2.2.1. UV-Vis Absorption
The absorption spectra of
3 were acquired in all of the solvents of
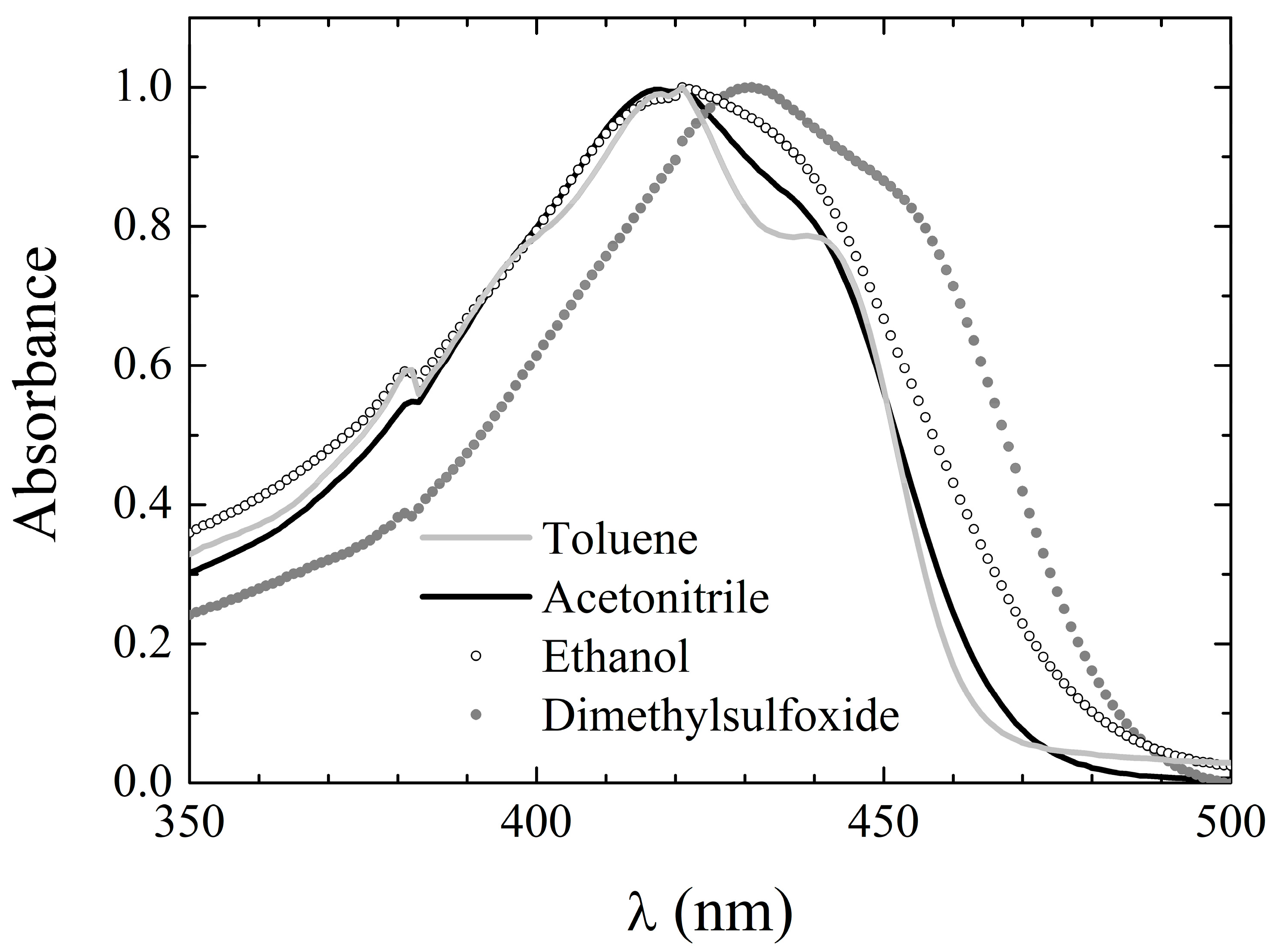
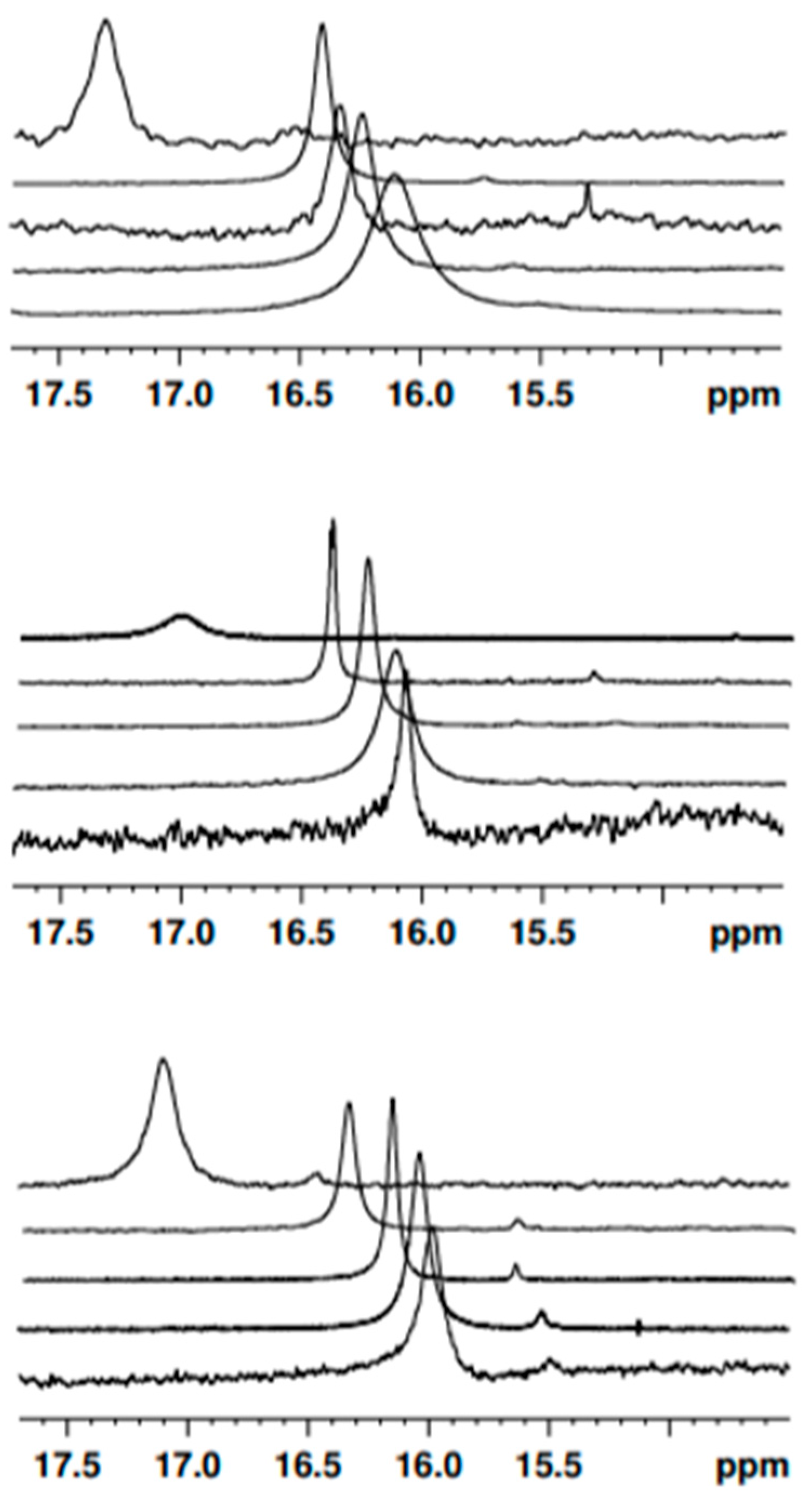
Table 1. The absorption spectrum consists of a single broad band peaking at around 410 nm, occasionally with a shoulder at longer wavelengths. Thus, also in the case of
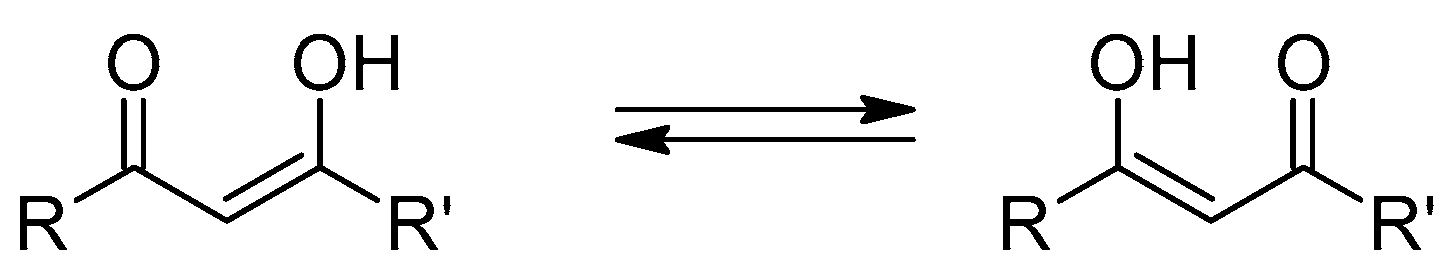
3, the keto–enol equilibrium seems to be shifted towards the enol tautomers, with the diketo tautomers appearing at most in traces, as confirmed by both the NMR and in silico calculations (vide infra). The absorption peak wavelength in the various solvents is reported in
Table 6, while exemplary spectra in selected solvents are shown in
Figure 4. The absorption bands of
3 are slightly, though systematically, blue-shifted with respect to those of both
1 and
2, which might indicate a reduced charge conjugation in
3. Interestingly, the same observation pertained to dicinnamoylmethane (DCMeth), a symmetric, non-phenyl substituted curcuminoid [
39], whose absorption peak wavelength is reported as λ
abs,DCMeth in the third column of
Table 6.
