
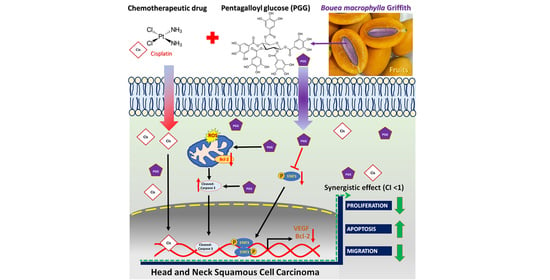
Pentagalloyl Glucose and Cisplatin Combination Treatment Exhibits a Synergistic Anticancer Effect in 2D and 3D Models of Head and Neck Carcinoma

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
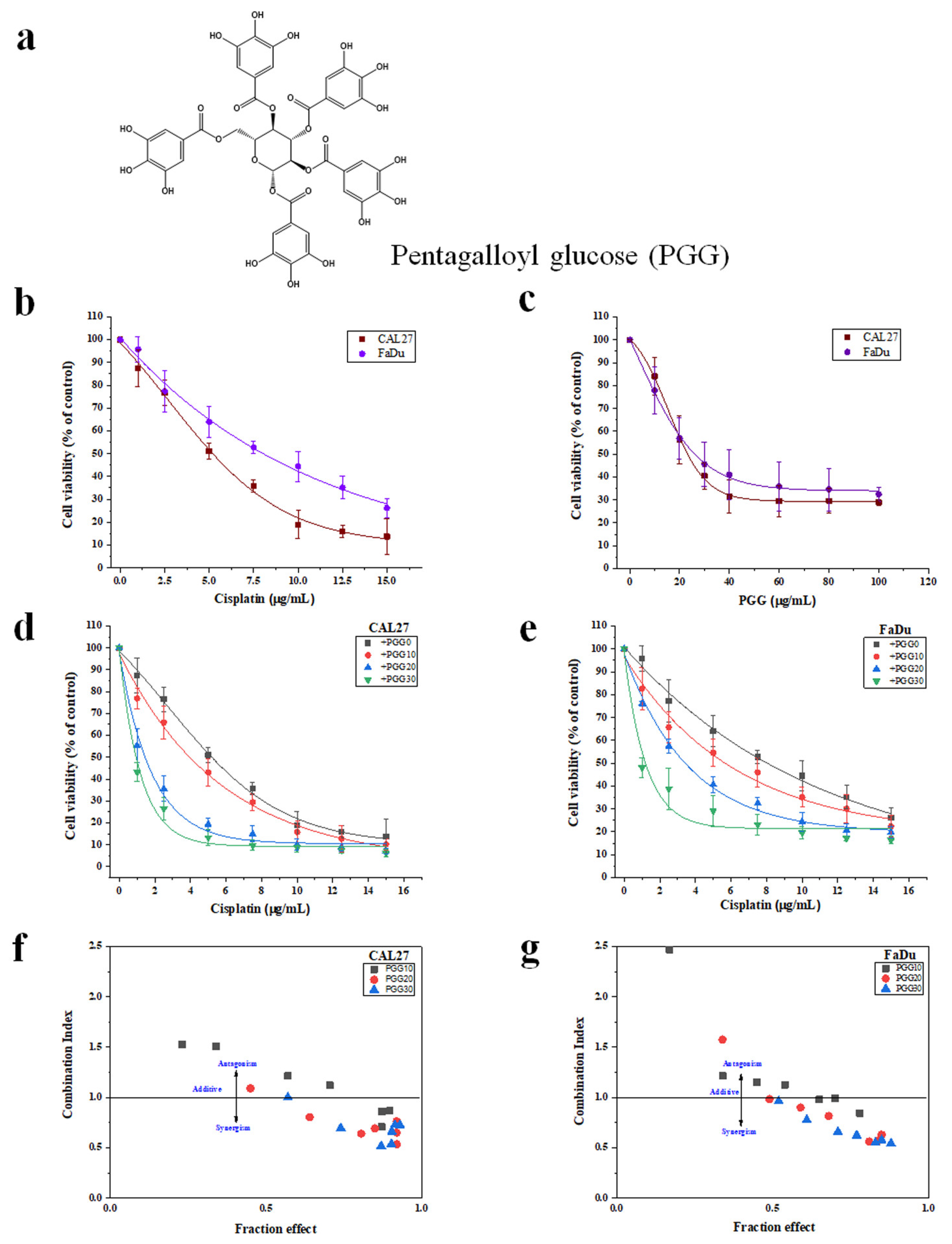
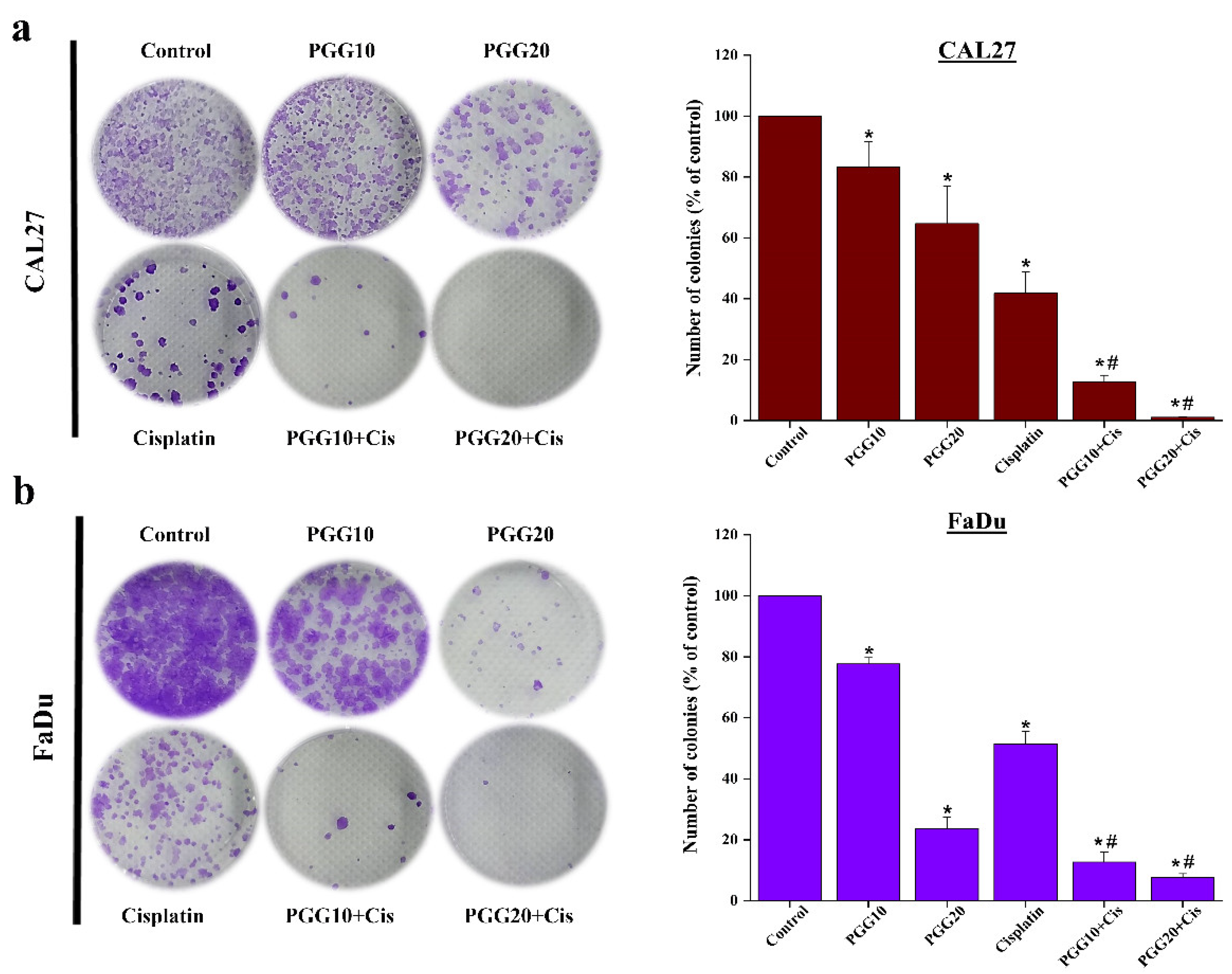
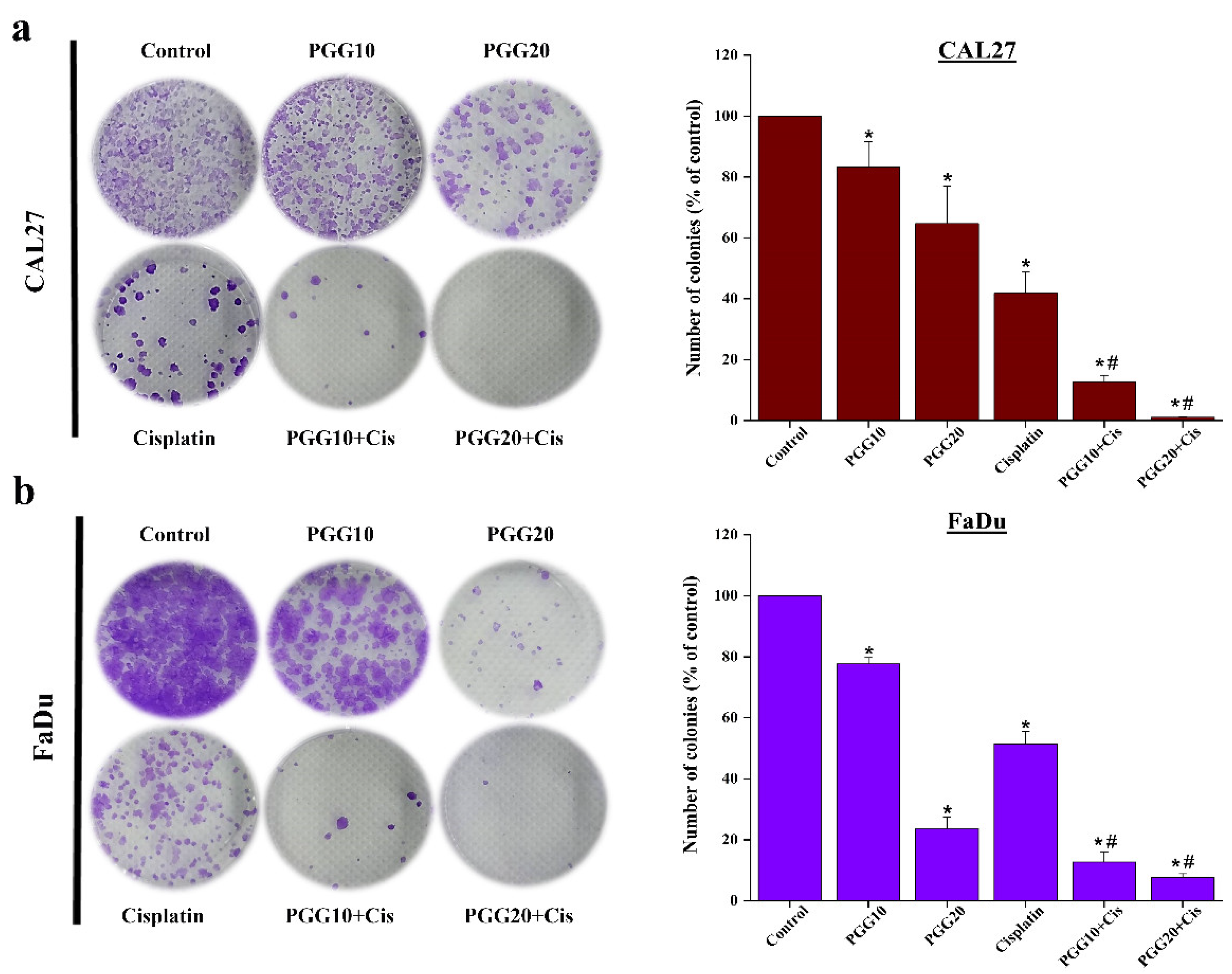
2.1. The Combination of PGG with Cisplatin Synergistically Inhibits Cell Proliferation and Clonogenic Survival of CAL27 and FaDu Cells in 2D Culture
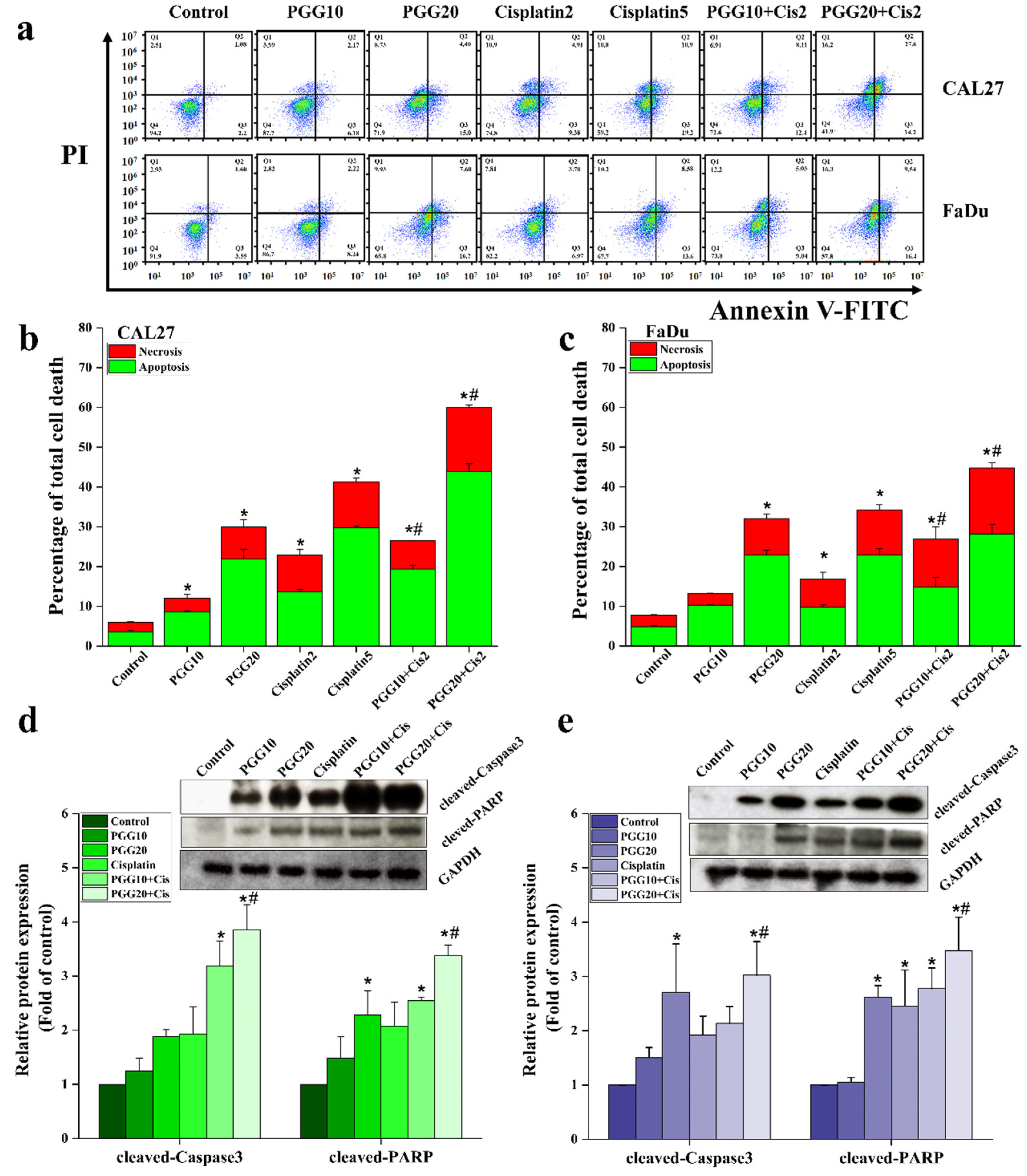
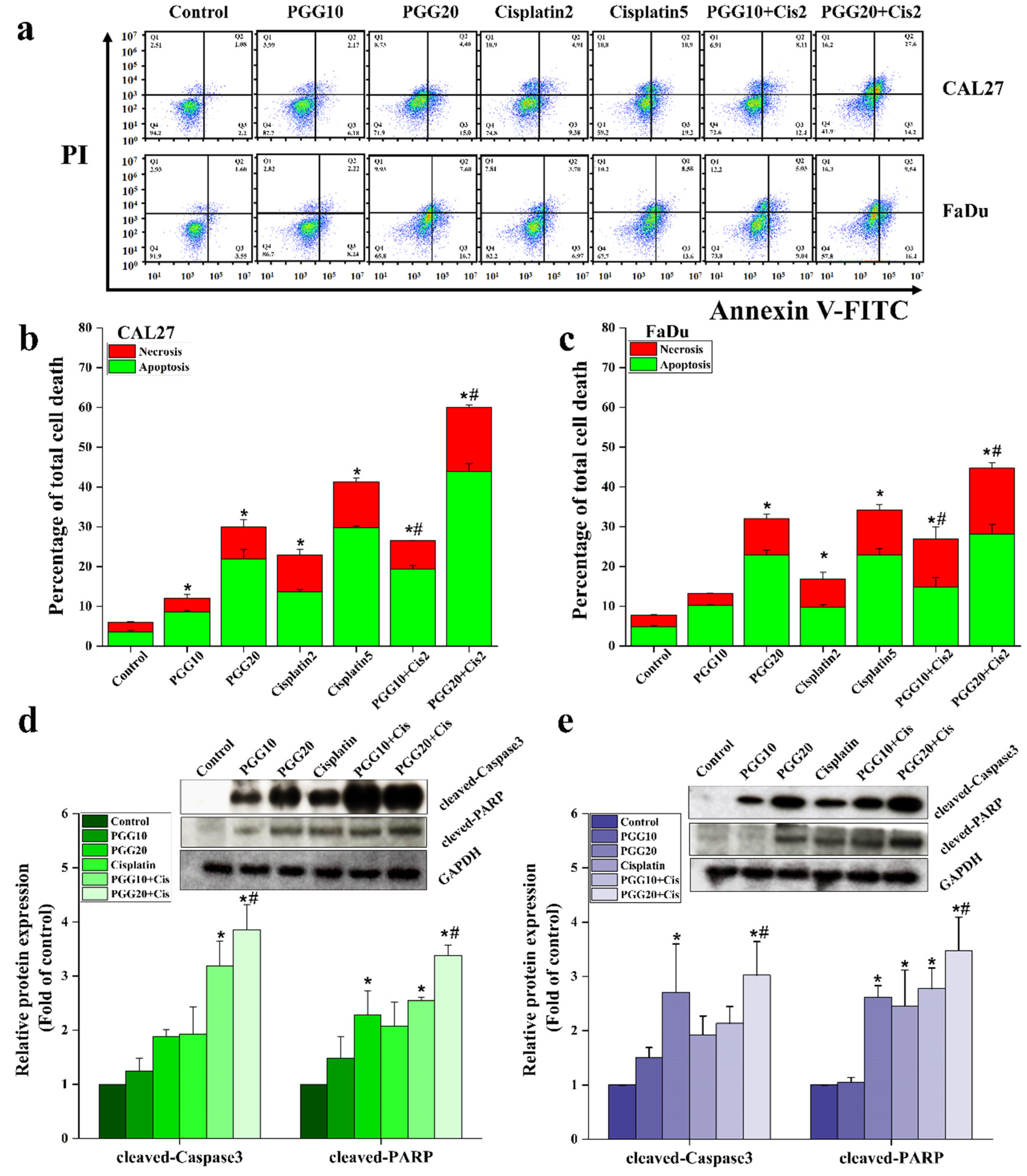
2.2. PGG Enhanced the Apoptosis Induction Effect of Cisplatin in CAL27 and FaDu Cells
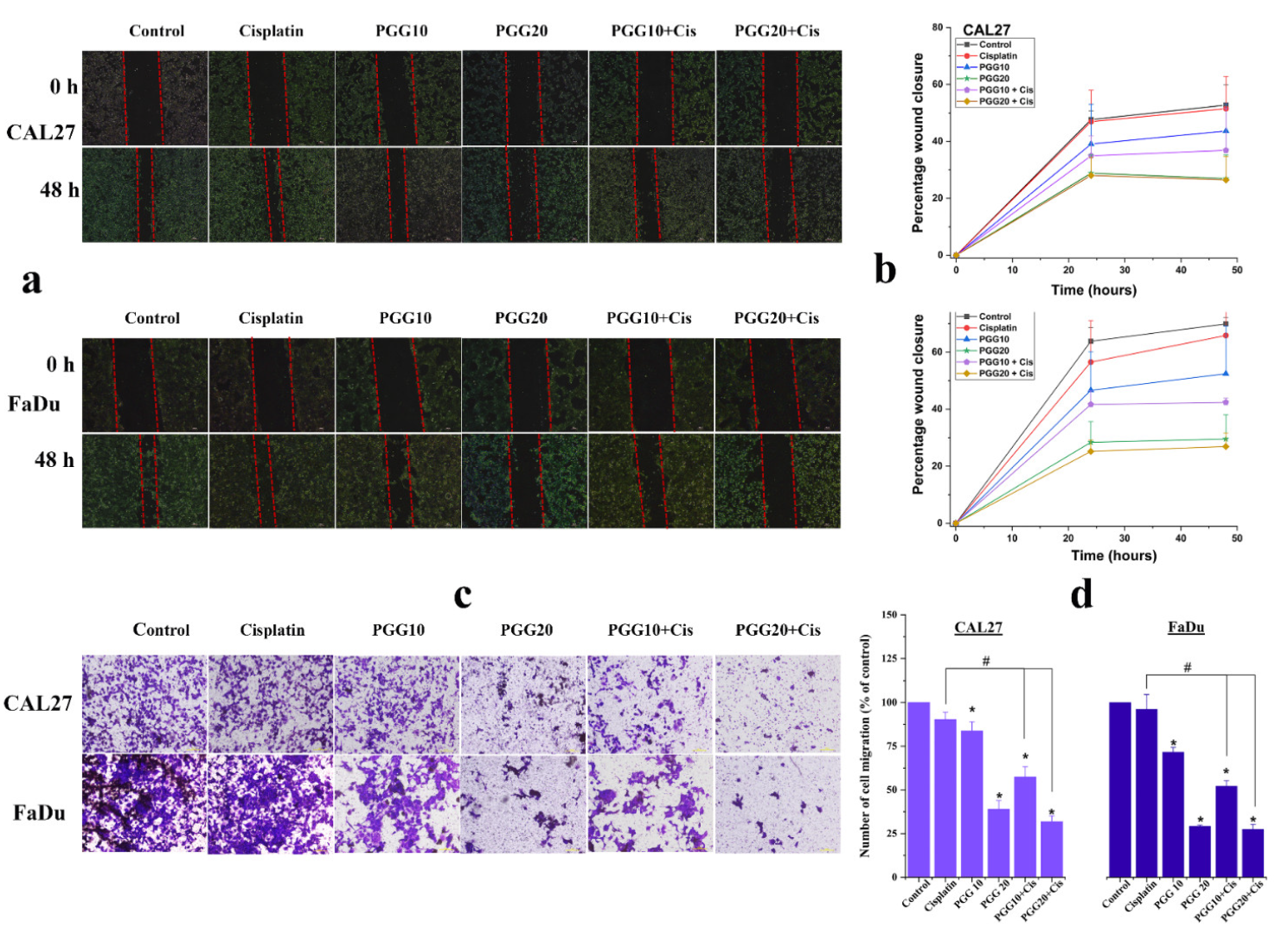
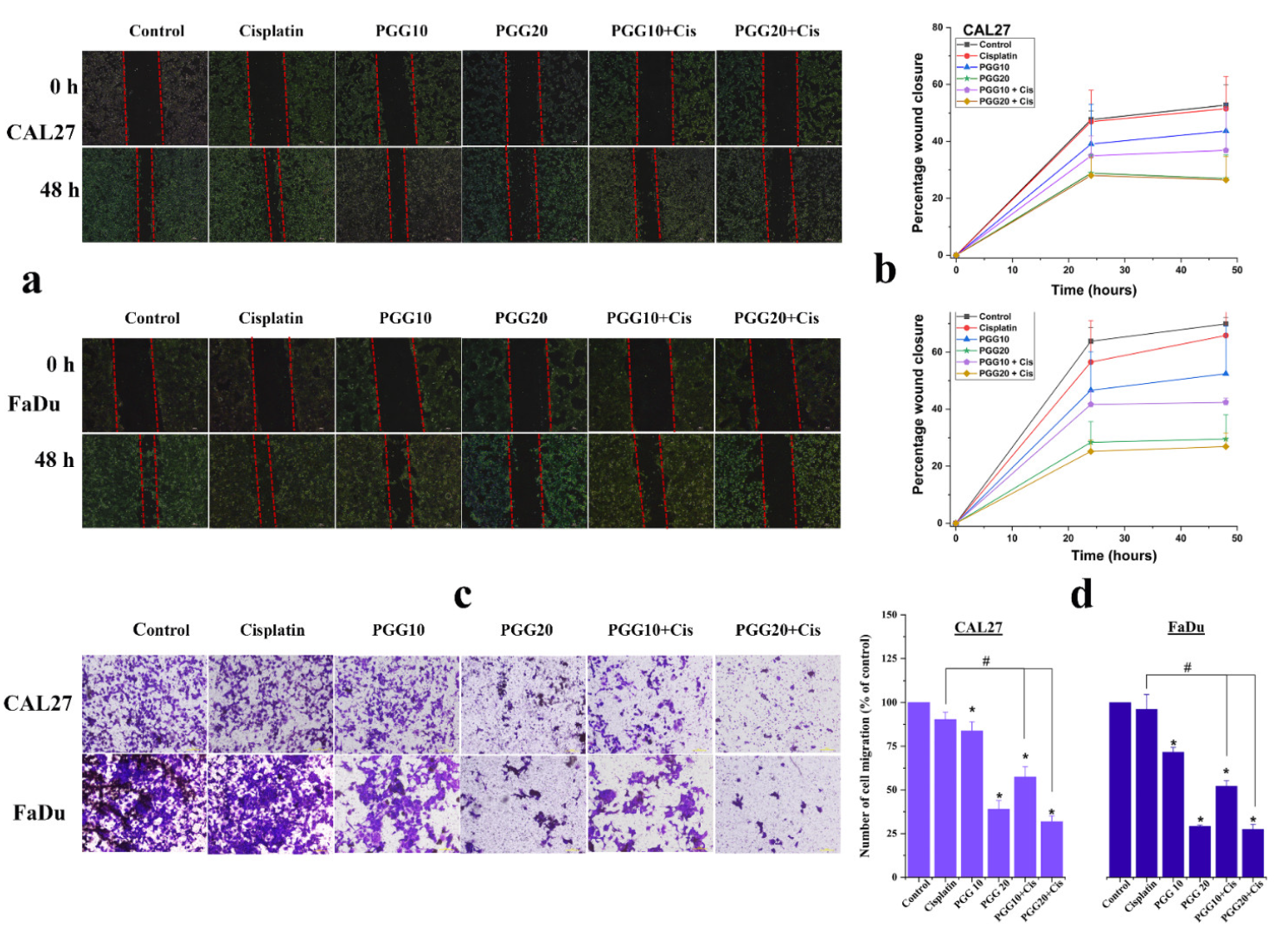
2.3. PGG Inhibits Migration and Promotes the Anti-Migration Effect of Cisplatin in CAL27 and FaDu Cells
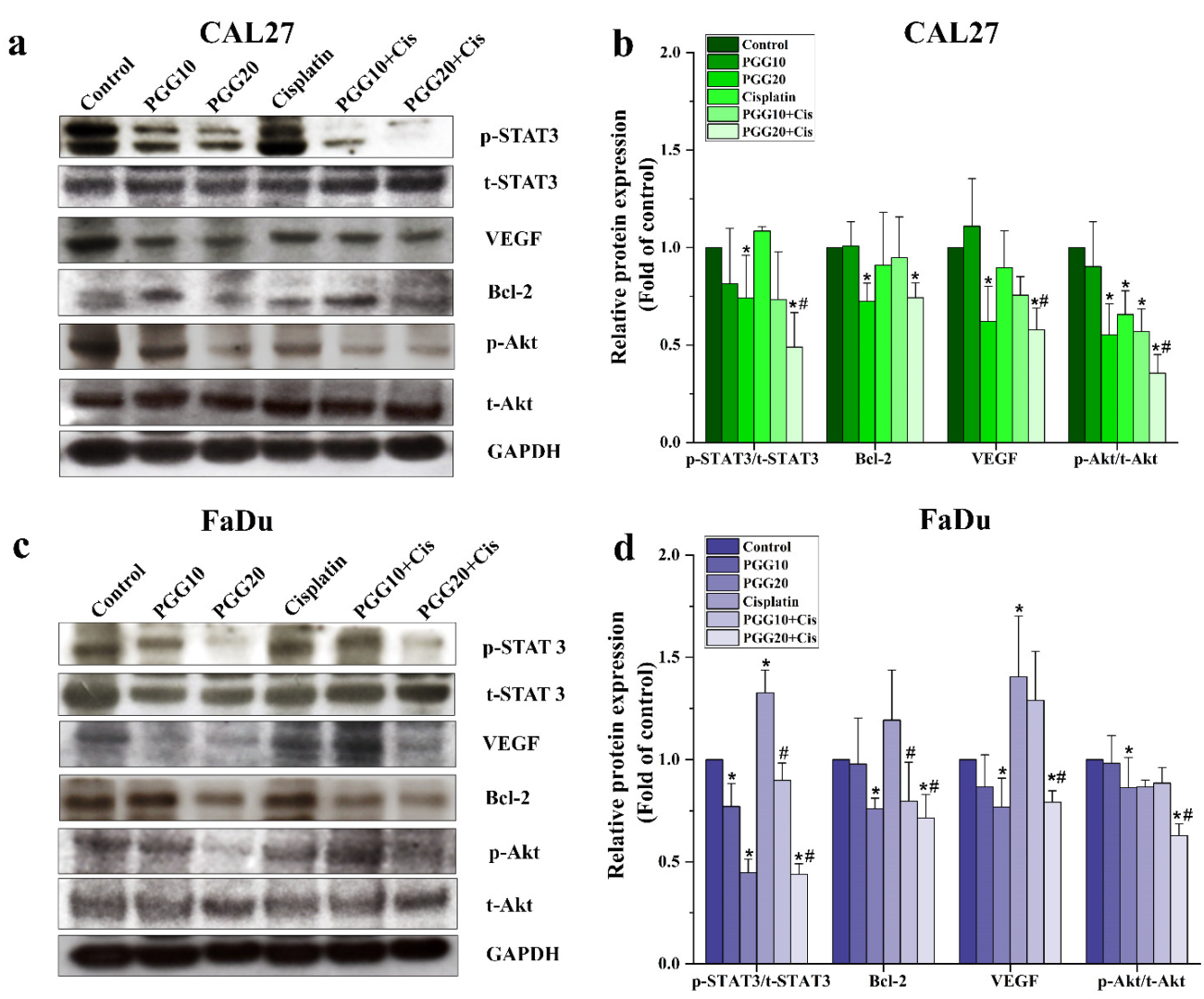
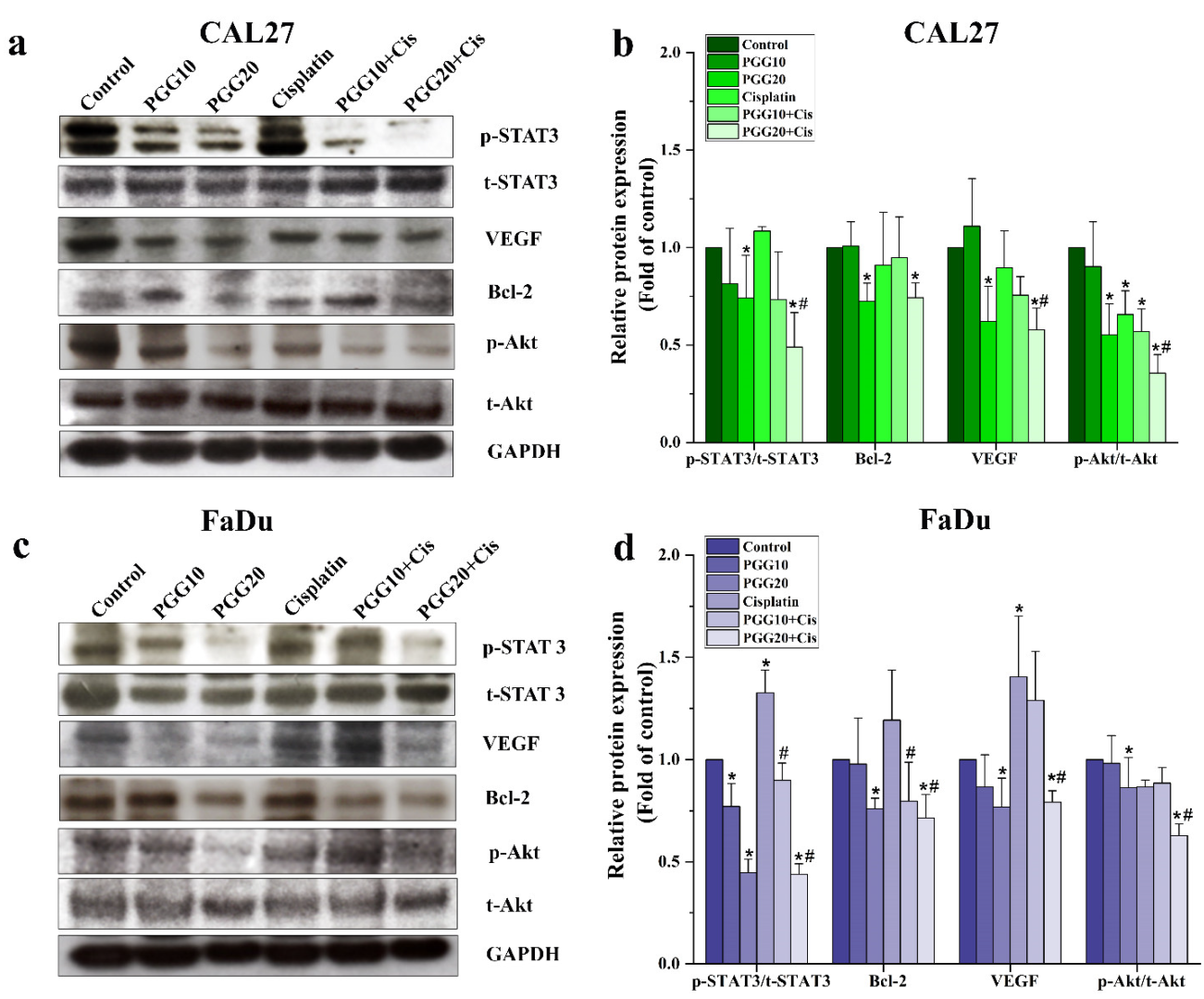
2.4. Combined Treatments of PGG and Cisplatin Abolished STAT3 and Akt Activation and Enhanced Drug Sensitivity of HNSCC Cells
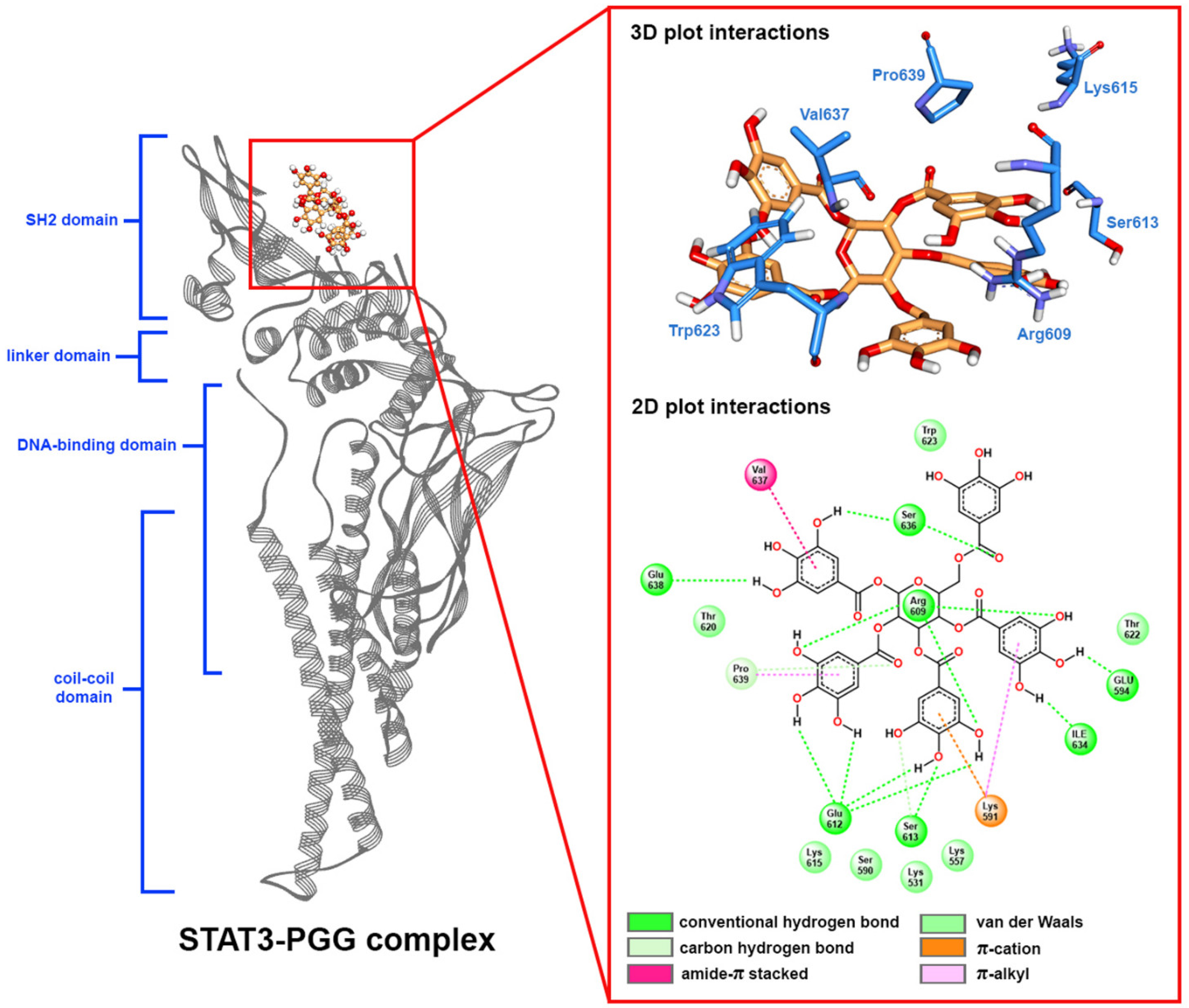
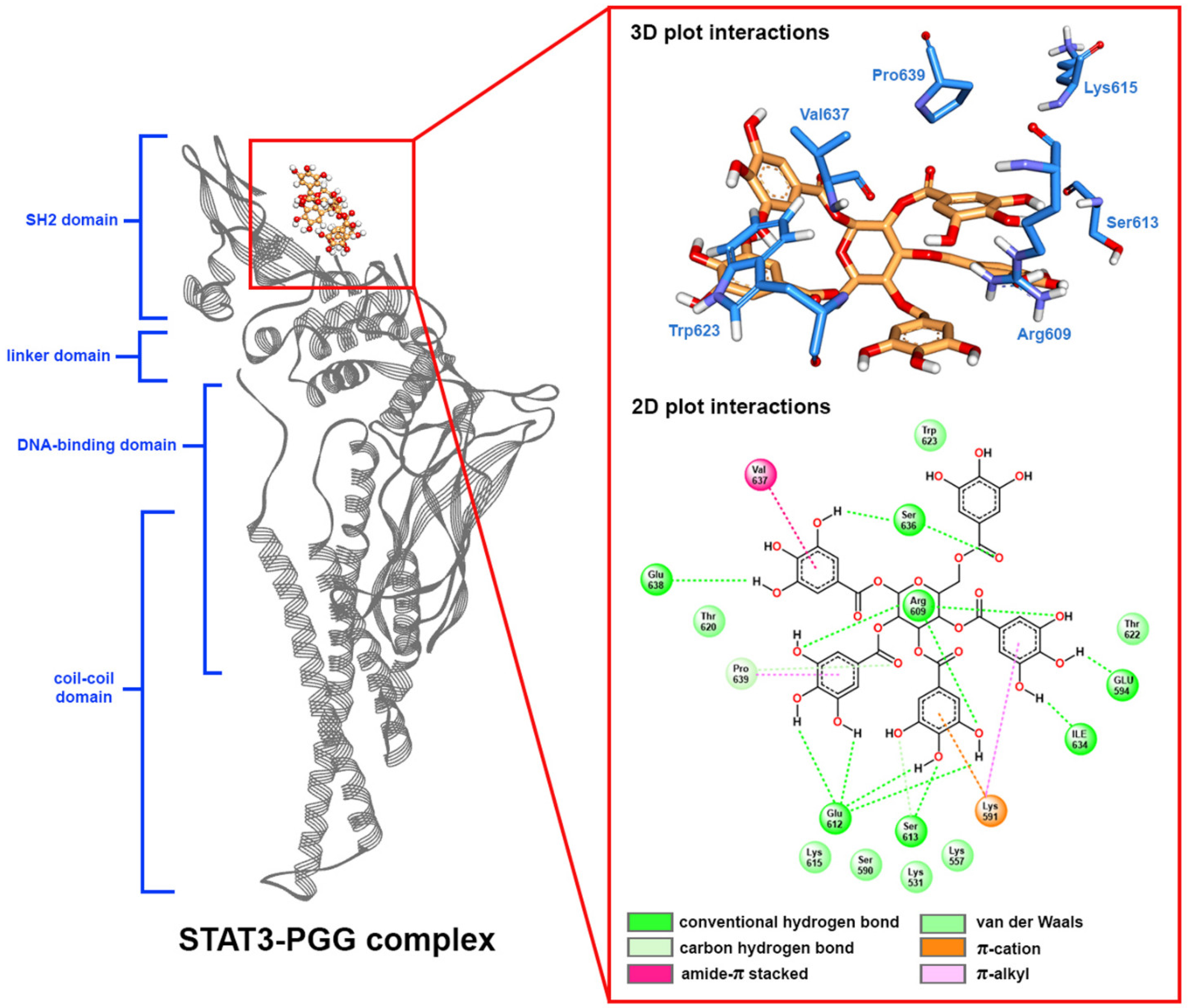
2.5. Molecular Docking
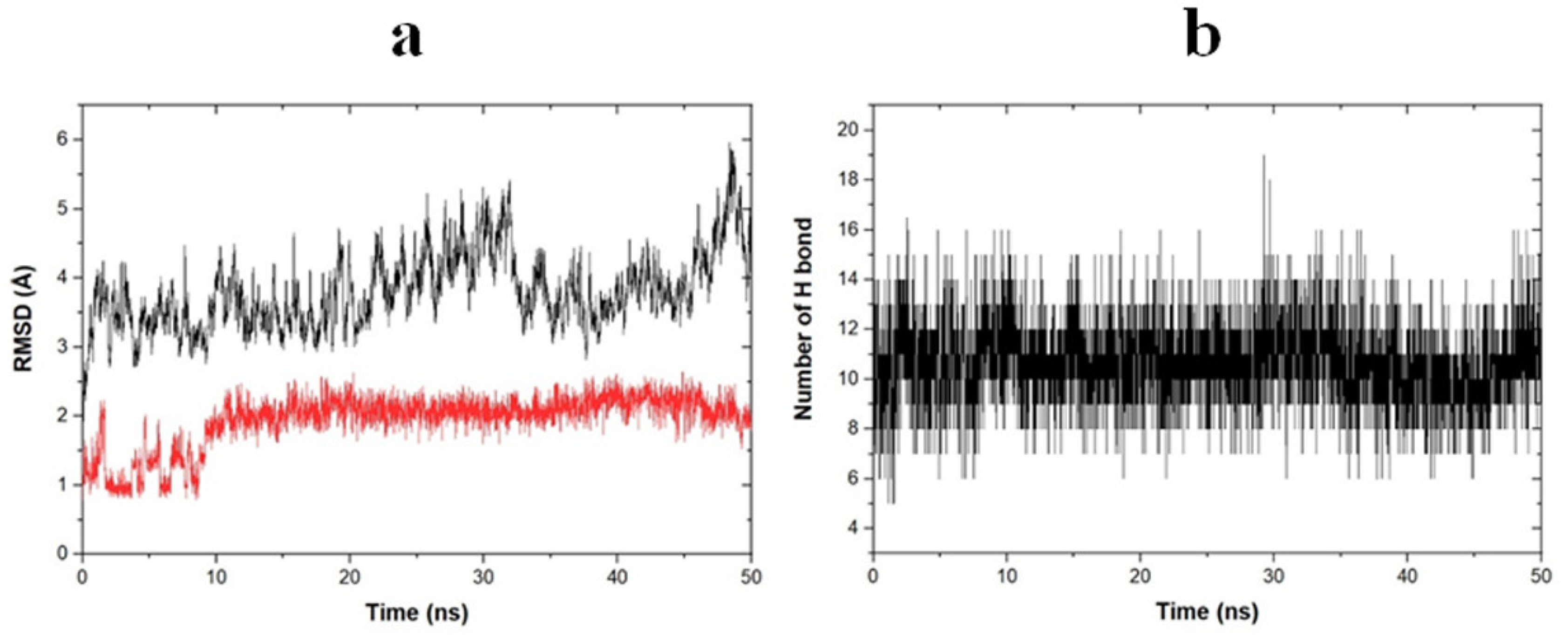
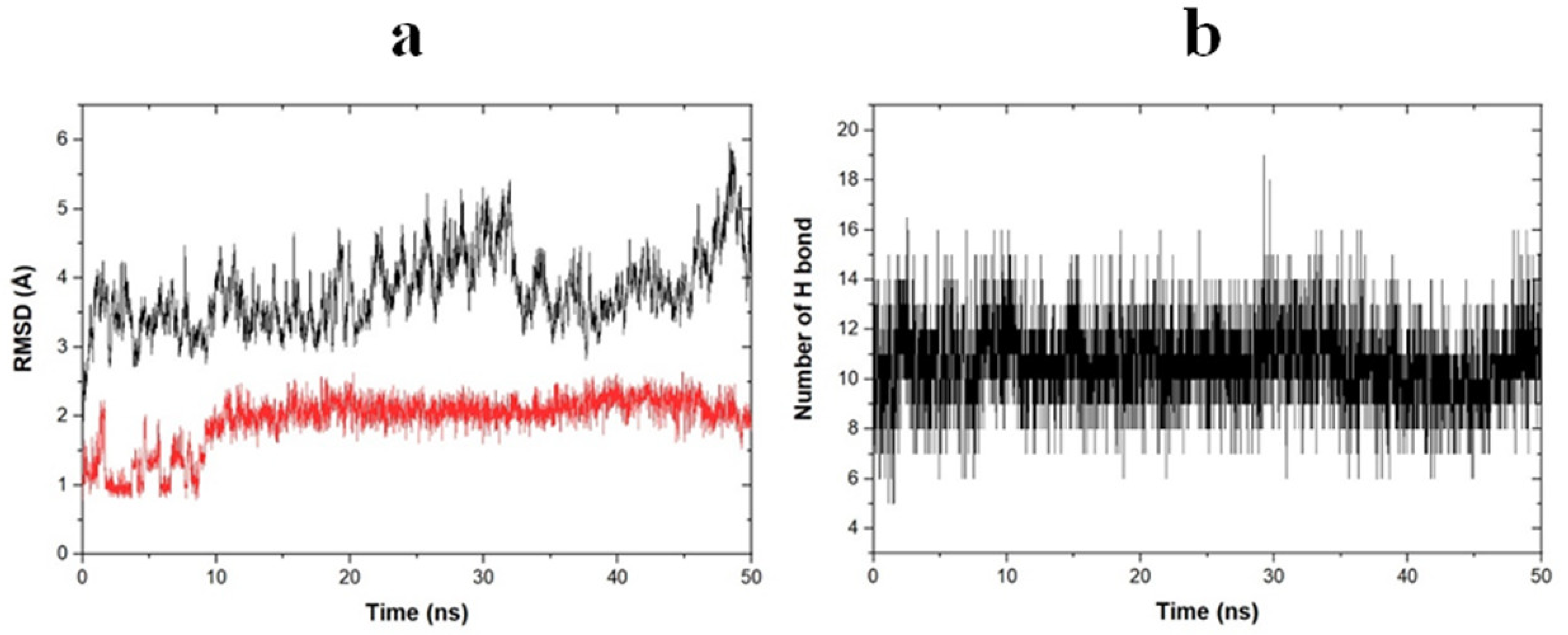
2.6. Molecular Dynamic Simulation
2.7. Free Binding Energy Calculation
2.8. The Combination of PGG and Cisplatin Exerts a Strong Cytotoxic Effect on HNSCC Cancer Cell Lines in the 3D Cell Culture
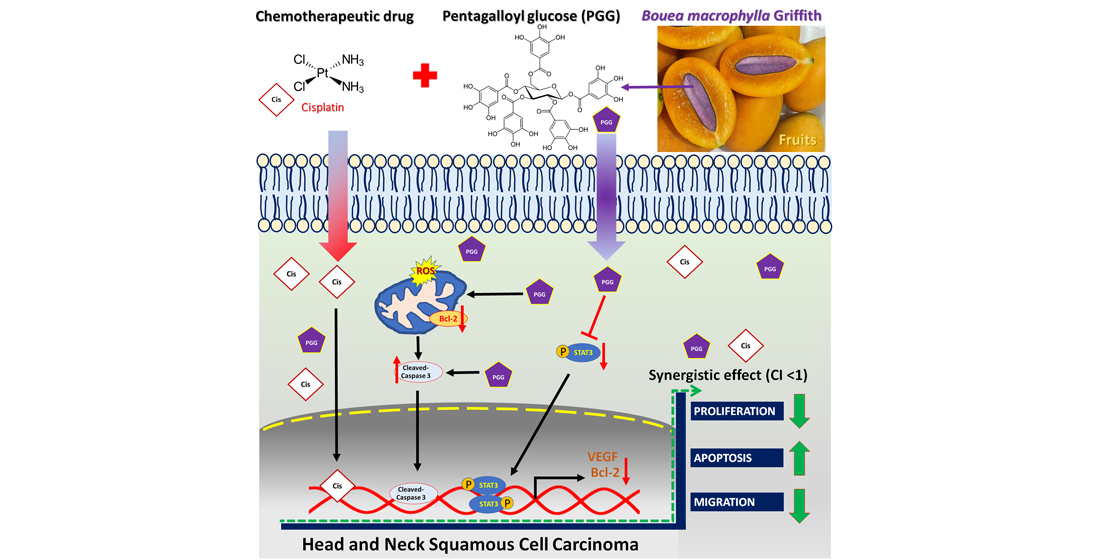
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. PGG Isolation and High-Performance Liquid Chromatography (HPLC) Quantification
4.3. Cell Lines and Cell Culture Conditions
4.4. Cell Viability Assay
4.5. Colony-Formation Assay
4.6. Apoptosis Assay in Monolayer Tumor Cells by Annexin V-FITC/PI Double Staining
4.7. Scratch Wound Healing Assay
