef]
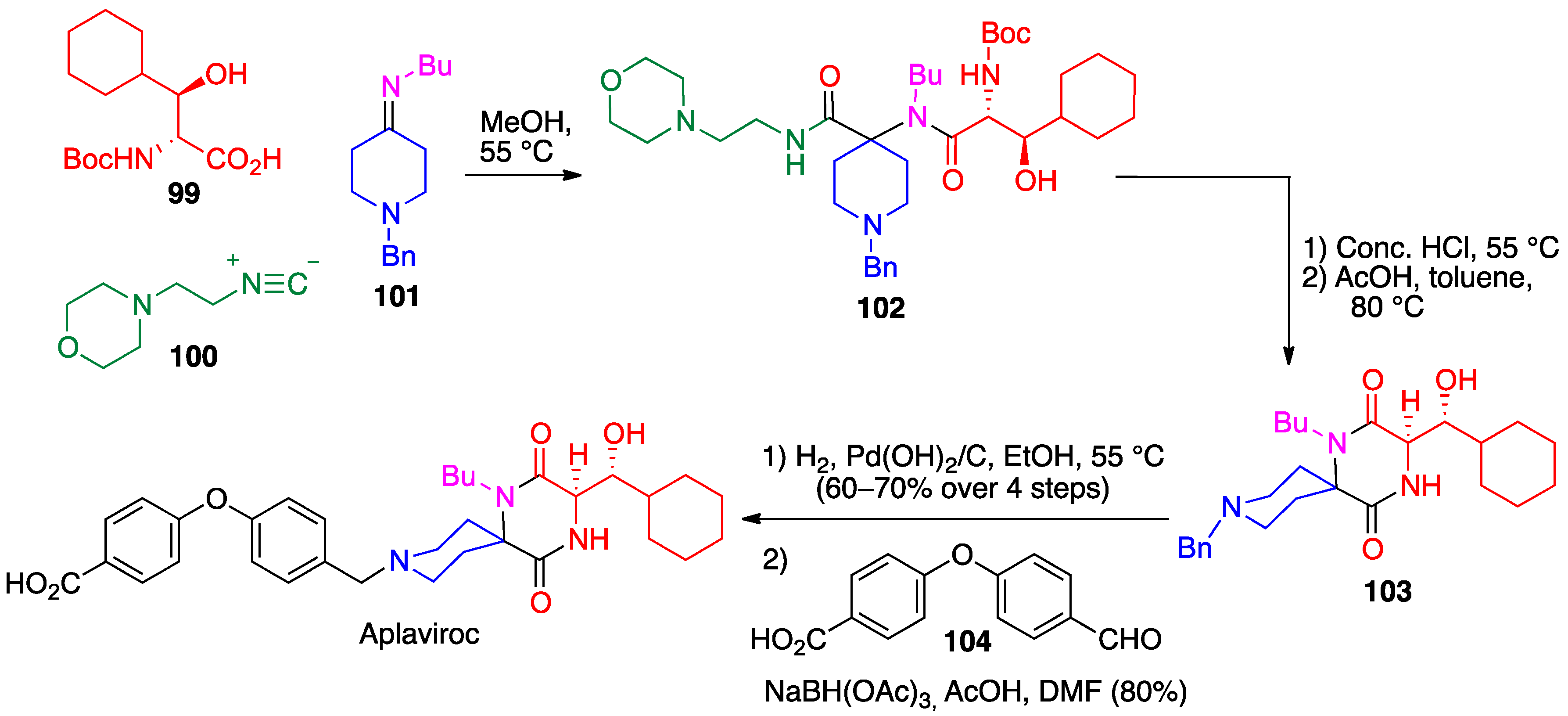
- Nishizawa, R.; Nishiyama, T.; Hisaichi, K.; Matsunaga, N.; Minamoto, C.; Habashita, H.; Takaoka, Y.; Toda, M.; Shibayama, S.; Tada, H.; et al. Spirodiketopiperazine-based CCR5 antagonists: Lead optimization from biologically active metabolite. Bioorg. Med. Chem. Lett. 2007, 17, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Sollis, S.L. Short and novel stereospecific synthesis of trisubstituted 2,5-diketopiperazines. J. Org. Chem. 2005, 70, 4735–4740. [Google Scholar] [CrossRef] [PubMed]
- Liddle, J.; Allen, M.J.; Borthwick, A.D.; Brooks, D.P.; Davies, D.E.; Edwards, R.M.; Exall, A.M.; Hamlett, C.; Irving, W.R.; Mason, A.M.; et al. The discovery of GSK221149A: A potent and selective oxytocin antagonist. Bioorg. Med. Chem. Lett. 2008, 18, 90–94. [Google Scholar] [CrossRef]
- Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. Total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 2002, 124, 6552–6554. [Google Scholar] [CrossRef]
