
The Protective Effect of Edaravone on TDP-43 Plus Oxidative Stress-Induced Neurotoxicity in Neuronal Cells: Analysis of Its Neuroprotective Mechanisms Using RNA Sequencing


Abstract
:1. Introduction
2. Results
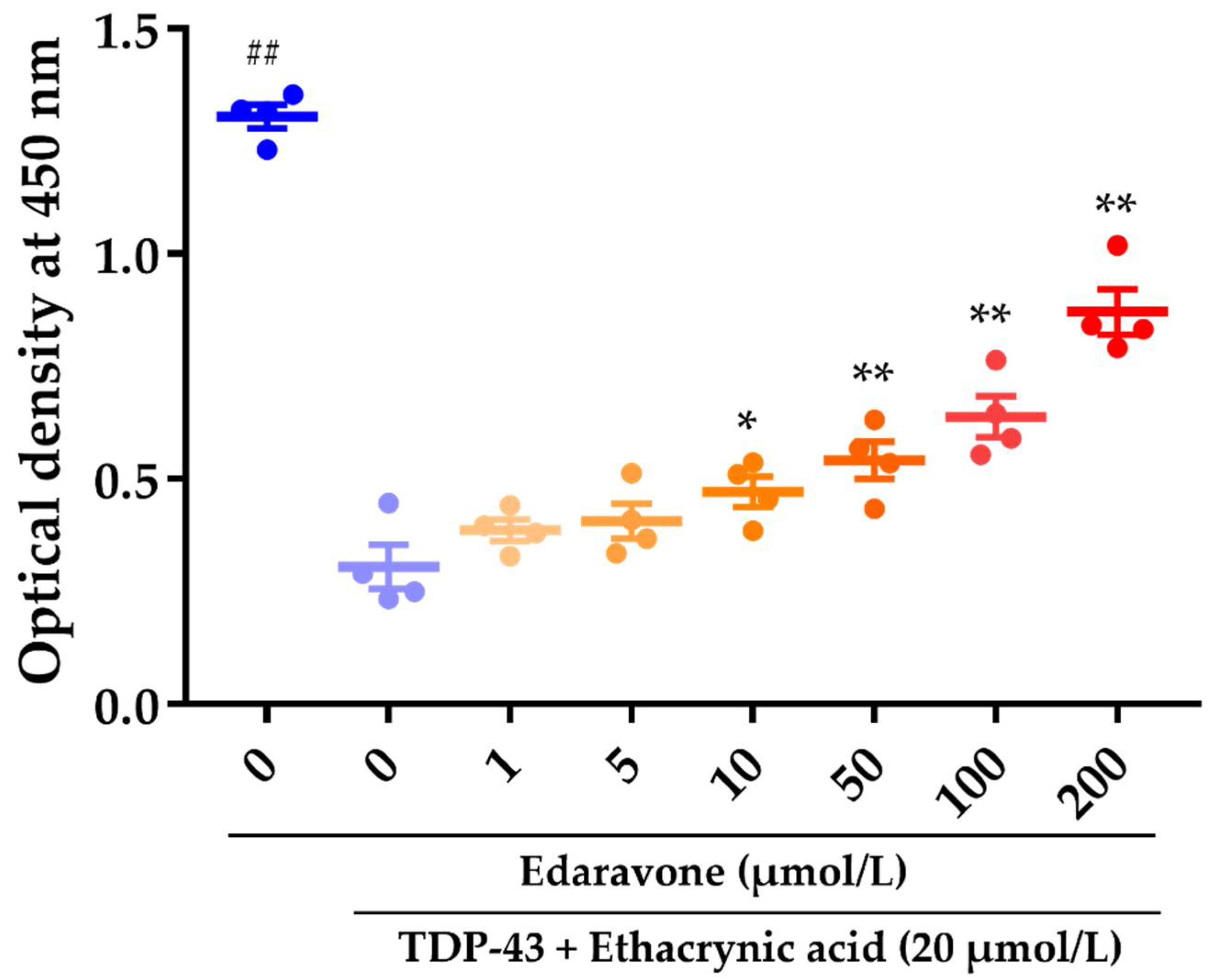
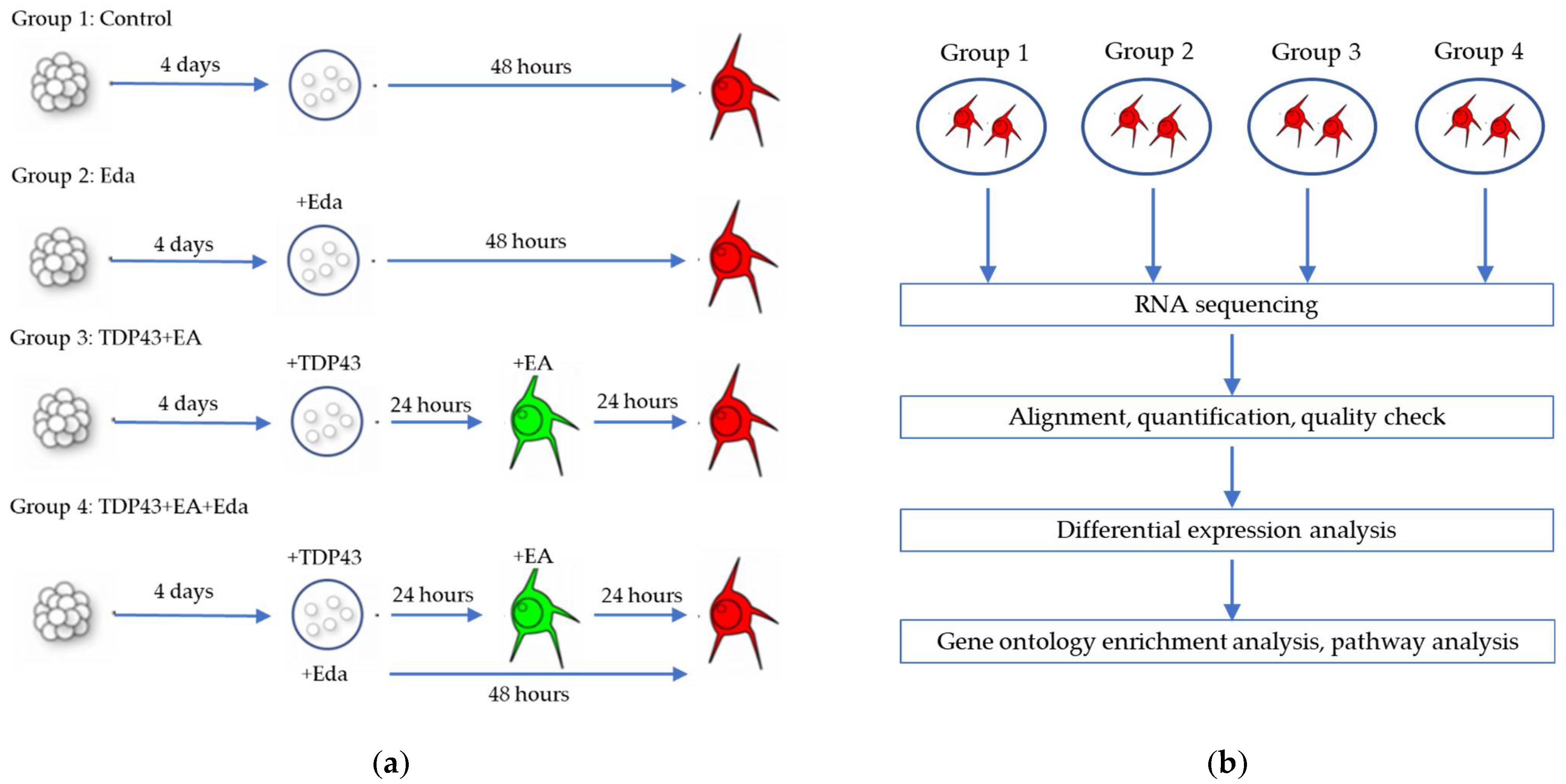
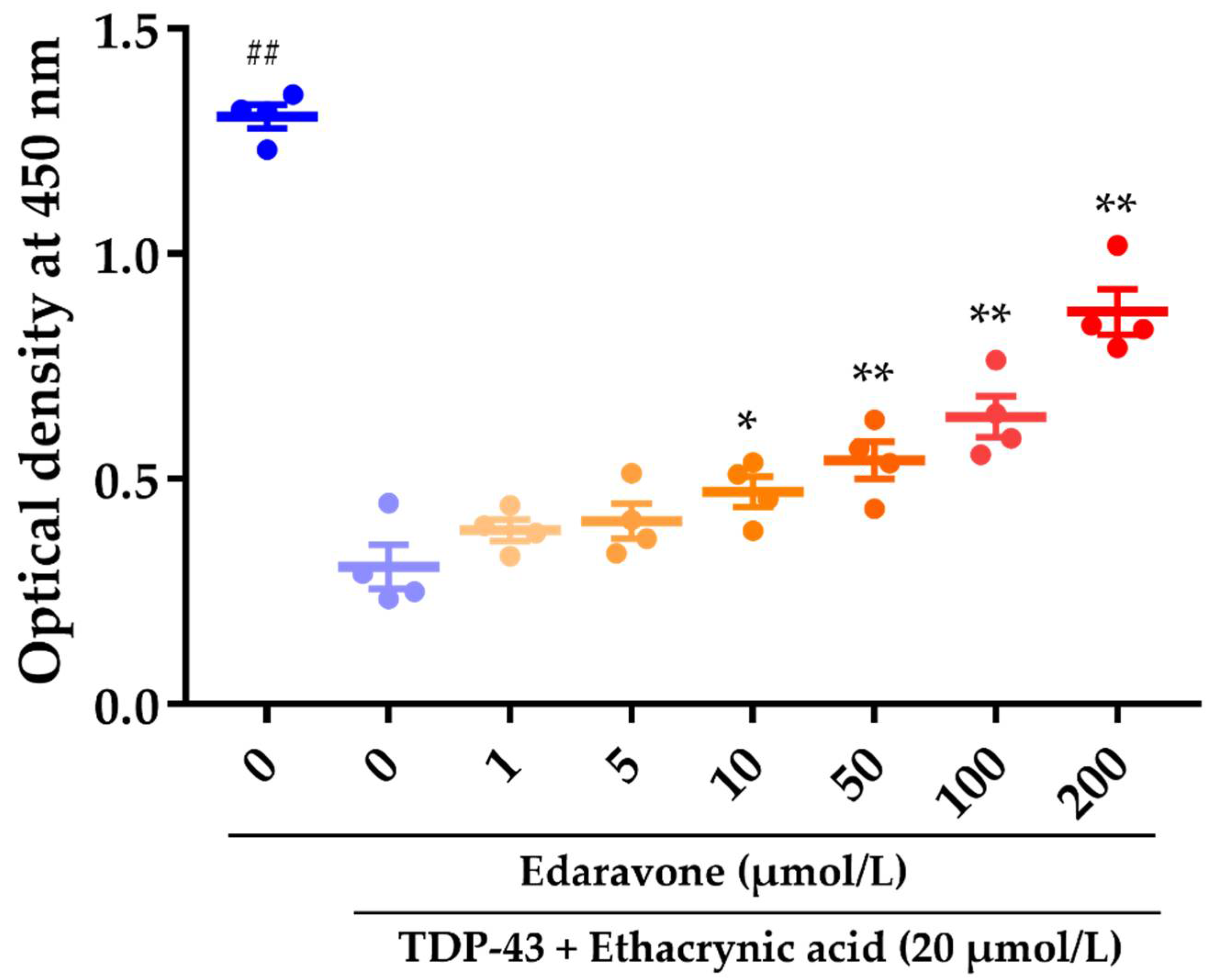
2.1. The Effect of Edaravone against Cell Death in Rat Neural Stem Cell-Derived Neurons Transduced with Adenoviruses Expressing TDP-43
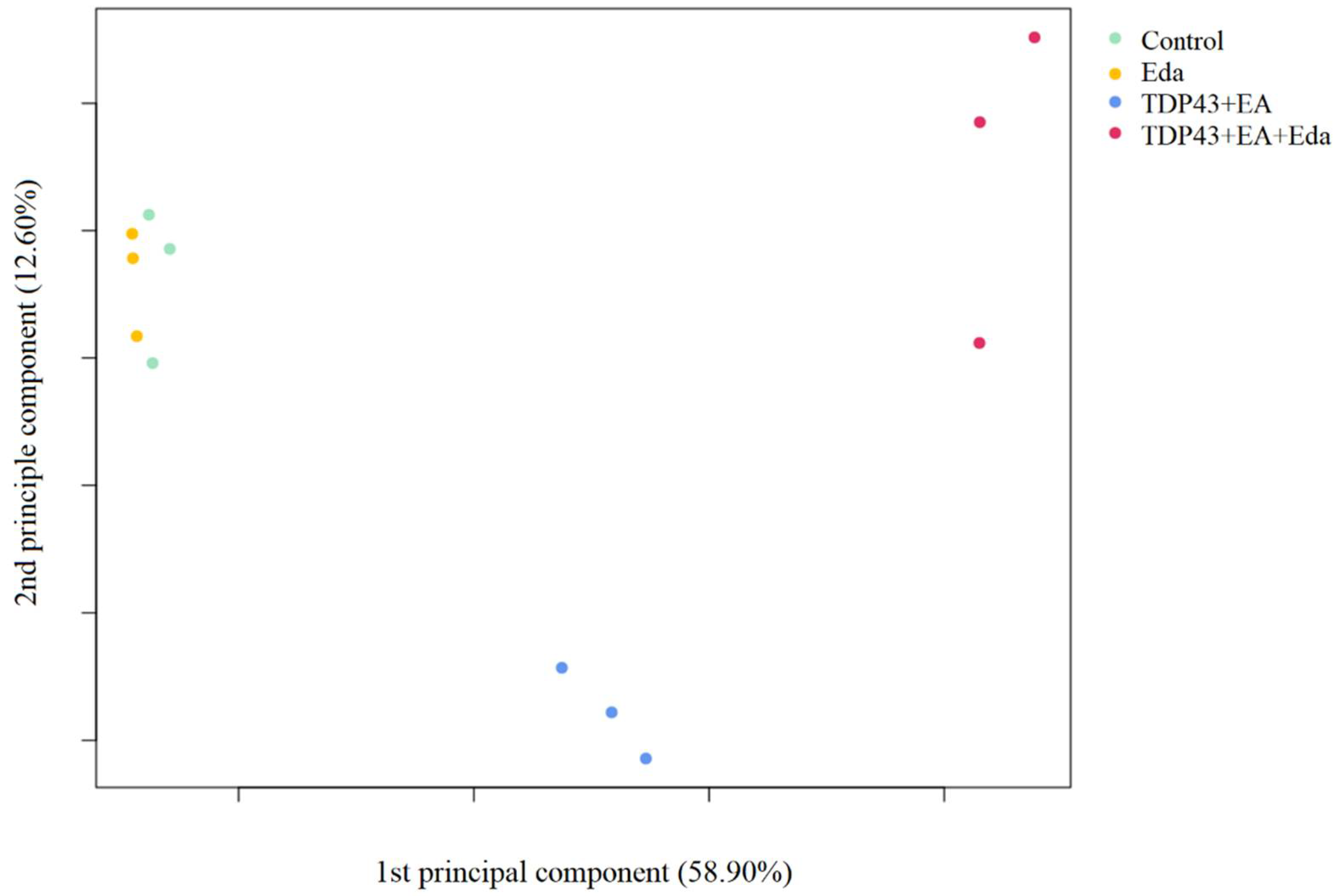
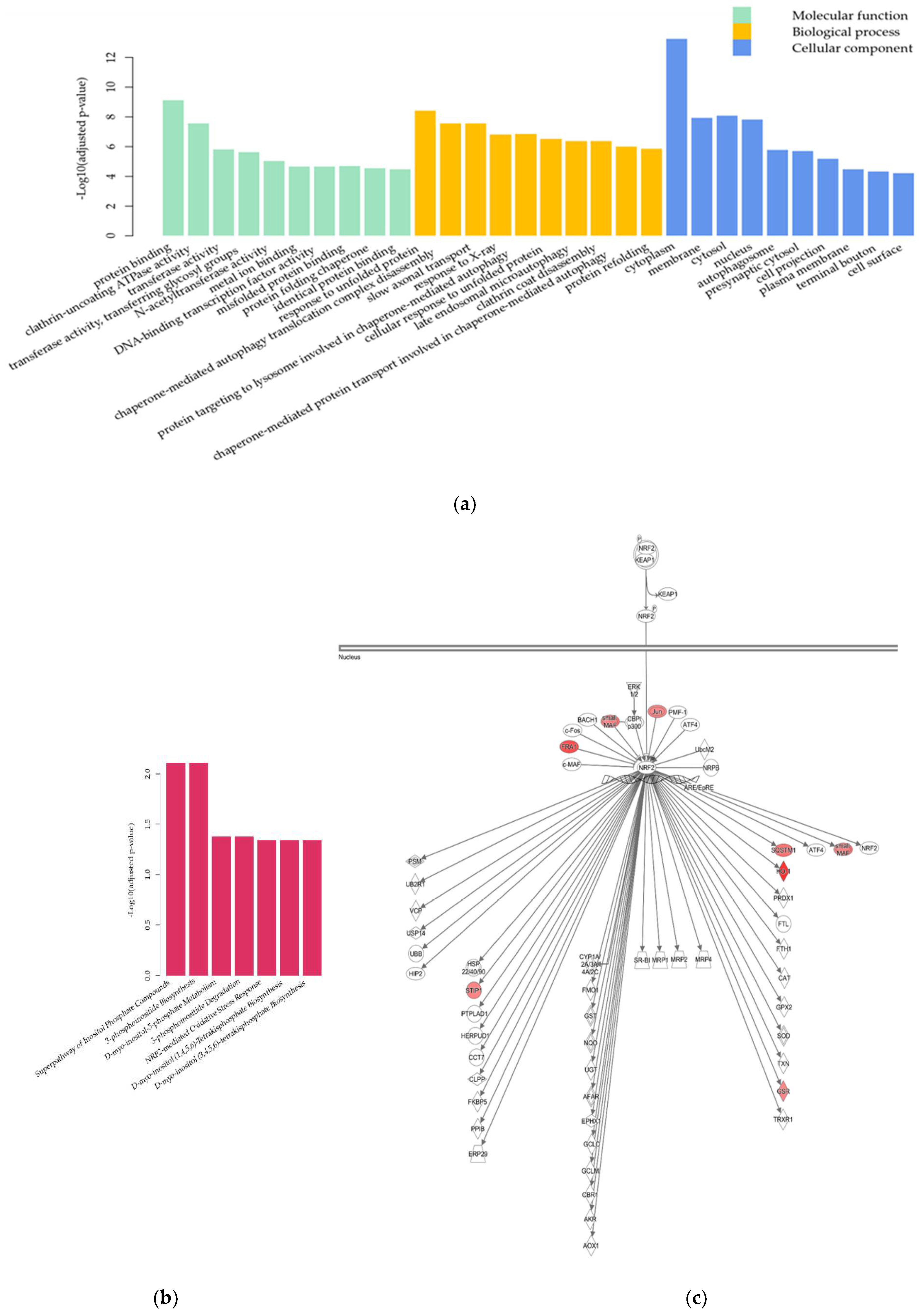
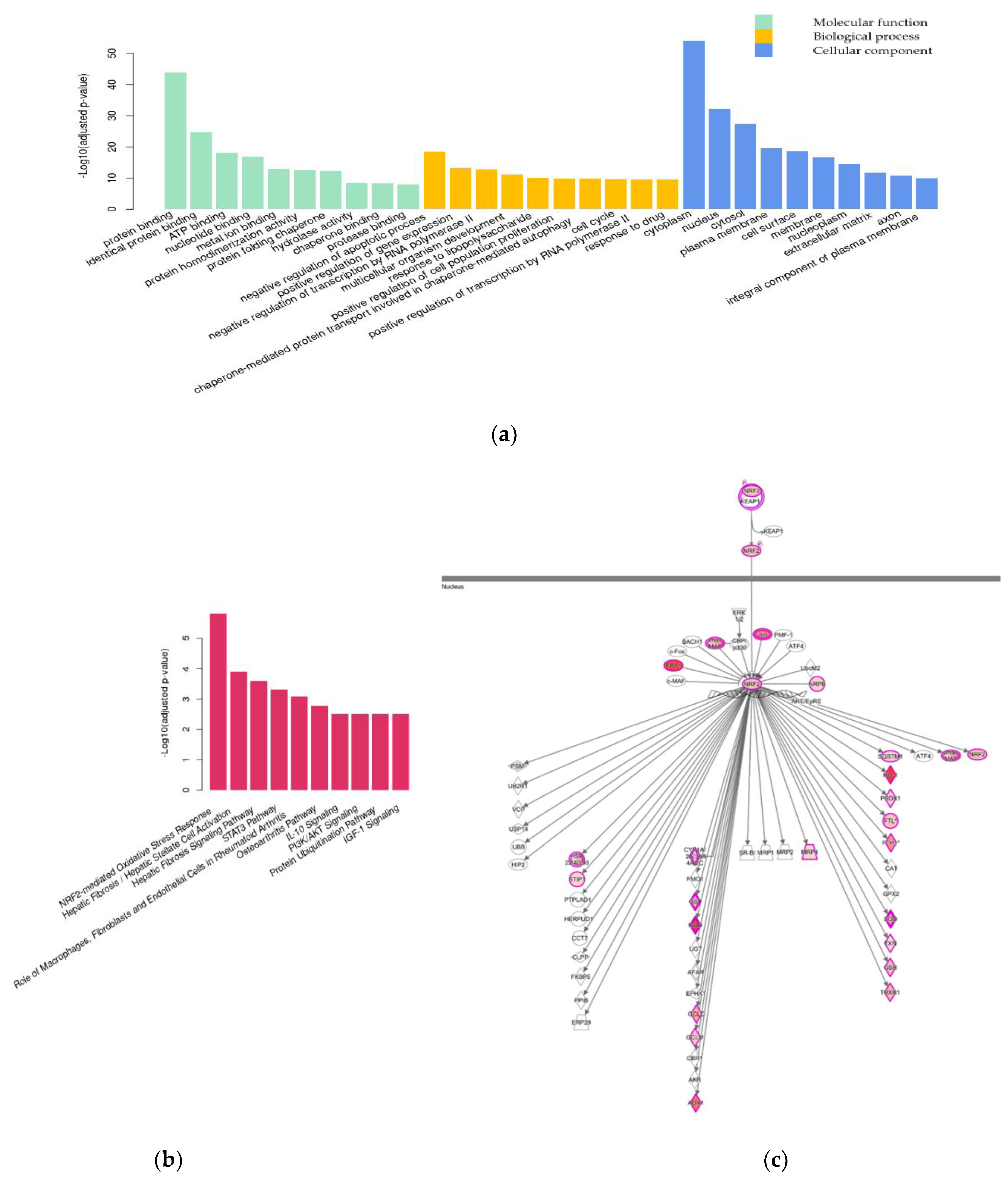
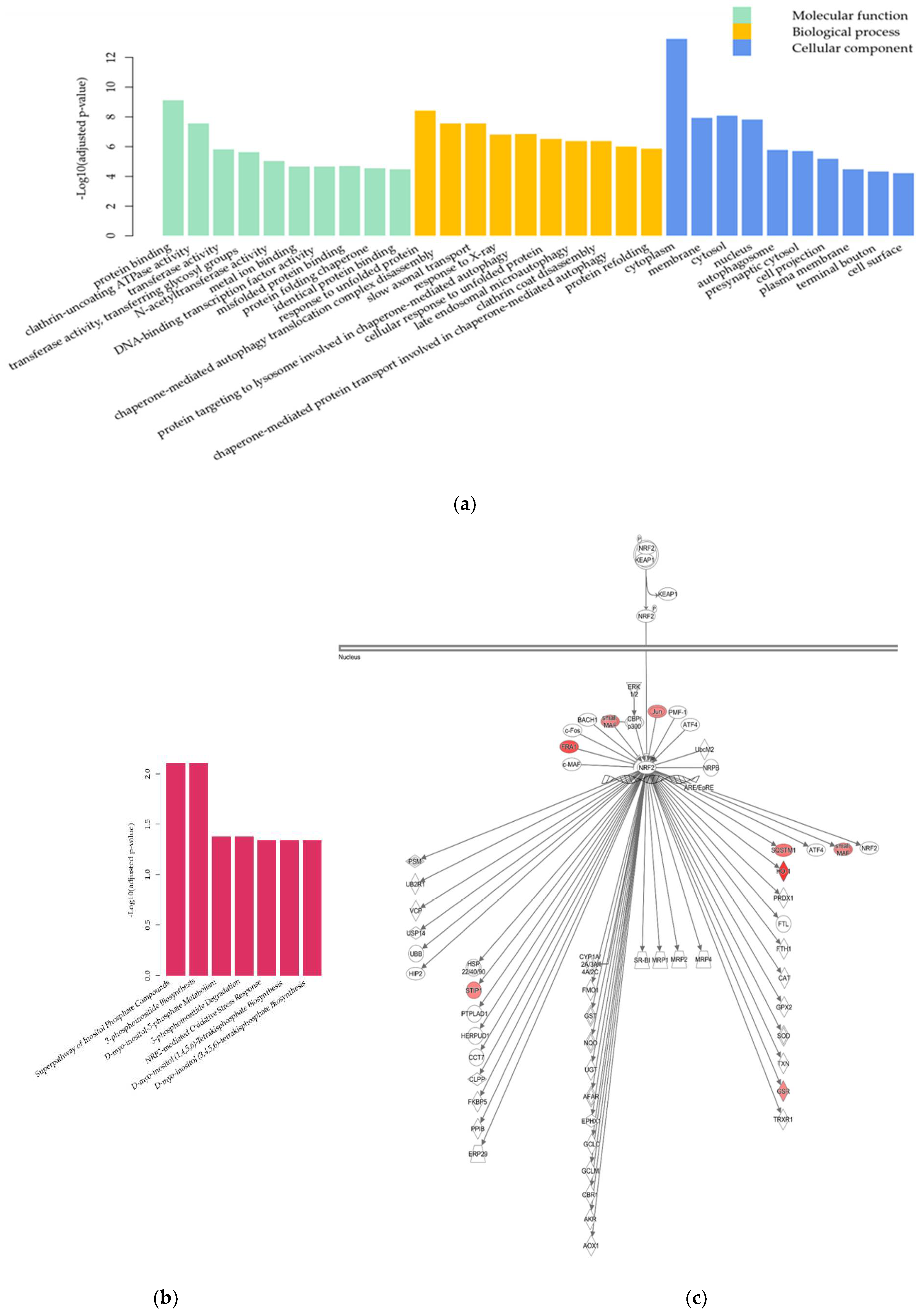
2.2. Transcriptome Analysis
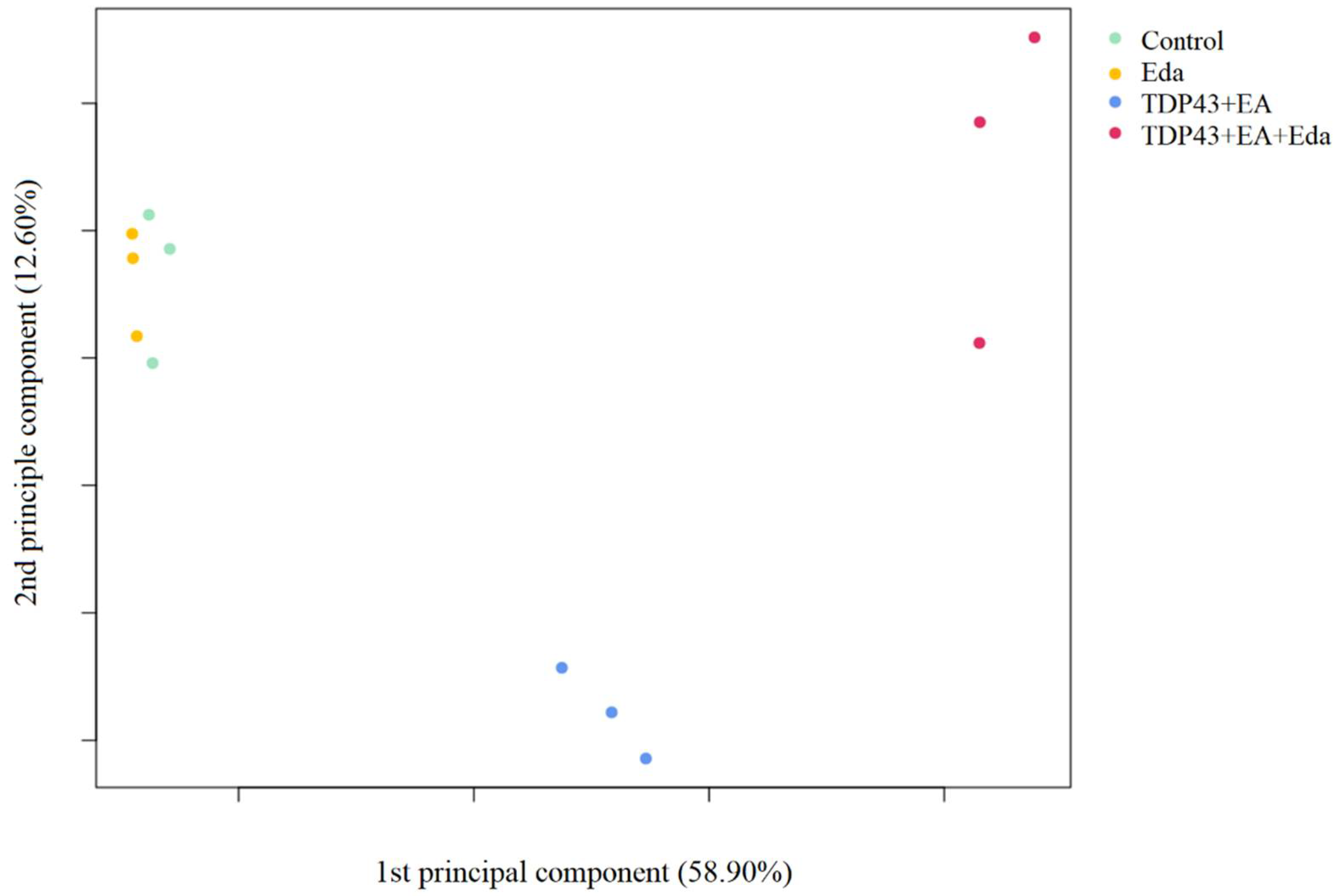
2.2.1. Principal Component Analysis
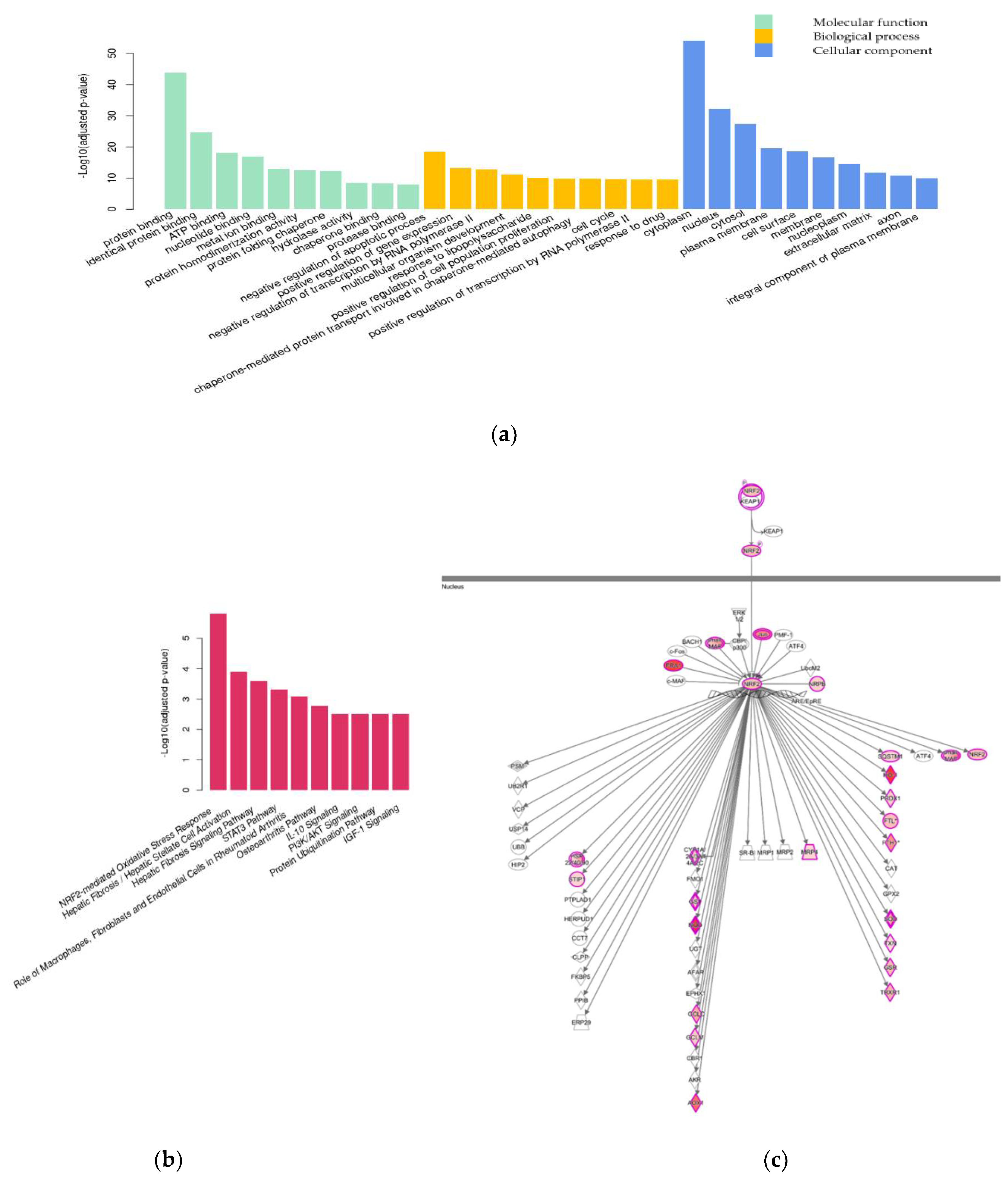
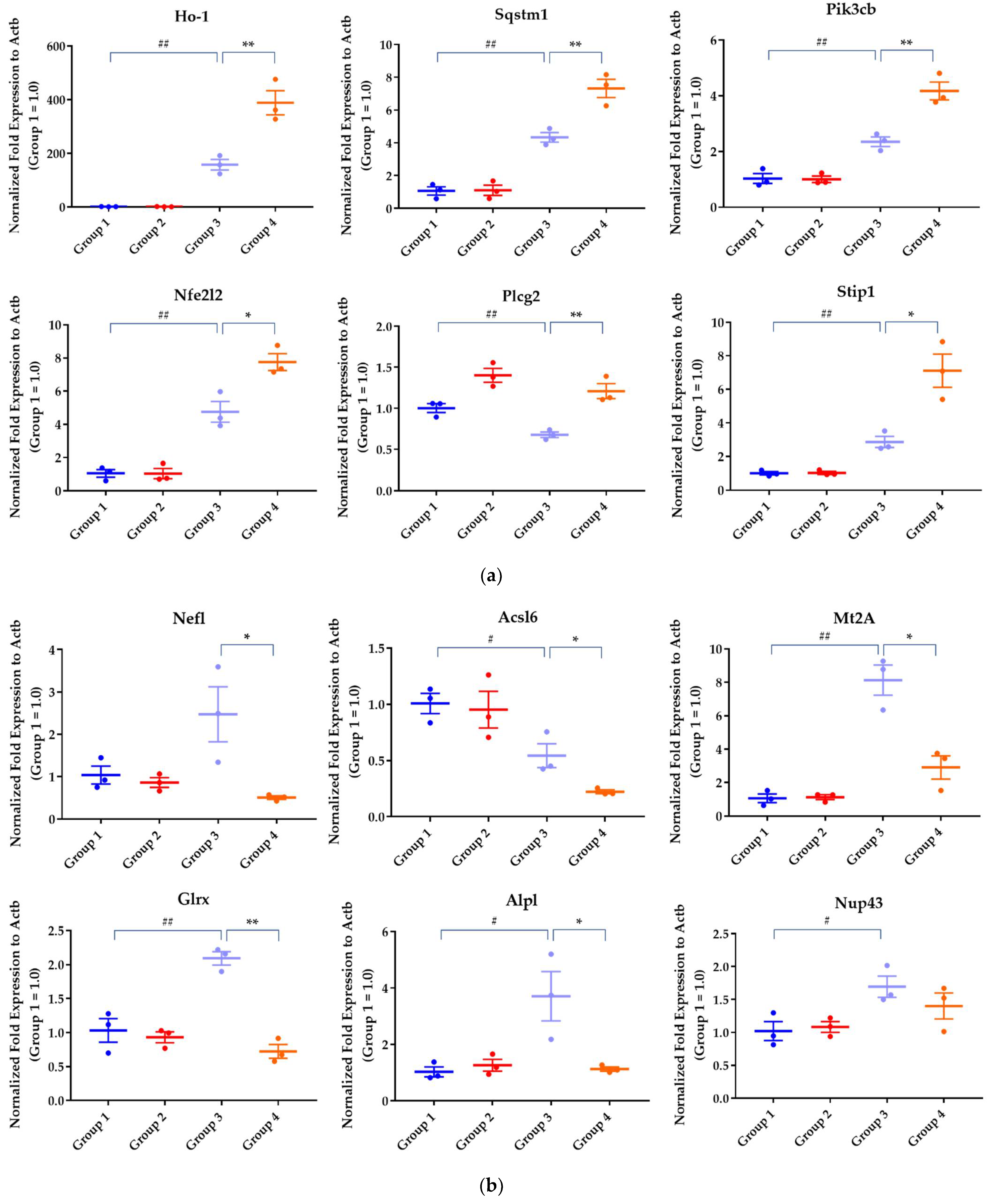
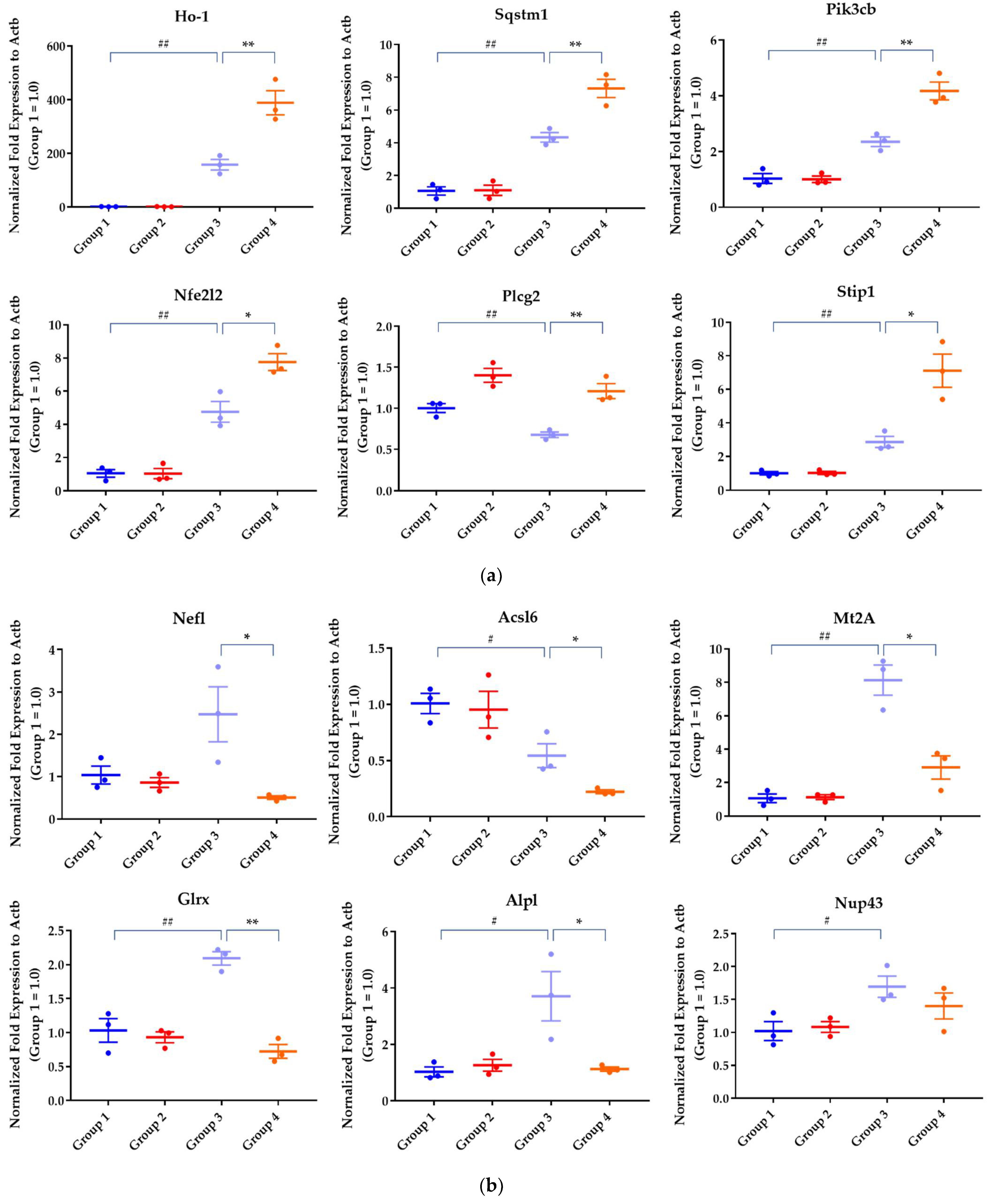
2.2.2. Differential Gene Expression Analysis
3. Discussion
3.1. The Effect of Edaravone against Cell Death in Rat Neural Stem Cell-Derived Neurons Transduced with Adenoviruses Expressing TDP-43
3.2. Transcriptome Analysis
3.2.1. Nrf2-Mediated Oxidative Stress Response
3.2.2. UPR and Autophagy
3.2.3. PI Metabolism
3.3. Mechanism of Action of Edaravone on the Neurotoxicity Induced by TDP-43 and Ethacrynic Acid
3.4. Limitation of the Study
4. Materials and Methods
4.1. Adult Rat Neural Stem Cell Line 1464R
4.2. Adenovirus Infection
4.3. Edaravone and/or Ethacrynic Acid Treatment
4.4. Cell Viability Assay
4.5. Double-Stranded cDNA Amplification
4.6. Library Preparation
4.7. Sequencing
4.8. Sequence Analysis
4.9. Principal Component Analysis
4.10. Differential Expression Analysis