4.8. Transwell Migration Assay
4.9. Western Blot Analysis
4.10. Three-Dimensional (3D) Tumor Model and Drug Response Assays
4.11. Cell Viability (Live/Dead) in 3-Dimensional Tumor Model by Propidium Iodide (PI)/Acridine Orange (AO) Staining
4.12. Molecular Docking
4.13. Molecular Dynamic Simulations
4.14. Free Binding Energy Calculations
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjoomataram, I.; Seiegel, R.L.; Torre, L.A.A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Lacas, B.; Carmel, A.; Landais, C.; Wong, S.J.; Licitra, L.; Tobias, J.S.; Burtness, B.; Ghi, M.G.; Cohen, E.E.W.; Grau, C.; et al. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 107 randomized trials and 19,805 patients, on behalf of MACH-NC Group. Radiother. Oncol. 2021, 156, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Machtay, M.; Moughan, J.; Trotti, A.; Garden, A.S.; Weber, R.S.; Cooper, J.S.; Forastiere, A.; Ang, K.K. Factors associated with severe late toxicity after concurrent chemoradiation for locally advanced head and neck cancer: An RTOG analysis. J. Clin. Oncol. 2008, 26, 3582–3589. [Google Scholar] [CrossRef]
- Pryor, D.I.; Porceddu, S.V.; Burmeister, B.H.; Guminski, A.; Thomson, D.B.; Shepherdson, K.; Poulsen, M. Enhanced toxicity with concurrent cetuximab and radiotherapy in head and neck cancer. Radiother. Oncol. 2009, 90, 172–176. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Gilad, Y.; Gellerman, G.; Lonard, D.M.; O’Malley, B.W. Drug Combination in Cancer Treatment-From Cocktails to Conjugated Combinations. Cancers 2021, 13, 669. [Google Scholar] [CrossRef]
- Garg, A.K.; Buchholz, T.A.; Aggarwal, B.B. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid. Redox Signal. 2005, 7, 1630–1647. [Google Scholar] [CrossRef]
- Bracci, L.; Fabbri, A.; Del Cornò, M.; Conti, L. Dietary Polyphenols: Promising adjuvants for colorectal cancer therapies. Cancers 2021, 13, 4499. [Google Scholar] [CrossRef]