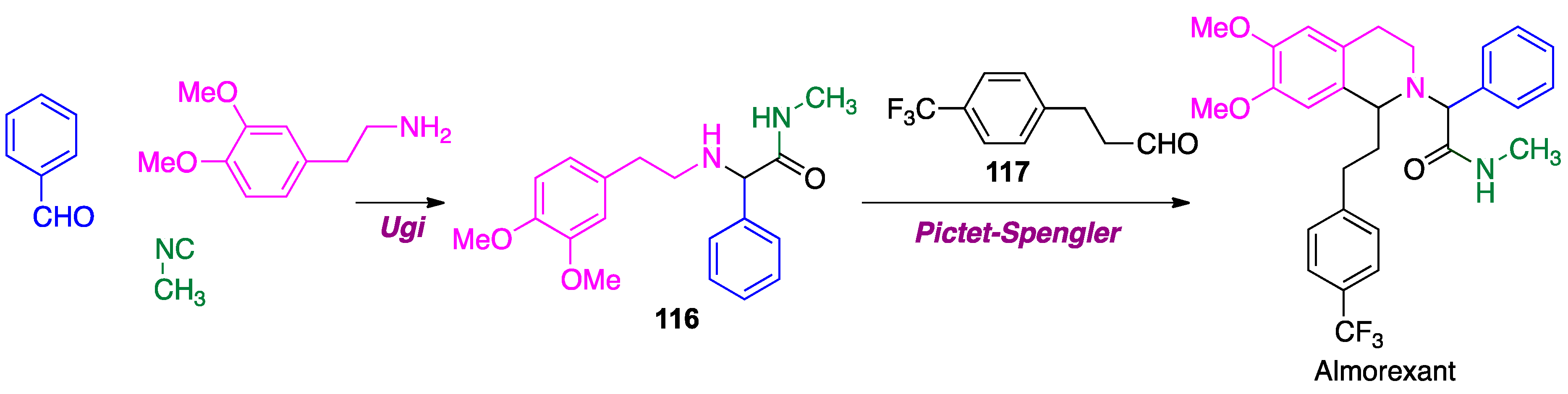
- Aissaoui, H.; Cappi, M.; Clozel, M.; Fischli, W.; Koberstein, R. 1,2,3,4-Tetrahydroisoquinoline Derivatives. US patent W0 01/68609 A1, 20 September 2001. [Google Scholar]
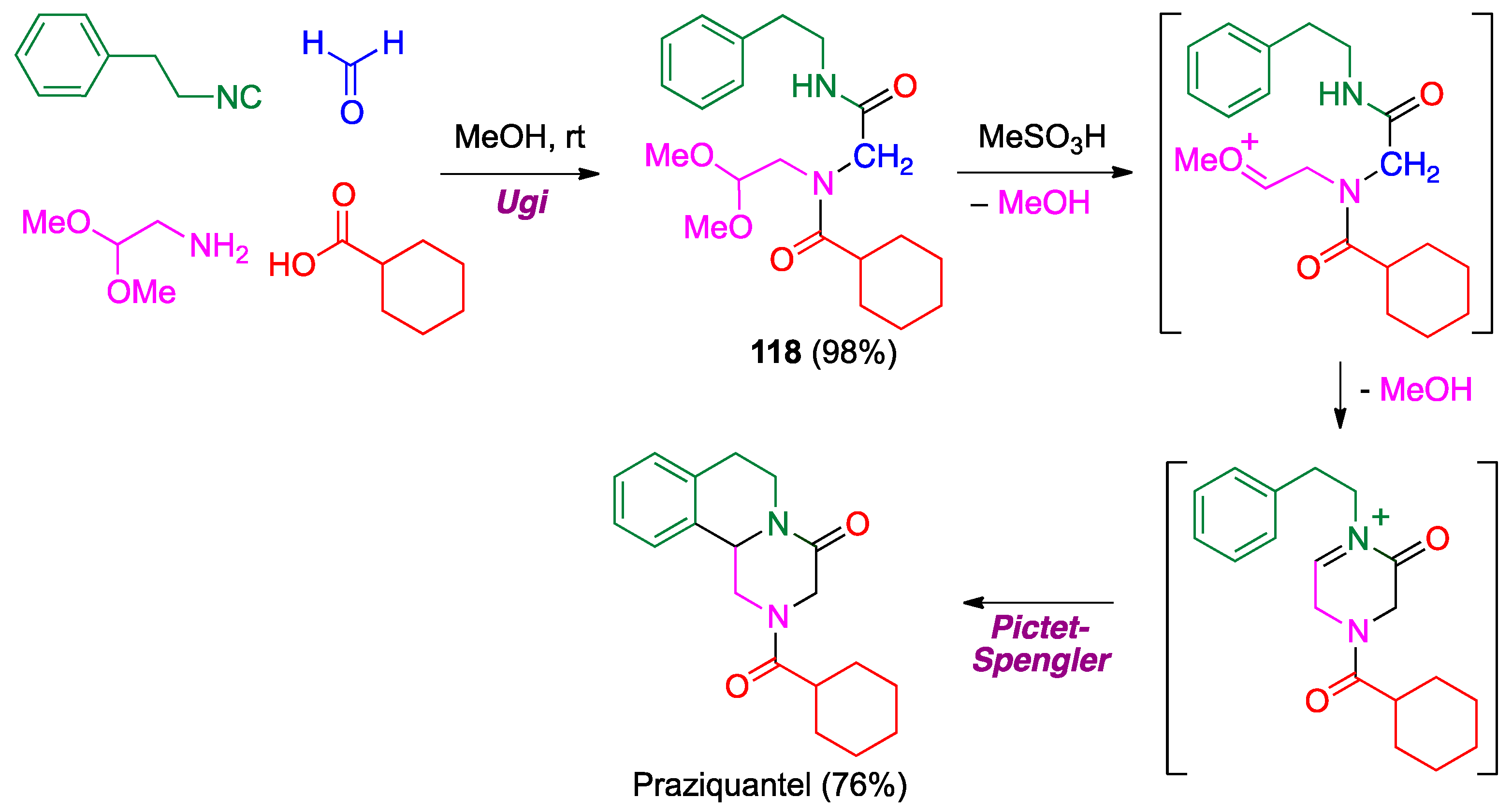
- Liu, H.; William, S.; Herdtweck, E.; Botros, S.; Dömling, A. MCR synthesis of praziquantel derivatives. Chem. Biol. Drug Des. 2012, 79, 470–477. [Google Scholar] [CrossRef] [PubMed]
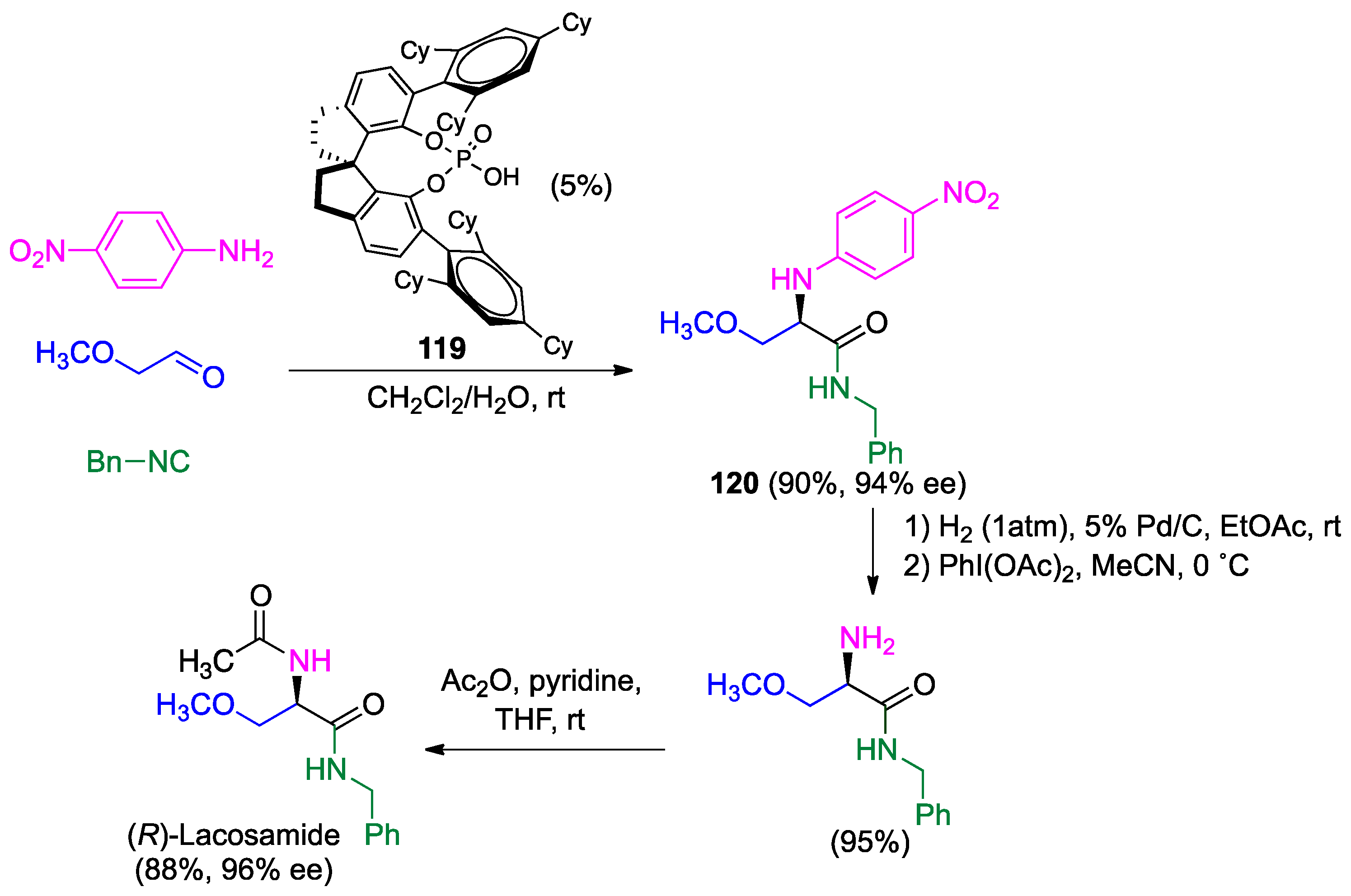
- Wehlan, H.; Oehme, J.; Schäfer, A.; Rossen, K. Development of Scalable Conditions for the Ugi reaction-Application to the synthesis of (R)-lacosamide. Org. Process Res. Dev. 2015, 19, 1980–1986. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.-Y.; Sun, H.; Li, S.-Y.; Xiang, S.-H.; Tan, B. Enantioselective three-component Ugi reaction catalyzed by chiral phosphoric acid. Sci. China: Chem. 2020, 63, 47–54. [Google Scholar] [CrossRef]
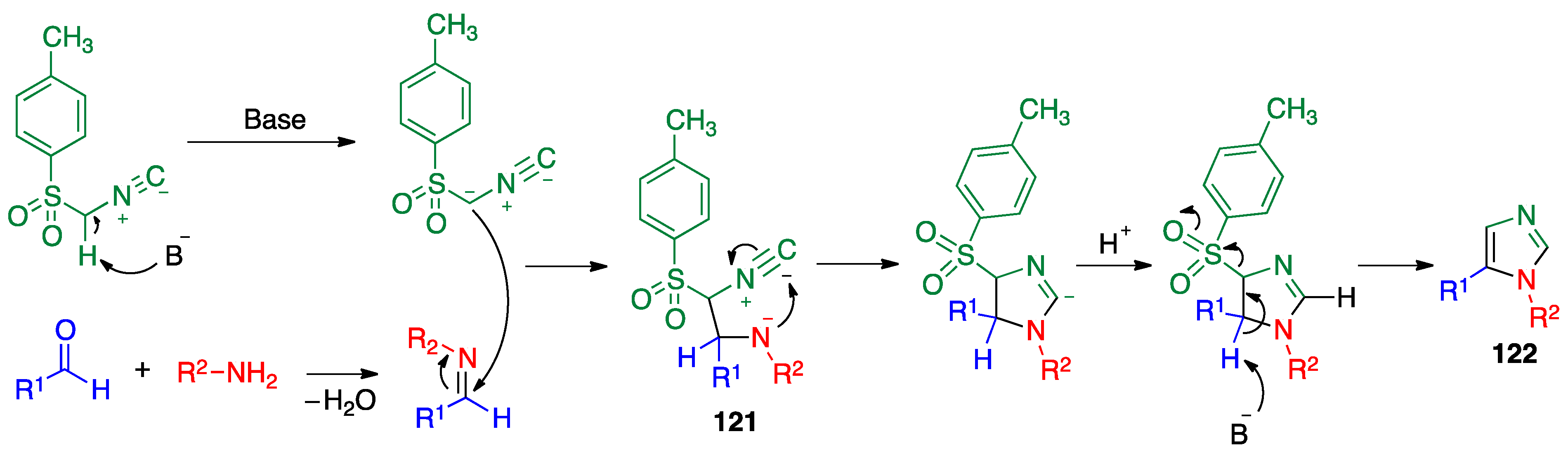