4.11. GO Enrichment Analysis
4.12. qRT-PCRStatistical Analysis
4.13. Pathway Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Shukla, S. Role of Edaravone as a Treatment Option for Patients with Amyotrophic Lateral Sclerosis. Pharmaceuticals 2020, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Spencer, D.M.; Halloran, M.A.; Soo, K.Y.; Atkin, J.D. Redox regulation in amyotrophic lateral sclerosis. Oxid. Med. Cell. Longev. 2013, 2013, 408681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Aoki, M.; Warita, H.; Mizuno, H.; Suzuki, N.; Yuki, S.; Itoyama, Y. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011, 1382, 321–325. [Google Scholar] [CrossRef]
- Ikeda, K.; Iwasaki, Y. Edaravone, a Free Radical Scavenger, Delayed Symptomatic and Pathological Progression of Motor Neuron Disease in the Wobbler Mouse. PLoS ONE 2015, 10, e0140316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watabe, K.; Akiyama, K.; Kawakami, E.; Ishii, T.; Endo, K.; Yanagisawa, H.; Sango, K.; Tsukamoto, M. Adenoviral expression of TDP-43 and FUS genes and shRNAs for protein degradation pathways in rodent motoneurons in vitro and in vivo. Neuropathology 2014, 34, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate—A Practical And Powerful Approach To Multiple Testing. J. Royal Statist. Soc. Series B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, D.; Ayala, V.; Granado-Serrano, A.B.; Jové, M.; Torres, P.; Boada, J.; Cabré, R.; Ramírez-Núñez, O.; Gonzalo, H.; Soler-Cantero, A.; et al. Interplay between TDP-43 and docosahexaenoic acid-related processes in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 88, 148–160. [Google Scholar] [CrossRef]