- Maleki Dana, P.; Sadoughi, F.; Asemi, Z.; Yousefi, B. The role of polyphenols in overcoming cancer drug resistance: A comprehensive review. Cell Mol. Biol. Lett. 2022, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Kantapan, J.; Paksee, S.; Chawapun, P.; Sangthong, P.; Dechsupa, N. Pentagalloyl glucose- and ethyl gallate-rich extract from Maprang seeds induce apoptosis in MCF-7 breast cancer cells through mitochondria-mediated pathway. Evid. Based Complement Altern. Med. 2020, 2020, 5686029. [Google Scholar] [CrossRef] [PubMed]
- Kantapan, J.; Dechsupa, N.; Tippanya, D.; Nobnop, W.; Chitapanarux, I. Gallotannin from Bouea macrophylla seed extract suppresses cancer stem-like cells and radiosensitizes head and neck cancer. Int. J. Mol. Sci. 2021, 22, 9253. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Lee, H.J.; Shaik, A.A.; Nkhata, K.; Xing, C.; Zhang, J.; Jeong, S.J.; Kim, S.H.; Lu, J. Penta-O-galloyl-beta-D-glucose induces G1 arrest and DNA replicative S-phase arrest independently of cyclin-dependent kinase inhibitor 1A, cyclin-dependent kinase inhibitor 1B and P53 in human breast cancer cells and is orally active against triple negative xenograft growth. Breast Cancer Res. 2010, 12, R67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Seo, N.J.; Jeong, S.J.; Park, Y.; Jung, D.B.; Koh, W.; Lee, H.J.; Lee, E.O.; Ahn, K.S.; Ahn, K.S.; et al. Oral administration of penta-O-galloyl- -D-glucose suppresses triple-negative breast cancer xenograft growth and metastasis in strong association with JAK1-STAT3 inhibition. Carcinogenesis 2011, 32, 804–811. [Google Scholar] [CrossRef]