- Van Leusen, A.M.; Wildeman, J.; Oldenziel, O.H. Chemistry of sulfonylmethyl isocyanides. Base-induced cycloaddition of sulfonylmethyl isocyanides to carbon, nitrogen double bonds. Synthesis of 1, 5-disubstituted and 1, 4, 5-trisubstituted imidazoles from aldimines and imidoyl chlorides. J. Org. Chem. 1977, 42, 1153–1159. [Google Scholar]
- Sisko, J. A one-pot synthesis of 1-(2,2,6,6-tetramethyl-4-piperidinyl)-4-(4-fluorophenyl)-5-(2-amino-4-pyrimidinyl)-imidazole: A potent inhibitor of P38 MAP kinase. J. Org. Chem. 1998, 63, 4529–4531. [Google Scholar] [CrossRef]
- Sisko, J.; Mellinger, M. Development of a general process for the synthesis of highly substituted imidazoles. Pure Appl. Chem. 2002, 74, 1349–1357. [Google Scholar] [CrossRef]
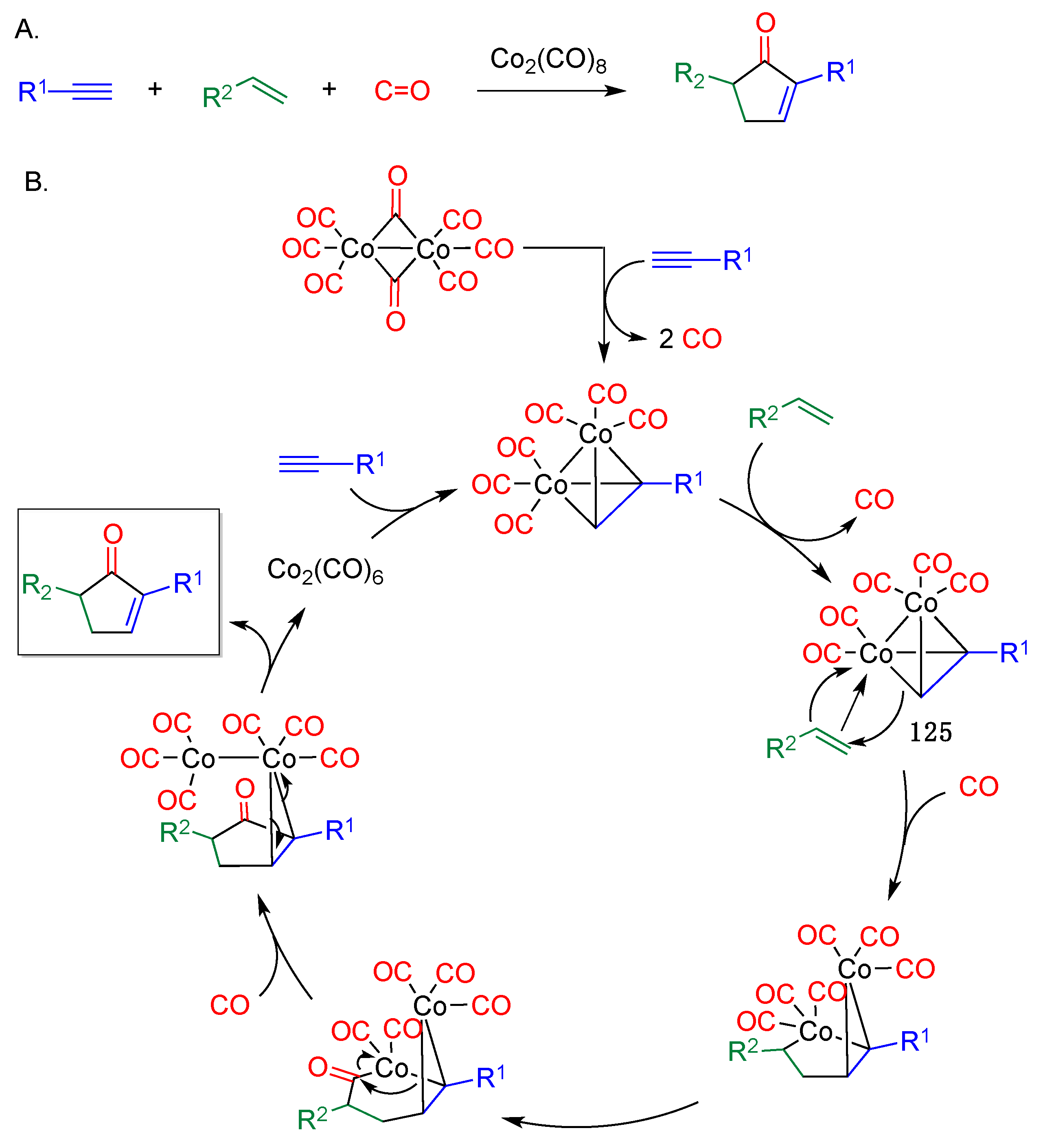
- Pauson, P.L.; Khand, I.U. Uses of cobalt-carbonyl acetylene complexes in organic synthesis. Ann. N. Y. Acad. Sci. 1977, 295, 2–14. [Google Scholar] [CrossRef]