- Harley, J.; Patani, R. Stress-Specific Spatiotemporal Responses of RNA-Binding Proteins in Human Stem-Cell-Derived Motor Neurons. Int. J. Mol. Sci. 2020, 21, 8346. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.K.; Chiang, M.H.; Toh, E.K.; Chang, C.F.; Huang, T.H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [Green Version]
- Harley, J.; Clarke, B.E.; Patani, R. The Interplay of RNA Binding Proteins, Oxidative Stress and Mitochondrial Dysfunction in ALS. Antioxidants 2021, 10, 552. [Google Scholar] [CrossRef]
- Kraus, R.L.; Pasieczny, R.; Lariosa-Willingham, K.; Turner, M.S.; Jiang, A.; Trauger, J.W. Antioxidant properties of minocycline: Neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J. Neurochem. 2005, 94, 819–827. [Google Scholar] [CrossRef]
- Hisano, K.; Watanabe, M.; Morimoto, Y. Protective effects of the free radical scavenger edaravone against glutamate neurotoxicity in nearly pure neuronal culture. J. Anesth. 2009, 23, 363–369. [Google Scholar] [CrossRef]
- Jami, M.S.; Salehi-Najafabadi, Z.; Ahmadinejad, F.; Hoedt, E.; Chaleshtori, M.H.; Ghatrehsamani, M.; Neubert, T.A.; Larsen, J.P.; Møller, S.G. Edaravone leads to proteome changes indicative of neuronal cell protection in response to oxidative stress. Neurochem. Int. 2015, 90, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Ito, K.; Sekine, S.; Tajima, A.; Horie, T. Ethacrynic-acid-induced glutathione depletion and oxidative stress in normal and Mrp2-deficient rat liver. Free Radic. Biol. Med. 2004, 37, 1718–1729. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp. Cell Res. 2019, 384, 111592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Minj, E.; Yadav, R.K.; Mehan, S. Targeting abnormal Nrf2/HO-1 signaling in amyotrophic lateral sclerosis: Current Insights on drug targets and influences on neurological disorders. Curr. Mol. Med. 2021, 21, 630–644. [Google Scholar] [CrossRef]
- Tian, Y.P.; Che, F.Y.; Su, Q.P.; Lu, Y.C.; You, C.P.; Huang, L.M.; Wang, S.G.; Wang, L.; Yu, J.X. Effects of mutant TDP-43 on the Nrf2/ARE pathway and protein expression of MafK and JDP2 in NSC-34 cells. Genet. Mol. Res. 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Foster, A.; Scott, D.; Layfield, R.; Rea, S.L. An FTLD-associated SQSTM1 variant impacts Nrf2 and NF-κB signalling and is associated with reduced phosphorylation of p62. Mol. Cell. Neurosci. 2019, 98, 32–45. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, D.; Liu, X.; Liu, X.; He, J.; Zhang, N.; Tang, L. hTBK1-c.978T>A mutation promotes the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 signaling. Am. J. Transl. Res. 2020, 12, 7386–7394. [Google Scholar]
- Zhang, D.; Xiao, Y.; Lv, P.; Teng, Z.; Dong, Y.; Qi, Q.; Liu, Z. Edaravone attenuates oxidative stress induced by chronic cerebral hypoperfusion injury: Role of ERK/Nrf2/HO-1 signaling pathway. Neurol. Res. 2018, 40, 1–10. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.; Chen, Z.; Geng, J.; Wang, Y.; Feng, H.; Zhu, G.; Chen, Q. Edaravone Reduces Iron-Mediated Hydrocephalus and Behavioral Disorder in Rat by Activating the Nrf2/HO-1 Pathway. J. Stroke Cerebrovasc. Dis. 2018, 27, 3511–3520. [Google Scholar] [CrossRef]