Moreover, the spectral features of
3 are less dependent on the environment than those of both
1 and
2, although a loss in structure can be still observed by changing from inert (cyclohexane and toluene) to reactive environments. This behavior is, again, similar to the one exhibited by DCMeth [
39].
The molar extinction coefficient of 3 in all tested solvents is in the range of 30,000 M−1cm−1 and does not appear to depend systematically on the solvent properties. Moreover, it is slightly, but systematically, lower than that of 2, stemming from further support of a reduced charge conjugation in 3.
2.2.2. Steady-State Fluorescence
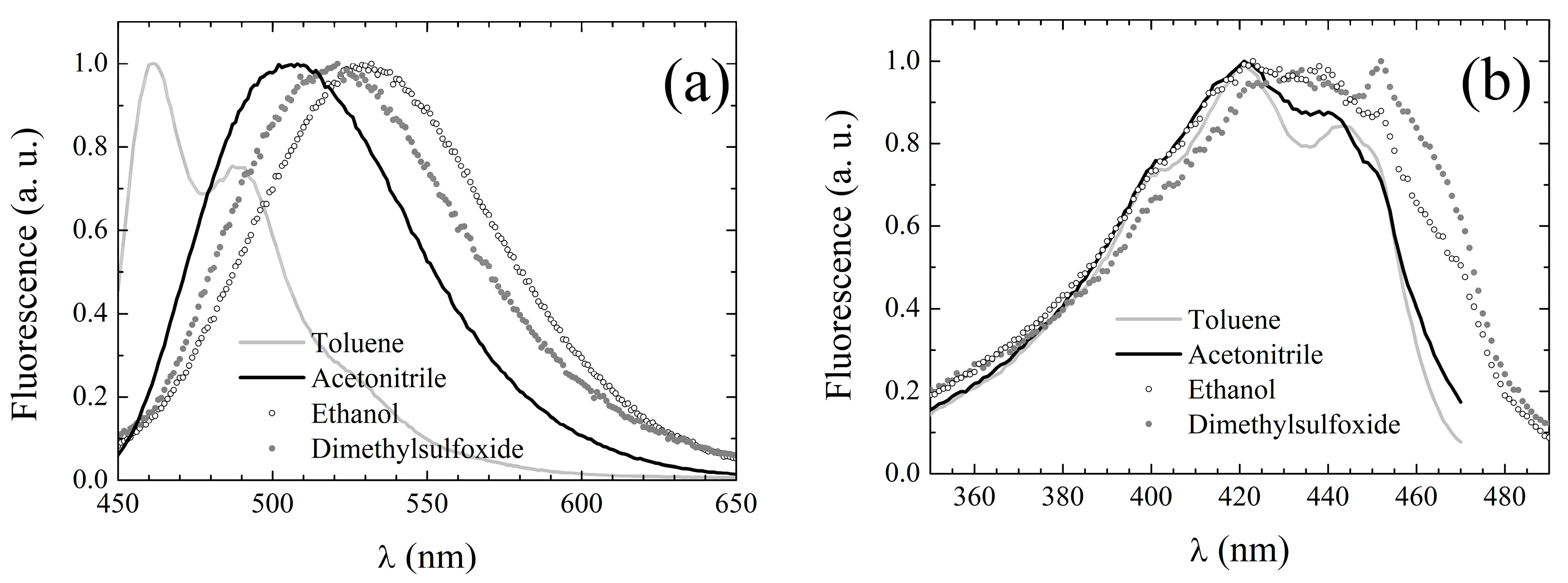
The peak fluorescence emission wavelengths of
3 in the different solvents of
Table 1, obtained upon excitation at the pertaining λ
abs, are reported in
Table 7, while exemplary spectra in selected solvents can be inspected in
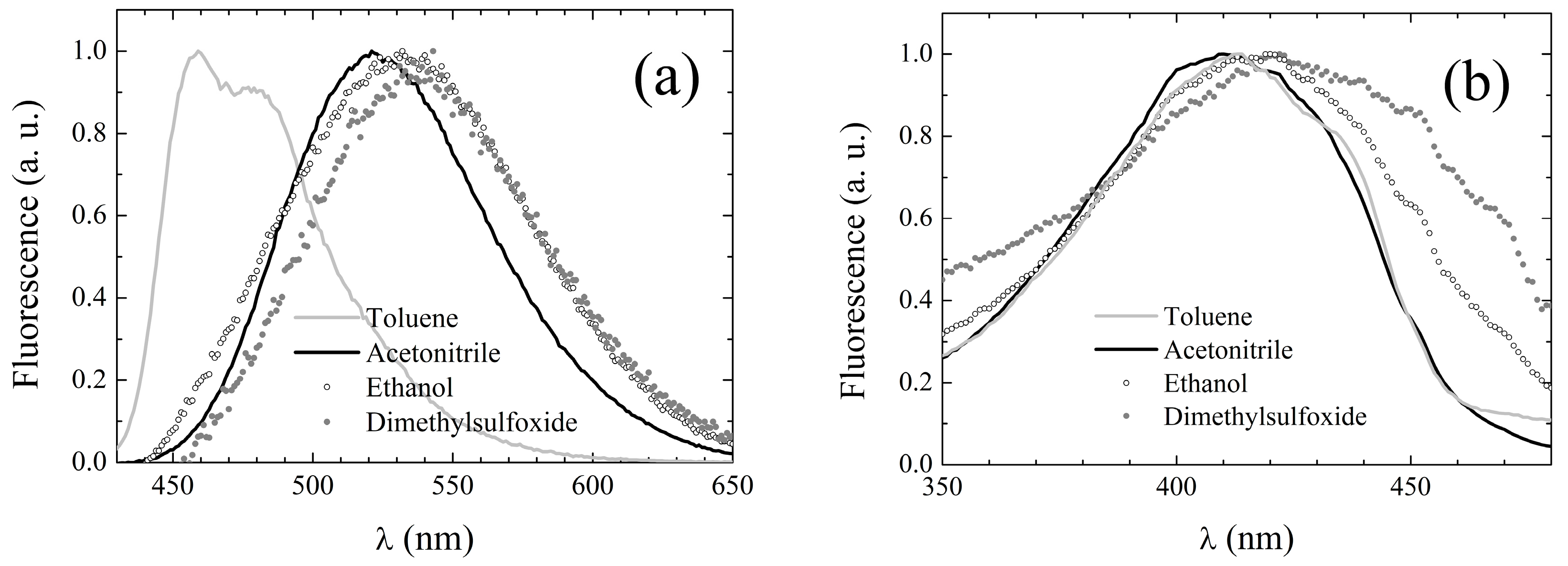
Figure 5a.
Unlike the absorption spectra, the emission spectra appear to resemble those of
1 rather than of DCMeth. First, the spectra of
3 are broad and structureless, with the exception of those acquired in non-polar solvents. In cyclohexane, the spectrum exhibits two peaks with a further shoulder at long wavelengths, while in toluene, the spectrum consists of a main peak and a red-shifted shoulder. Conversely, those of DCMeth were double-peaked in all of the tested solvents (see third column of
Table 7). Moreover, the fingerprints of the solvent dependence of the fluorescence emission typical of
1 are also exhibited by
3, even more neatly than by
2. Although a direct correlation between peak emission wavelength and polarity is not observed, in the polar solvents, the emission spectrum is generally peaked at longer wavelengths than in inert ones. Furthermore, in the H-bonding solvents (both H-bond acceptors and alcohols), the fluorescence is systematically red-shifted with respect to polar weakly H-bonding solvents of comparable polarity. The fluorescence quantum yield of
3 (see fourth column in
Table 3) is notably higher than that of
1 in non-polar (cyclohexane and toluene) and slightly polar (chloroform and dichloromethane) solvents, while it is comparable in highly polar environments and sizably lower in H-bonding solvents with the only exception of ethylene glycol. The comparison with
2 unravels a systematic reduction in the quantum yield for
3 with respect to the other asymmetric compounds, which might correlate with the specific phenolic substituents. Indeed, DCMeth was characterized by a particularly low quantum yield in all solvents [
39] (the values are reported in the fifth line of
Table 7 for the sake of straightforward comparison).