- Ricker, J.D.; Geary, L.M. Recent advances in the Pauson–Khand reaction. Top. Catal. 2017, 60, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Magnus, P.; Exon, C.; Albaugh-Robertson, P. Dicobaltoctacarbonyl-alkyne complexes as intermediates in the synthesis of bicyclo [3.3.0] octenones for the synthesis of coriolin and hirsutic acid. Tetrahedron 1985, 41, 5861–5869. [Google Scholar] [CrossRef]
- Lanver, A.; Schmalz, H.-G. A Pauson–Khand approach to new carbocyclic nucleoside analogs. Eur. J. Org. Chem. 2005, 2005, 1444–1458. [Google Scholar] [CrossRef]
- Vázquez-Romero, A.; Rodríguez, J.; Lledó, A.; Verdaguer, X.; Riera, A. Enantioselective syntheses of carbanucleosides from the Pauson-Khand adduct of trimethylsilylacetylene and norbornadiene. Org. Lett. 2008, 10, 4509–4512. [Google Scholar] [CrossRef]
- Huang, Y.; Dömling, A. The Gewald multicomponent reaction. Mol. Divers. 2011, 15, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Jana, S.; Sonawane, M.; Fiey, B.; Balzarini, J.; Liekens, S.; Dehaen, W. A New four-component rection involving Michael addition and Gewald reaction leading to diverse biologically active 2-aminotiophenes. Org. Biomol. Chem. 2017, 15, 3892–3900. [Google Scholar] [CrossRef]