- Zhang, M.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Xiao, J.; Zhang, H.Y.; Chen, D.Q. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp. Ther. Med. 2019, 18, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Wu, Z.; Wang, X.; Gao, C.; Liu, R.; Kang, F.; Dai, M. Protective effects of combined treatment with mild hypothermia and edaravone against cerebral ischemia/reperfusion injury via oxidative stress and Nrf2 pathway regulation. Int. J. Oncol. 2020, 57, 500–508. [Google Scholar] [CrossRef]
- Ohta, Y.; Nomura, E.; Shang, J.; Feng, T.; Huang, Y.; Liu, X.; Shi, X.; Nakano, Y.; Hishikawa, N.; Sato, K.; et al. Enhanced oxidative stress and the treatment by edaravone in mice model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2019, 97, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Guo, Y.; Wang, H.; Zhao, L.; Ma, Z.; Li, T.; Liu, J.; Sun, M.; Jian, Y.; Yao, L.; et al. Edaravone reduces Aβ-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci. 2019, 221, 259–266. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.T.; Razzaq, A.; Di Gregorio, S.E.; Hong, S.; Charles, B.; Lopes, M.H.; Beraldo, F.; Prado, V.F.; Prado, M.A.M.; Duennwald, M.L. Hsp90 and its co-chaperone Sti1 control TDP-43 misfolding and toxicity. FASEB J. 2021, 35, e21594. [Google Scholar] [CrossRef]
- Kalmar, B.; Greensmith, L. Cellular Chaperones As Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- de Mena, L.; Lopez-Scarim, J.; Rincon-Limas, D.E. TDP-43 and ER Stress in Neurodegeneration: Friends or Foes? Front. Mol. Neurosci. 2021, 14, 772226. [Google Scholar] [CrossRef]