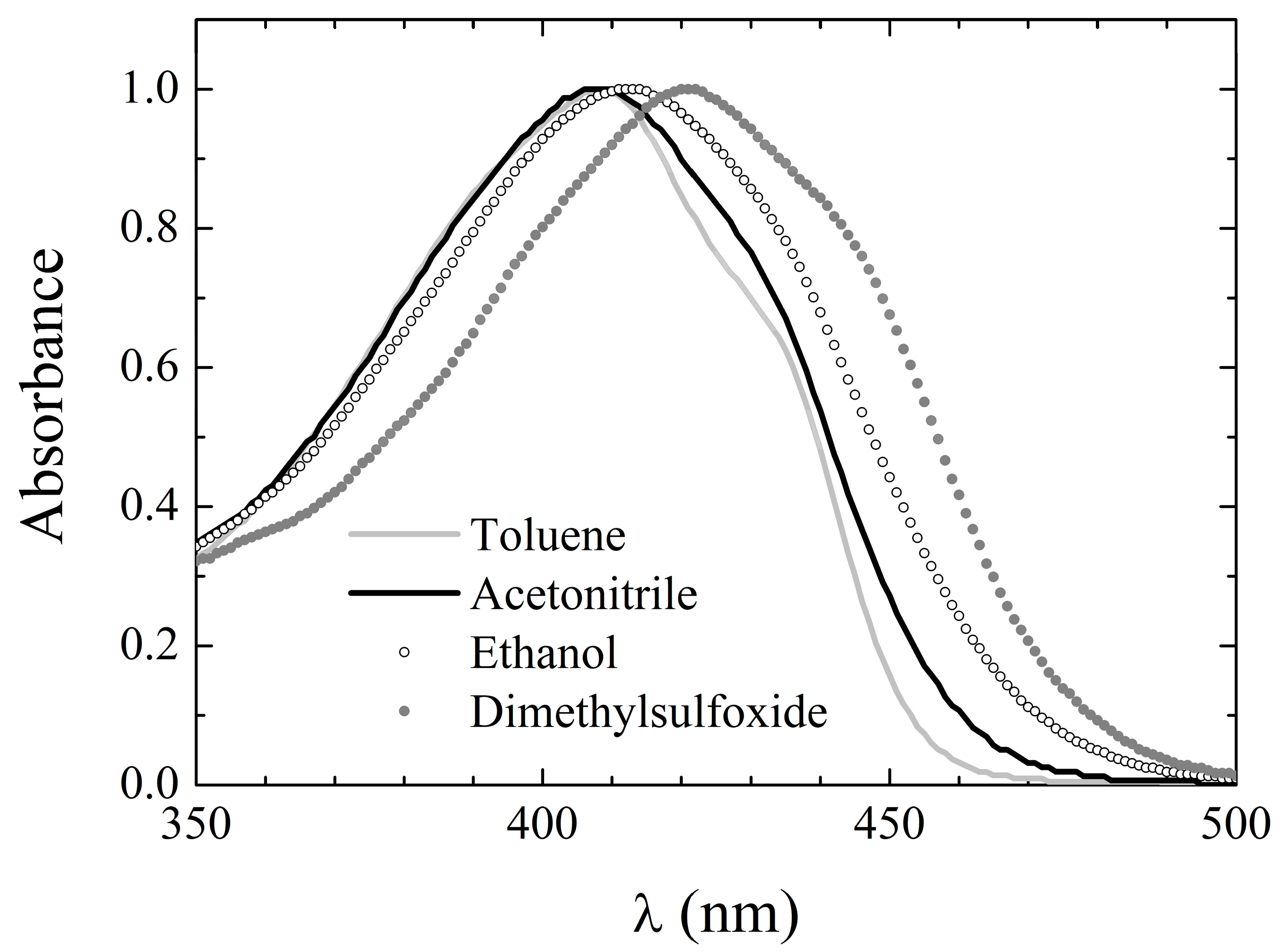
Fluorescence excitation spectra were also recorded, setting the observation wavelength at the emission peak whenever it was >500 nm, and at 500 nm otherwise. The excitation peak wavelengths are listed in
Table 7, column 6, and appear to be only slightly red-shifted with respect to the corresponding absorption peak wavelengths (see
Table 6). Moreover, the excitation spectral line shapes conserve the main features of the corresponding UV-Vis absorption spectra, as evidenced by the exemplary spectra displayed in
Figure 5b. These features suggest that for
3, S
1 deactivation also simply occurs through inverse transition backwards to the ground state with no involvement of additional electronic energy levels.