- Chakrabarti, J.K.; Hotten, T.M.; Tupper, D.E. Process for Preparing 2-Methylthienobenzodiazepine. U.S. Patent US6008216, 12 December 1999. [Google Scholar]
- Kim, J.H.; Kim, J.H.; Lee, J.G.; Jung, H.S.; Han, N.S.; Park, Y.; Kang, S.H.; Jung, H.J.; Lee, G.T.; Choi, H.E.; et al. Compounds Having Angiogenesis Inhibitory Activity and Method for Preparing Same, and Pharmaceutical Composition Comprising Same. U.S. Patent US9227955, 5 January 2016. [Google Scholar]
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef]
- Oliverio, M.; Costanzo, P.; Nardi, M.; Rivalta, I.; Procopio, A. Facile ecofriendly synthesis of monastrol and its structural isomers via Biginelli reaction. ACS Sustain. Chem. Eng. 2014, 2, 1228–1233. [Google Scholar] [CrossRef]
- Kumar, D.; Jadhavar, P.S.; Nautiyal, M.; Sharma, H.; Meena, P.K.; Adane, L.; Pancholia, S.; Chakraborti, A.K. Convenient synthesis of 2,3-disubstituted quinazolin-4(3H)-ones and 2-styryl-3-substituted quinazolin-4(3H)-ones: Applications towards the synthesis of drugs. RSC Adv. 2015, 5, 30819–30825. [Google Scholar] [CrossRef]
- Hayashi, Y. Domino and one-pot syntheses of biologically active compounds using diphenylprolinol silyl ether. Phys. Sci. Rev. 2020, 5, 20180088. [Google Scholar] [CrossRef]
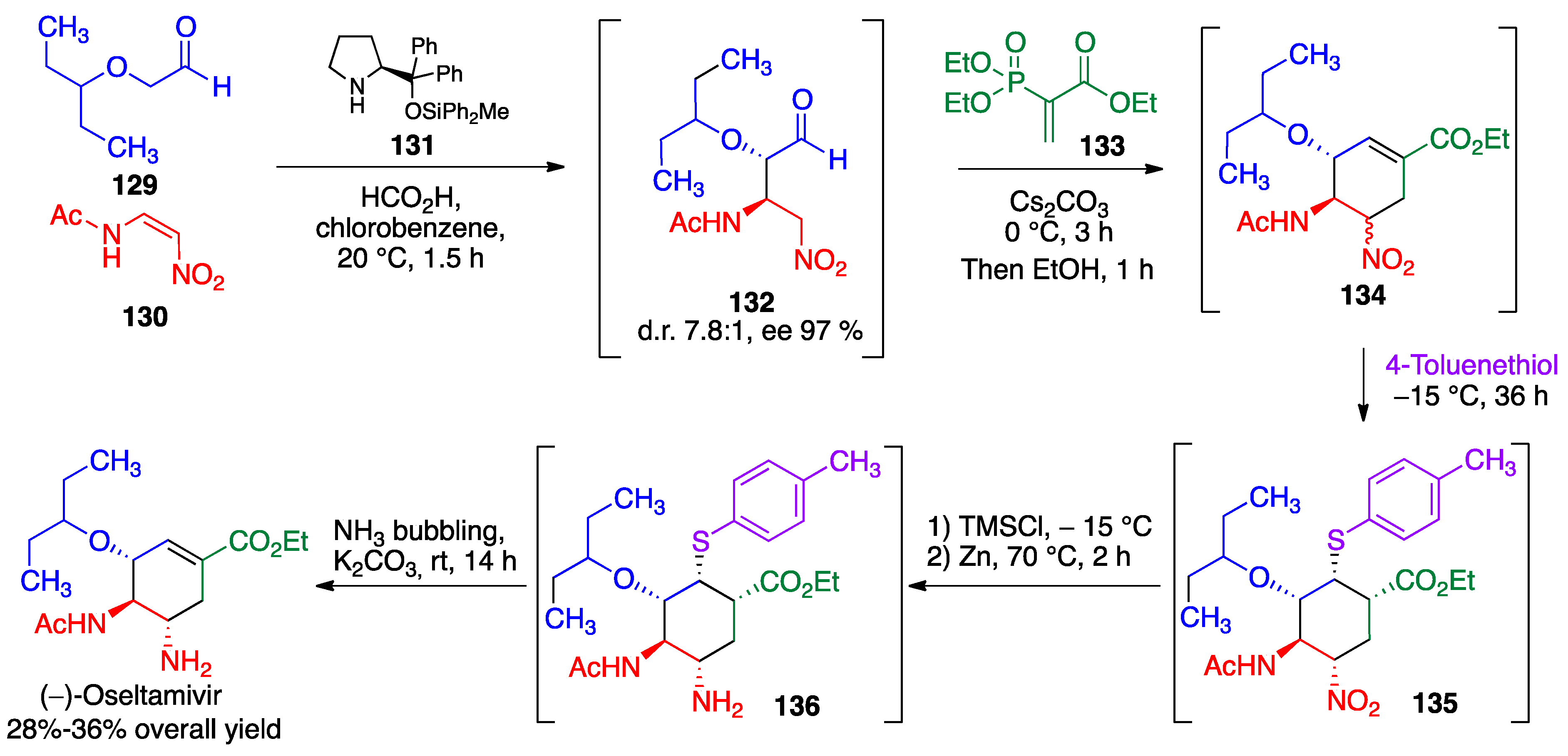
- Mukaiyama, T.; Ishikawa, H.; Koshino, H.; Hayashi, Y. One-pot synthesis of (−)-oseltamivir and mechanistic insights into the organocatalyzed Michael reaction. Chem. Eur. J. 2013, 19, 17789–17800. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Ogasawara, S. Time economical total synthesis of (−)-oseltamivir. Org Lett. 2016, 18, 3426–3429. [Google Scholar] [CrossRef]
- Ruijter, E.; Sheffelaar, R.; Orru, R. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew. Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Zarganes-Tzitzikas, T.; Chandgude, A.L.; Dömling, A. Multicomponent reactions, union of MCRs and beyond. Chem. Rec. 2015, 15, 981–996. [Google Scholar] [CrossRef]