- Tseeleesuren, D.; Kant, R.; Yen, C.H.; Hsiao, H.H.; Chen, Y.A. 1,2,3,4,6-Penta-O-Galloyl-Beta-D-Glucopyranoside inhibits proliferation of multiple myeloma cells accompanied with suppression of MYC expression. Front. Pharm. 2018, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Chai, Y.; Wang, L.; Zhang, J.; Lee, H.J.; Kim, S.H.; Lü, J. Pentagalloylglucose induces autophagy and caspase-independent programmed deaths in human PC-3 and mouse TRAMP-C2 prostate cancer cells. Mol. Cancer Ther. 2009, 8, 2833–2843. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.T.; Lin, T.P.; Liu, L.C.; Huang, C.H.; Lin, J.K.; Kao, J.Y.; Way, T.D. Penta-O-galloyl-beta-D-glucose suppresses prostate cancer bone metastasis by transcriptionally repressing EGF-induced MMP-9 expression. J. Agric. Food Chem. 2009, 57, 3331–3339. [Google Scholar] [CrossRef]
- Hu, H.; Lee, H.J.; Jiang, C.; Zhang, J.; Wang, L.; Zhao, Y.; Xiang, Q.; Lee, E.O.; Kim, S.H.; Lu, J. Penta-1,2,3,4,6-O-galloyl-beta-D-glucose induces p53 and inhibits STAT3 in prostate cancer cells in vitro and suppresses prostate xenograft tumor growth in vivo. Mol. Cancer Ther. 2008, 7, 2681–2691. [Google Scholar] [CrossRef] [Green Version]
- Kantapan, J.; Paksee, S.; Duangya, A.; Sangthong, P.; Roytrakul, S.; Krobthong, S.; Suttana, W.; Dechsupa, N. A radiosensitizer, gallotannin-rich extract from Bouea macrophylla seeds, inhibits radiation-induced epithelial-mesenchymal transition in breast cancer cells. BMC Complement Med. Ther. 2021, 21, 189. [Google Scholar] [CrossRef]
- Ryu, H.G.; Jeong, S.J.; Kwon, H.Y.; Lee, H.J.; Lee, E.O.; Lee, M.H.; Choi, S.H.; Ahn, K.S.; Kim, S.H. Penta-O-galloyl-β-D-glucose attenuates cisplatin-induced nephrotoxicity via reactive oxygen species reduction in renal epithelial cells and enhances antitumor activity in Caki-2 renal cancer cells. Toxicol. Vitr. 2012, 26, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Zhao, S.; Wang, J.Y.; Zheng, H.C.; Ma, C.M. Inhibitory effects and molecular mechanisms of pentagalloyl glucose in combination with 5-FU on aggressive phenotypes of HepG2 cells. Nat. Prod. Res. 2021, 35, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. 