2.2.3. Excited-State Dynamics
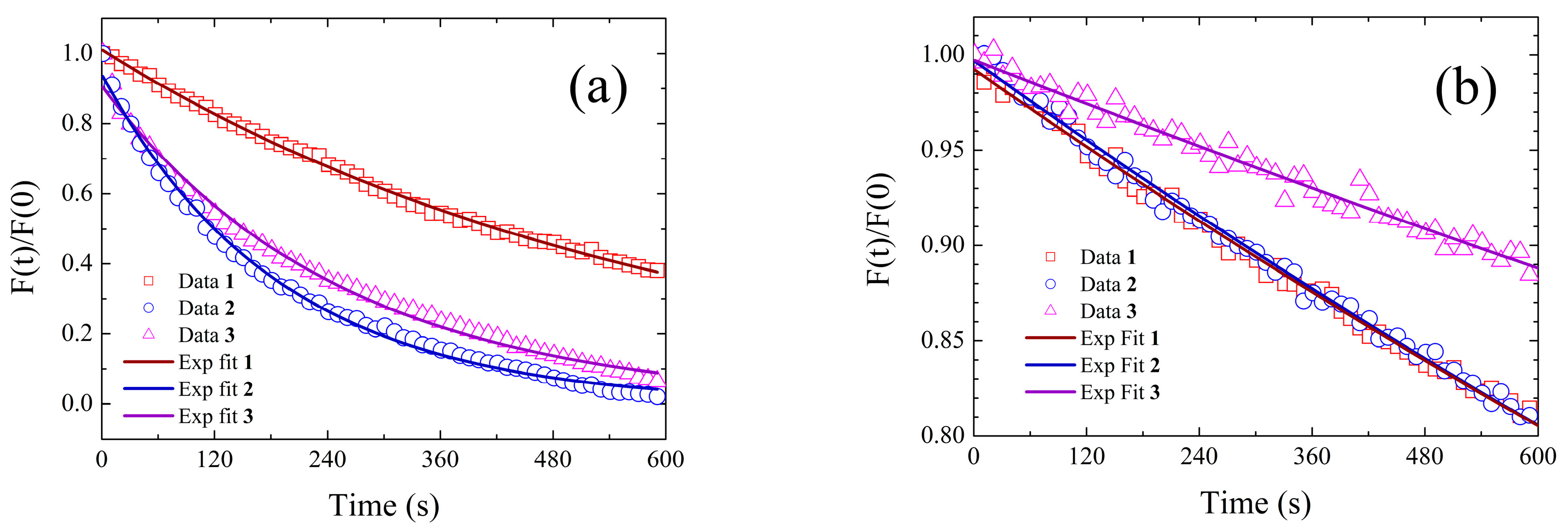
The best-fitting parameters retrieved from the fit of the experimental decay distributions measured for
3 in the different solvents are reported in
Table 8. The values of the average S
1 state lifetime, defined according to Equation (1), are also listed in the fifth column.