- Zhao, Z.Y.; Luan, P.; Huang, S.X.; Xiao, S.H.; Zhao, J.; Zhang, B.; Gu, B.B.; Pi, R.B.; Liu, J. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci. Ther. 2013, 19, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Long, H.; Li, Y.; Liu, Y.; Zhou, W.; Li, Q.; Yin, G.; Zhang, N.; Cai, W. Edaravone protects against glutamate-induced PERK/EIF2α/ATF4 integrated stress response and activation of caspase-12. Brain Res. 2013, 1519, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.D.; Rea, S.L. The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Neural Regen. Res. 2020, 15, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Volpatti, J.R.; Al-Maawali, A.; Smith, L.; Al-Hashim, A.; Brill, J.A.; Dowling, J.J. The expanding spectrum of neurological disorders of phosphoinositide metabolism. Dis. Model. Mech. 2019, 12, dmm038174. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Millán, J.L. What Can We Learn About the Neural Functions of TNAP from Studies on Other Organs and Tissues? Subcell. Biochem. 2015, 76, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 2019, 9, 9968. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Mitchell, J.C. Mechanisms of TDP-43 Proteinopathy Onset and Propagation. Int. J. Mol. Sci. 2021, 22, 6004. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Shaw, P.J.; De Vos, K.J. Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front. Mol. Neurosci. 2017, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, G.; Milani, P. Endoplasmic reticulum, oxidative stress and their complex crosstalk in neurodegeneration: Proteostasis, signaling pathways and molecular chaperones. AIMS Mol. Sci. 2017, 4, 424–444. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.r-project.org/ (accessed on 17 November 2021).
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Group 1 | Group 2 | Group 3 | Group 4 |