2013, 46, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bei, R.; Budillon, A.; Masuelli, L.; Cereda, V.; Vitolo, D.; Di Gennaro, E.; Ripavecchia, V.; Palumbo, C.; Ionna, F.; Losito, S.; et al. Frequent overexpression of multiple ErbB receptors by head and neck squamous cell carcinoma contrasts with rare antibody immunity in patients. J. Pathol. 2004, 204, 317–325. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural biology of STAT3 and its implications for anticancer therapies development. Int. J. Mol. Sci. 2018, 19, 1591. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ren, X.; Deng, C.; Yang, L.; Yan, E.; Guo, T.; Li, Y.; Xu, M.X. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol. Rep. 2013, 30, 2691–2696. [Google Scholar] [CrossRef] [Green Version]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [Green Version]
- Maiuthed, A.; Chanvorachote, P. Cisplatin at sub-toxic levels mediates integrin switch in lung cancer cells. Anticancer Res. 2014, 34, 7111–7117. [Google Scholar]
- Liu, Y.Q.; Zhang, G.A.; Zhang, B.C.; Wang, Y.; Liu, Z.; Jiao, Y.L.; Liu, N.; Zhao, Y.R. Short low concentration cisplatin treatment leads to an epithelial mesenchymal transition-like response in DU145 prostate cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 1025–1028. [Google Scholar] [CrossRef]
- Kalyankrishna, S.; Grandis, J.R. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. The role of STAT3 signaling in mediating tumor resistance to cancer therapy. Curr. Drug Targets 2014, 15, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in cancer immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-κB and stat3 transcription factor signatures differentiate