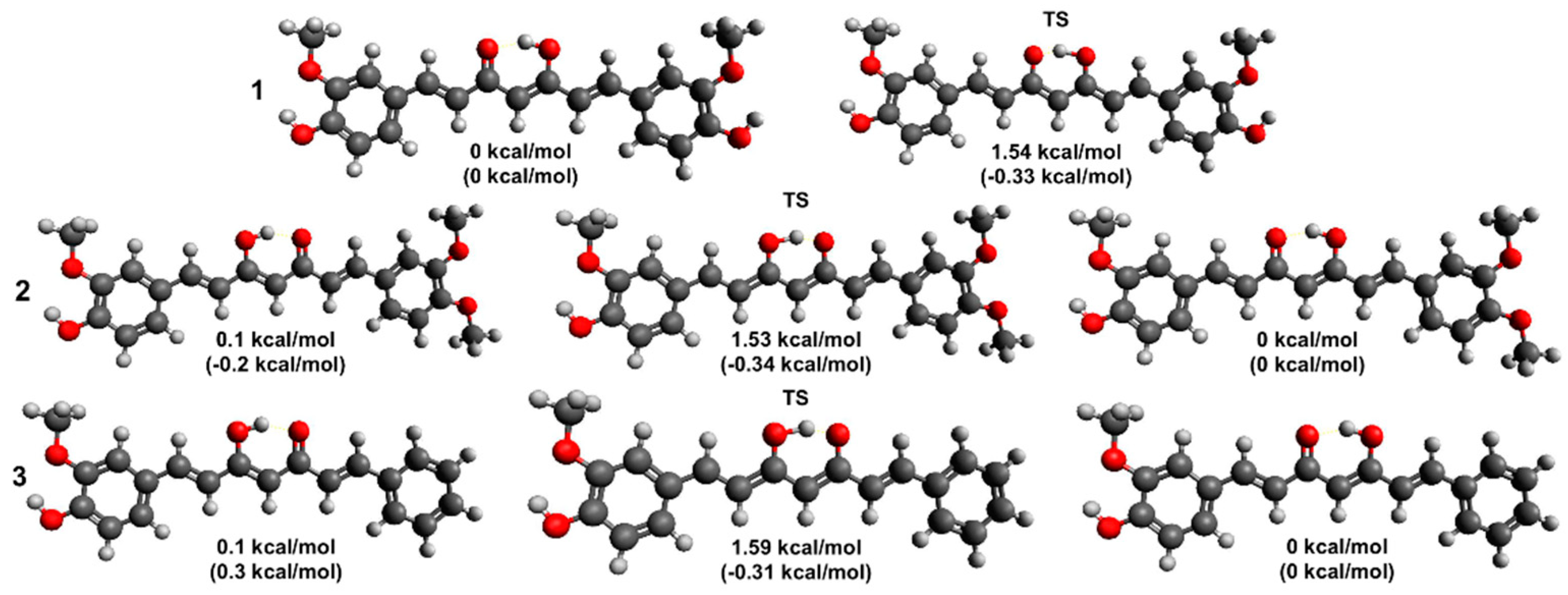
In inert environments, a behavior similar to that of 2 is observed, with 3 displaying a three-exponential decay pattern in cyclohexane and an essentially single-exponential decay in toluene. However, in cyclohexane, the ESIPT rate appears to be much faster for 3 than for both 1 and 2, and is comparable with that measured in DCMeth. Moreover, the relative probability of 3 to decay by means of direct ESIPT is comparable with that measured for 1, and is much higher than that of 2. Reketonization occurs on timescales similar to those typical of both 1 and 2, and faster than for DCMeth.
The net effect is that the overall stability of the excited state of
3 is comparable to that of
1 and inferior to that of
2 (see the fifth column of
Table 8 and the last column of
Table 4 for the average lifetimes of
3 and
1, respectively). Conversely, in toluene, like for
2, the main decay transient has the time constant typical of reketonization dynamics. The effect is a two-fold increase in the average S
1-state lifetime of
3 with respect to
1 in this solvent. In both non-polar solvents, the presence of traces of the non-polar trans-diketo isomer is denounced by the longest-lived component.
In polar, weakly H-bonding solvents, 3 seems to exhibit a behavior similar to that of 1. The decay patterns are essentially single-exponential, suggesting that solvent rearrangement-moderated ESIPT is the main decay mechanism. This latter occurs systematically on faster timescales with respect to both 1 and 2. The residual, long-lived component resolved in the weakly polar solvents chloroform and dichloromethane (ε < 10) can be ascribed to traces of anti-diketo isomer, which were also observed in DCMeth in solvents of comparable polarity. The survival of traces of 3 in the trans-diketo isomer even in mildly polar environments agrees with a reduced polarity difference between the latter and the enol tautomers with respect to 1 and 2.