- Ugi, I. From isocyanides via four-component condensations to antibiotic syntheses. Angew. Chem. Int. Ed. Engl. 1982, 21, 810–819. [Google Scholar] [CrossRef]
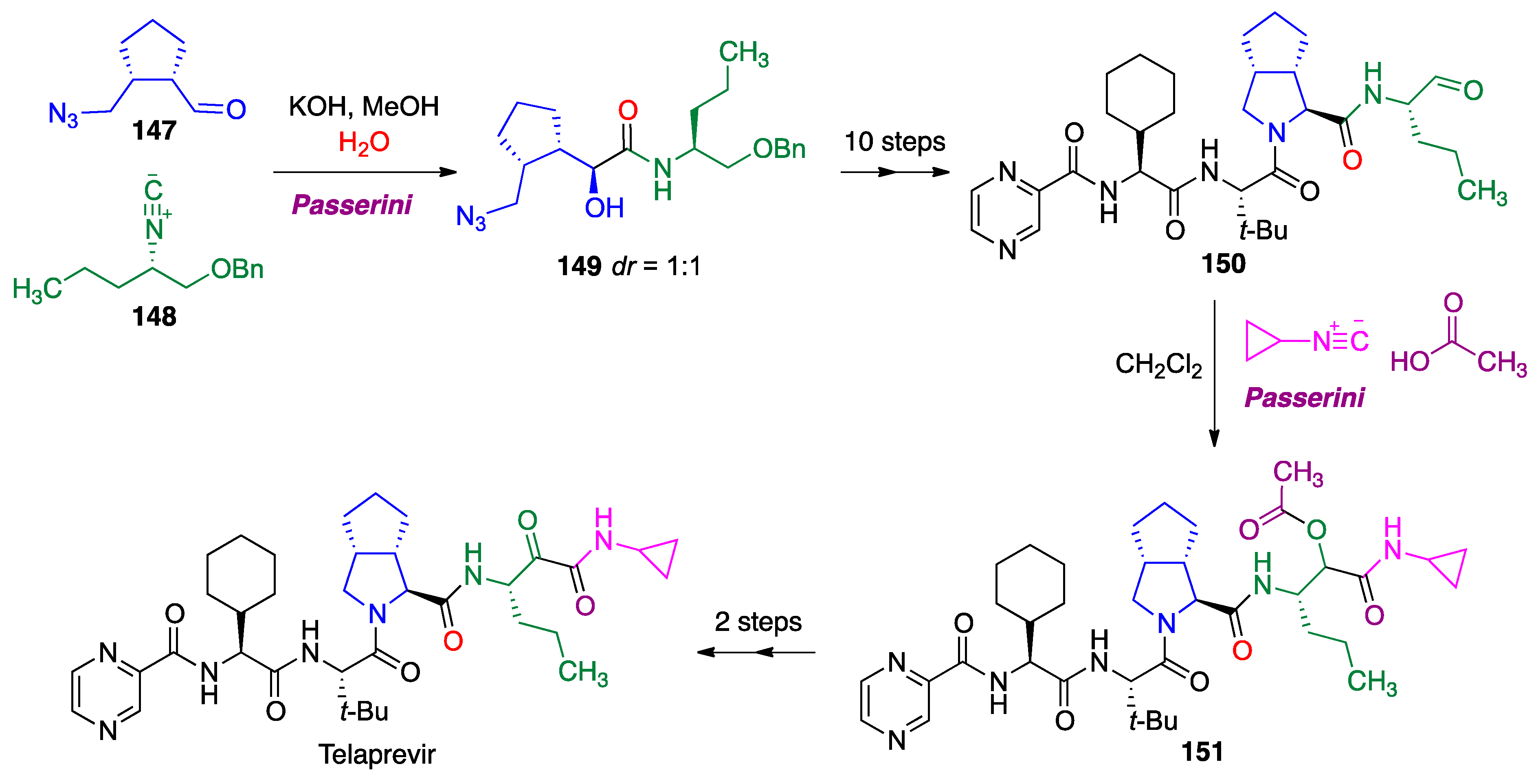
- Znabet, A.; Polak, M.M.; Janssen, E.; de Kanter, F.J.; Turner, N.J.; Orru, R.V.; Ruijter, E. A highly efficient synthesis of telaprevir by strategic use of biocatalysis and multicomponent reactions. Chem. Commun. 2010, 46, 7918–7920. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Dömling, A. Modern multicomponent reactions for better drug syntheses. Org. Chem. Front. 2014, 1, 834–837. [Google Scholar] [CrossRef]
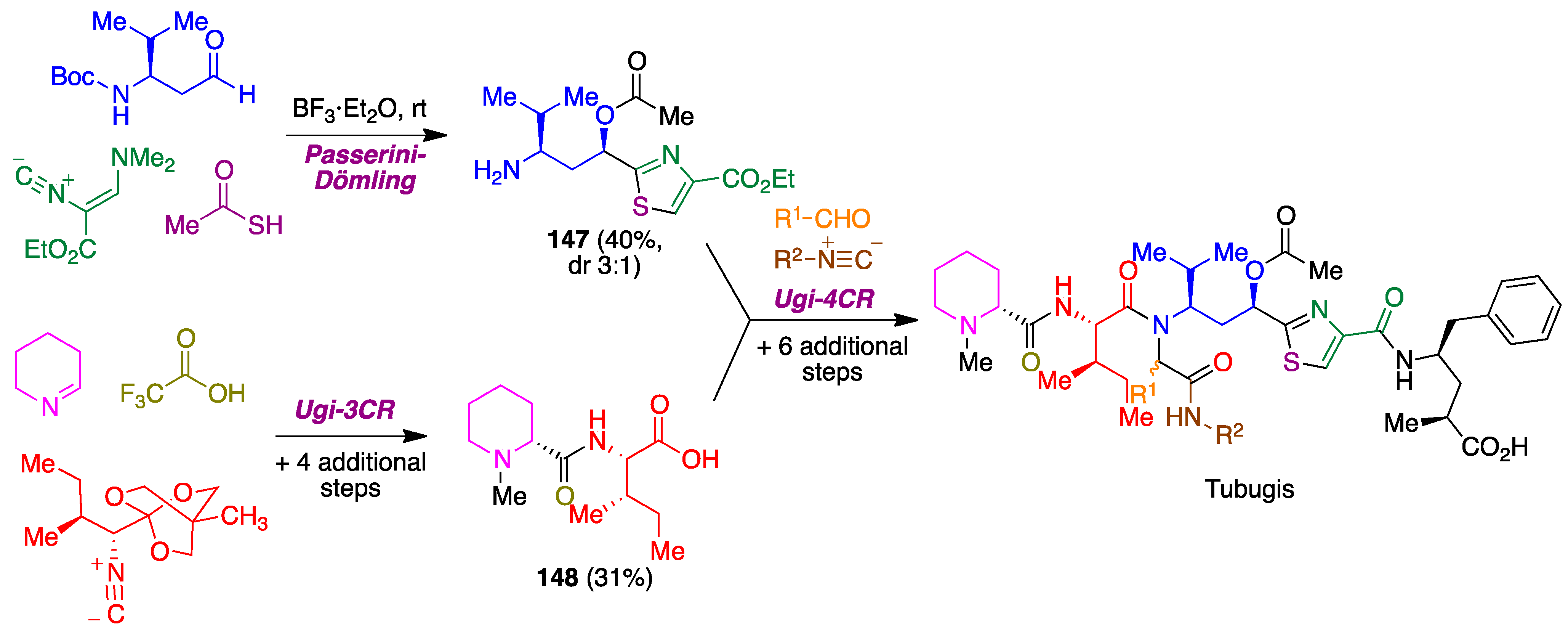
- Pando, O.; Stark, S.; Denkert, A.; Porzel, A.; Preusentanz, R.; Wessjohann, L.A. The multiple multicomponent approach to natural product mimics: Tubugis, N-substituted anticancer peptides with picomolar activity. J. Am. Chem. Soc. 2011, 133, 7692–7695. [Google Scholar] [CrossRef]




