HPV-positive and HPV-negative head and neck squamous cell carcinoma. Int. J. Cancer Res. 2015, 137, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.T.; Way, T.D.; Lin, J.K. Pentagalloylglucose inhibits estrogen receptor alpha by lysosome-dependent depletion and modulates ErbB/PI3K/Akt pathway in human breast cancer MCF-7 cells. Mol. Carcinog. 2006, 45, 551–560. [Google Scholar] [CrossRef]
- Barbosa, M.; Xavier, C.; Pereira, R.F.; Petrikaitė, V.; Vasconcelos, M.H. 3D cell culture models as recapitulators of the tumor microenvironment for the screening of anti-cancer drugs. Cancers 2021, 14, 190. [Google Scholar] [CrossRef]
- Ekert, J.E.; Johnson, K.; Strake, B.; Pardinas, J.; Jarantow, S.; Perkinson, R.; Colter, D.C. Three-dimensional lung tumor microenvironment modulates therapeutic compound responsiveness in vitro--implication for drug development. PLoS ONE 2014, 9, e92248. [Google Scholar] [CrossRef]
- Hoffmann, O.I.; Ilmberger, C.; Magosch, S.; Joka, M.; Jauch, K.W.; Mayer, B. Impact of the spheroid model complexity on drug response. J. Biotechnol. 2015, 205, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nature reviews. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Dechsupa, N.; Kantapan, J.; Tungjai, M.; Intorasoot, S. Maprang “Bouea macrophylla Griffith” seeds: Proximate composition, HPLC fingerprint, and antioxidation, anticancer and antimicrobial properties of ethanolic seed extracts. Heliyon 2019, 5, e02052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. Gauss View, Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. Open Source Drug Discovery Consortium. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | CAL27 | FaDU | ||||
|---|---|---|---|---|---|---|
| IC50 (µg/mL) | CI at IC50 | DRI | IC50 (µg/mL) | CI at IC50 | DRI | |
| Cisplatin alone | 5.4 ± 0.4 | 7.6 ± 1.4 | ||||