|---|---|---|---|---|
| Group 1 (Control) | 0/0 | 853/467 | 1654/1095 | |
| Group 2 (Eda) | 876/478 | 1661/1130 | ||
| Group 3 (TDP-43 + EA) | 169/260 | |||
| Group 4 (TDP-43 + EA + Eda) |
| Gene Name | Description | Group 3 to Group 1 | Group 4 to Group 3 | ||
|---|---|---|---|---|---|
| log2 Ratio | p Values | log2 Ratio | p Values | ||
| Upregulated genes a | |||||
| Ho-1 | heme oxygenase 1 | 6.601 | 0.000 | 1.755 | 4.712 × 10−44 |
| Cebpb | CCAAT/enhancer binding protein beta | 2.930 | 4.358 × 10−164 | 1.661 | 1.309 × 10−28 |
| Plpp3 | phospholipid phosphatase 3 | 0.751 | 7.027 × 10−4 | 1.307 | 3.287 × 10−10 |
| Sqstm1 | sequestosome 1 | 1.366 | 1.227 × 10−55 | 1.225 | 2.665 × 10−26 |
| Pik3cb | phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit beta | 0.879 | 8.668 × 10−11 | 1.157 | 2.285 × 10−18 |
| Txndc2 | thioredoxin domain-containing 2 | 0.917 | 4.981 × 10−2 | 1.153 | 5.690 × 10−3 |
| Hspa8 | heat shock protein family A (Hsp70) member 8 | 1.931 | 1.714 × 10−79 | 1.151 | 4.385 × 10−13 |
| Hsph1 | heat shock protein family H (Hsp110) member 1 | 4.000 | 0.000 | 1.027 | 2.124 × 10−7 |
| Ptgds | prostaglandin D2 synthase | −1.115 | 1.478 × 10−4 | 0.954 | 2.334 × 10−3 |
| Nfe2l2 | nuclear factor erythroid 2-like 2 | 1.898 | 1.063 × 10−43 | 0.923 | 9.336 × 10−11 |
| Plcg2 | phospholipase C, gamma 2 | −1.570 | 3.015 × 10−11 | 0.881 | 4.049 × 10−3 |
| Downregulated genes b | |||||
| Nefl | neurofilament light | 0.738 | 1.168 × 10−1 | −1.778 | 5.902 × 10−7 |
| Acsl6 | acyl-CoA synthetase long-chain family member 6 | −1.225 | 2.073 × 10−6 | −1.582 | 1.693 × 10−5 |