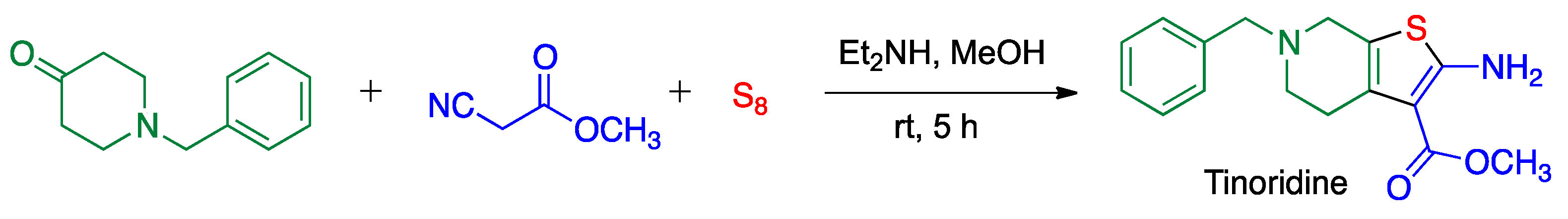

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cores, Á.; Clerigué, J.; Orocio-Rodríguez, E.; Menéndez, J.C. Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients. Pharmaceuticals 2022, 15, 1009. https://doi.org/10.3390/ph15081009
Cores Á, Clerigué J, Orocio-Rodríguez E, Menéndez JC. Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients. Pharmaceuticals. 2022; 15(8):1009. https://doi.org/10.3390/ph15081009
Chicago/Turabian StyleCores, Ángel, José Clerigué, Emmanuel Orocio-Rodríguez, and J. Carlos Menéndez. 2022. "Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients" Pharmaceuticals 15, no. 8: 1009. https://doi.org/10.3390/ph15081009
APA StyleCores, Á., Clerigué, J., Orocio-Rodríguez, E., & Menéndez, J. C. (2022). Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients. Pharmaceuticals, 15(8), 1009. https://doi.org/10.3390/ph15081009