| Cisplatin with 10 µg/mL PGG | 4.0 ± 0.1 | 1.2 | 1.35 | 5.4 ± 0.9 | 1.13 | 1.41 |
| Cisplatin with 20 µg/mL PGG | 1.4 ± 0.2 | 0.64 | 3.86 | 3.6 ± 1.7 | 0.81 | 2.11 |
| Cisplatin with 30 µg/mL PGG | 0.8 ± 0.6 | 0.51 | 6.75 | 1.2 ± 0.6 | 0.62 | 6.33 |
| No. | Hydrogen Bonding * | Distance (Å) | Occupancy (%) |
|---|---|---|---|
| 1 | Glu612-C=O····HO-PGG | 1.758 ± 0.181 | 92.91 |
| 2 | Glu612-C=O····HO-PGG | 1.669 ± 0.152 | 88.62 |
| 3 | Glu612-C=O····HO-PGG | 1.674 ± 0.156 | 85.43 |
| 4 | Glu612-C=O····HO-PGG | 1.665 ± 0.156 | 80.14 |
| 5 | Ser636-NH····O=C-PGG | 2.416 ± 0.404 | 60.08 |
| 6 | Ser636-C=O····HO-PGG | 2.463 ± 0.870 | 40.92 |
| 7 | Glu638-C=O····HO-PGG | 2.785 ± 0.538 | 23.15 |
| 8 | Arg609-NH·····O-PGG | 2.450 ± 0.382 | 19.66 |
| 9 | Arg609-NH····O-PGG | 2.653 ± 0.597 | 18.56 |
| 10 | Arg609-NH····O-PGG | 2.486 ± 0.372 | 10.58 |
| 11 | Ile634-C=O····HO-PGG | 2.706 ± 0.549 | 3.49 |
| Terms | Energy (kcal/mol) |
|---|---|
| van der Waals (ΔEvdw) | −46.858 ± 4.360 |
| electrostatic (ΔEelect) | −47.356 ± 4.681 |
| polar solvation (ΔEpolar) | 72.616 ± 7.008 |
| SASA (ΔESASA) | −5.861 ± 0.3566 |
| binding energy (ΔGbind) | −27.938 ± 5.224 |
| Residue | Energy (kcal/mol) | |||
|---|---|---|---|---|
| Potential Energy (ΔEMM) | Polar Solvation (ΔEpolar) | SASA (ΔESASA) | Binding Energy (ΔGbind) | |
| Lys531 | −0.916 ± 0.017 | −1.721 ± 0.024 | −0.001 ± 0.000 | −0.806 ± 0.015 |
| Lys557 | −1.522 ± 0.036 | 1.023 ± 0.075 | −0.075 ± 0.002 | −0.574 ± 0.057 |
| Arg609 | 0.515 ± 0.025 | −0.890 ± 0.052 | −0.142 ± 0.001 | −0.518 ± 0.043 |
| Ser613 | −0.091 ± 0.014 | −0.258 ± 0.017 | −0.146 ± 0.002 | −0.495 ± 0.016 |
| Lys615 | −0.882 ± 0.014 | 0.034 ± 0.011 | −0.001 ± 0.000 | −0.848 ± 0.010 |
| Thr620 | −0.928 ± 0.008 | 0.457 ± 0.009 | −0.054 ± 0.001 | −0.525 ± 0.010 |
| Thr622 | −0.327 ± 0.005 | −0.588 ± 0.006 | −0.009 ± 0.000 | −0.923 ± 0.006 |