| Mt2A | metallothionein 2A | 2.134 | 5.863 × 10−9 | −1.425 | 9.110 × 10−5 |
| Mt1 | metallothionein 1 | 0.866 | 8.031 × 10−6 | −1.392 | 3.600 × 10−9 |
| Glrx | glutaredoxin | 0.726 | 3.319 × 10−3 | −1.262 | 8.634 × 10−6 |
| Alpl | alkaline phosphatase, biomineralization associated | 1.344 | 1.392 × 10−4 | −1.098 | 1.057 × 10−3 |
| Nup43 | nucleoporin 43 | 0.947 | 6.178 × 10−5 | −0.888 | 1.606 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soejima-Kusunoki, A.; Okada, K.; Saito, R.; Watabe, K. The Protective Effect of Edaravone on TDP-43 Plus Oxidative Stress-Induced Neurotoxicity in Neuronal Cells: Analysis of Its Neuroprotective Mechanisms Using RNA Sequencing. Pharmaceuticals 2022, 15, 842. https://doi.org/10.3390/ph15070842
Soejima-Kusunoki A, Okada K, Saito R, Watabe K. The Protective Effect of Edaravone on TDP-43 Plus Oxidative Stress-Induced Neurotoxicity in Neuronal Cells: Analysis of Its Neuroprotective Mechanisms Using RNA Sequencing. Pharmaceuticals. 2022; 15(7):842. https://doi.org/10.3390/ph15070842
Chicago/Turabian StyleSoejima-Kusunoki, Aki, Kinya Okada, Ryuta Saito, and Kazuhiko Watabe. 2022. "The Protective Effect of Edaravone on TDP-43 Plus Oxidative Stress-Induced Neurotoxicity in Neuronal Cells: Analysis of Its Neuroprotective Mechanisms Using RNA Sequencing" Pharmaceuticals 15, no. 7: 842. https://doi.org/10.3390/ph15070842
APA StyleSoejima-Kusunoki, A., Okada, K., Saito, R., & Watabe, K. (2022). The Protective Effect of Edaravone on TDP-43 Plus Oxidative Stress-Induced Neurotoxicity in Neuronal Cells: Analysis of Its Neuroprotective Mechanisms Using RNA Sequencing. Pharmaceuticals, 15(7), 842. https://doi.org/10.3390/ph15070842