The decays of
3 are multi-exponential in most of the other solvents, revealing complex and hardly interpretable deactivation photophysics. In H-bond-accepting solvents, benchmarks of the DCMeth excited-state dynamics are recognizable in the decays of
3. Indeed, for both
3 and DCMeth, the fluorescence decay patterns in DMF and DMSO are dominated by a transient with time constant in the range typical of direct ESIP mechanisms. Some valuable hints can be extracted from this analogy. Indeed, in the case of DCMeth, the enhanced proneness to decay by direct ESIPT in any environment was ascribed to the formation of a particular variant of the closed enol tautomer having c
2ν symmetry, in which the enol proton resulted in being delocalized and equally shared between the two keto oxygens, together with the closed enol structure with the enol proton localized in proximity to either one of the two keto oxygens adopted by
1 and several other curcuminoids. In particular, because in the c
2ν structure the enol proton is more acidic, the latter is expected to be dominant in H-bond-accepting solvents, where a transient transfer of the enol proton to the H-bond-accepting moieties might be responsible for fostering the exchange of the proton itself between the two keto oxygens. This mechanism was invoked in [
39] to explain the very complex fluorescence decay distributions measured for DCMeth. The observation of a transient typical of direct ESIPT in the decay of
3 in H-bond-accepting solvents suggests that, as in DCMeth, the keto–enolic ring is more likely to assume the symmetric c
2ν structure than in
1 and
2. Moreover, explicit-solvent simulations on
3’s interactions with DMF unraveled a sufficiently strong affinity between the enol proton of
3 and the solvent carbonylic oxygen (with an equilibrium distance of 2.43 Å) to distort the hydrogen-bonding structure, reducing the enolic O–H distance by 0.03 Å and increasing the distance between the proton and the ketonic oxygen by 0.22 Å. In our view, this finding suggests a reduced likelihood for the direct ESIPT and prefigures the transient solute–solvent proton transfer described above as being likely to take place for
3 as well. A second transient accounts for roughly 1/3 of the excited-state deactivation events and has a lifetime in the range of a few hundred picoseconds, typical of the solute/solvent charge/energy transfer dynamics observed in H-bonding solvents for DCMeth,
1, and
2. Finally, a third component is barely detectable, which we ascribe to decay through solvent rearrangement-moderated ESIPT. The latter is similar, in time and amplitude, to the homologous contributions measured in the decays of DCMeth and
2 in H-bond acceptors, while the same deactivation mechanism occurs on significantly shorter timescales for
1, suggesting the formation of a looser intermolecular H-bond between the solvents’ H-bond-accepting moieties and the enol proton in the parent compound.
In alcohols, the decay dynamics seem to depend on the specific solute/solvent interactions. Indeed, although the intermolecular charge/energy transfer dynamics occur in all alcohols on timescales similar to those observed for this mechanism in 1, 2, and DCMeth, as well as for 3 itself in H-bond acceptors, an additional decay component is resolved in methanol and ethylene glycol, whose time constant is in the range typical of direct ESIPT, which was observed in all alcoholic solvents for DCMeth. This suggests that, in the case of these solvents, a sort of bridging interaction between the enol proton, the H-bond-accepting moiety of the alcohol, the protic moiety, and the unshielded keto oxygen might be established. Interestingly, these solvents are those endowed with the highest Kamlet–Taft acidity parameters among those used in this study. Moreover, the bridging effect seems to be most effective in promoting the enol proton transfer in methanol, which is the smallest solvent and is thus more likely to juxtapose between the enol and keto moiety of 3.
The overall effect of these complex excited-state dynamics is to destabilize the excited state of
3 with respect of
2 and even of
1. However, the electron-withdrawing character of non-substituted phenyl rings in the molecular structure of a curcuminoid was postulated to have a highly destabilizing effect on the excited state [
53] through favoring ESIPT dynamics. In this respect, the introduction of asymmetry in the molecular structure produces a consistent stabilization of
3 compared to DCMeth, the most unstable curcuminoid we have examined until now, which bears non-substituted phenyl rings at both sides of the keto–enol system, and whose average excited-state lifetimes are reported in the last column of
Table 8 for the sake of straightforward comparison.
The excited-state dynamics can be further investigated by estimating the values of the radiative decay rate constant,
kfl, and the non-radiative decay rate constant,
knr, from the measured
τav and
ϕfluo, according to Equations (2) and (3). The calculated values of
kfl and
knr in the tested solvents are reported in
Table 9 (columns two and four, respectively) and compared with those calculated for DCMeth (columns three and five, respectively) [
39]. We recall that the homologous data for
1 are listed in
Table 5, columns three and five. The non-radiative rate constant is systematically lower for
3 than for DCMeth in polar, weakly H-bonding solvents, where it is in the same order of magnitude as that of
1, while in both inert and H-bonding environments, the tendency to decay through non-radiative pathways of
3 and DCMeth are comparable. In all of the solvents, the radiative decay rate of
3 is in the same order of magnitude of that of
1 and is much higher than that measured for DCMeth.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}