| Trp623 | −0.735 ± 0.007 | 0.288 ± 0.004 | −0.068 ± 0.001 | −0.514 ± 0.007 |
| Ile634 | −0.793 ± 0.009 | 0.435 ± 0.010 | −0.075 ± 0.001 | −0.433 ± 0.009 |
| Ser636 | −4.245 ± 0.021 | 4.300 ± 0.019 | −0.349 ± 0.001 | −0.296 ± 0.023 |
| Val637 | −2.886 ± 0.012 | 1.632 ± 0.013 | −0.147 ± 0.001 | −1.402 ± 0.014 |
| Pro639 | −1.269 ± 0.007 | 0.015 ± 0.002 | −0.118 ± 0.001 | −1.372 ± 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantapan, J.; Intachai, N.; Khamto, N.; Meepowpan, P.; Sangthong, P.; Wantanajittikul, K.; Dechsupa, N.; Chitapanarux, I. Pentagalloyl Glucose and Cisplatin Combination Treatment Exhibits a Synergistic Anticancer Effect in 2D and 3D Models of Head and Neck Carcinoma. Pharmaceuticals 2022, 15, 830. https://doi.org/10.3390/ph15070830
Kantapan J, Intachai N, Khamto N, Meepowpan P, Sangthong P, Wantanajittikul K, Dechsupa N, Chitapanarux I. Pentagalloyl Glucose and Cisplatin Combination Treatment Exhibits a Synergistic Anticancer Effect in 2D and 3D Models of Head and Neck Carcinoma. Pharmaceuticals. 2022; 15(7):830. https://doi.org/10.3390/ph15070830
Chicago/Turabian StyleKantapan, Jiraporn, Nuttawadee Intachai, Nopawit Khamto, Puttinan Meepowpan, Padchanee Sangthong, Kittichai Wantanajittikul, Nathupakorn Dechsupa, and Imjai Chitapanarux. 2022. "Pentagalloyl Glucose and Cisplatin Combination Treatment Exhibits a Synergistic Anticancer Effect in 2D and 3D Models of Head and Neck Carcinoma" Pharmaceuticals 15, no. 7: 830. https://doi.org/10.3390/ph15070830
APA StyleKantapan, J., Intachai, N., Khamto, N., Meepowpan, P., Sangthong, P., Wantanajittikul, K., Dechsupa, N., & Chitapanarux, I. (2022). Pentagalloyl Glucose and Cisplatin Combination Treatment Exhibits a Synergistic Anticancer Effect in 2D and 3D Models of Head and Neck Carcinoma. Pharmaceuticals, 15(7), 830. https://doi.org/10.3390/ph15070830