
Aryl Hydrocarbon Receptor (AhR)-Mediated Signaling in iPSC-Derived Human Motor Neurons

 ,
,  , ,
, ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
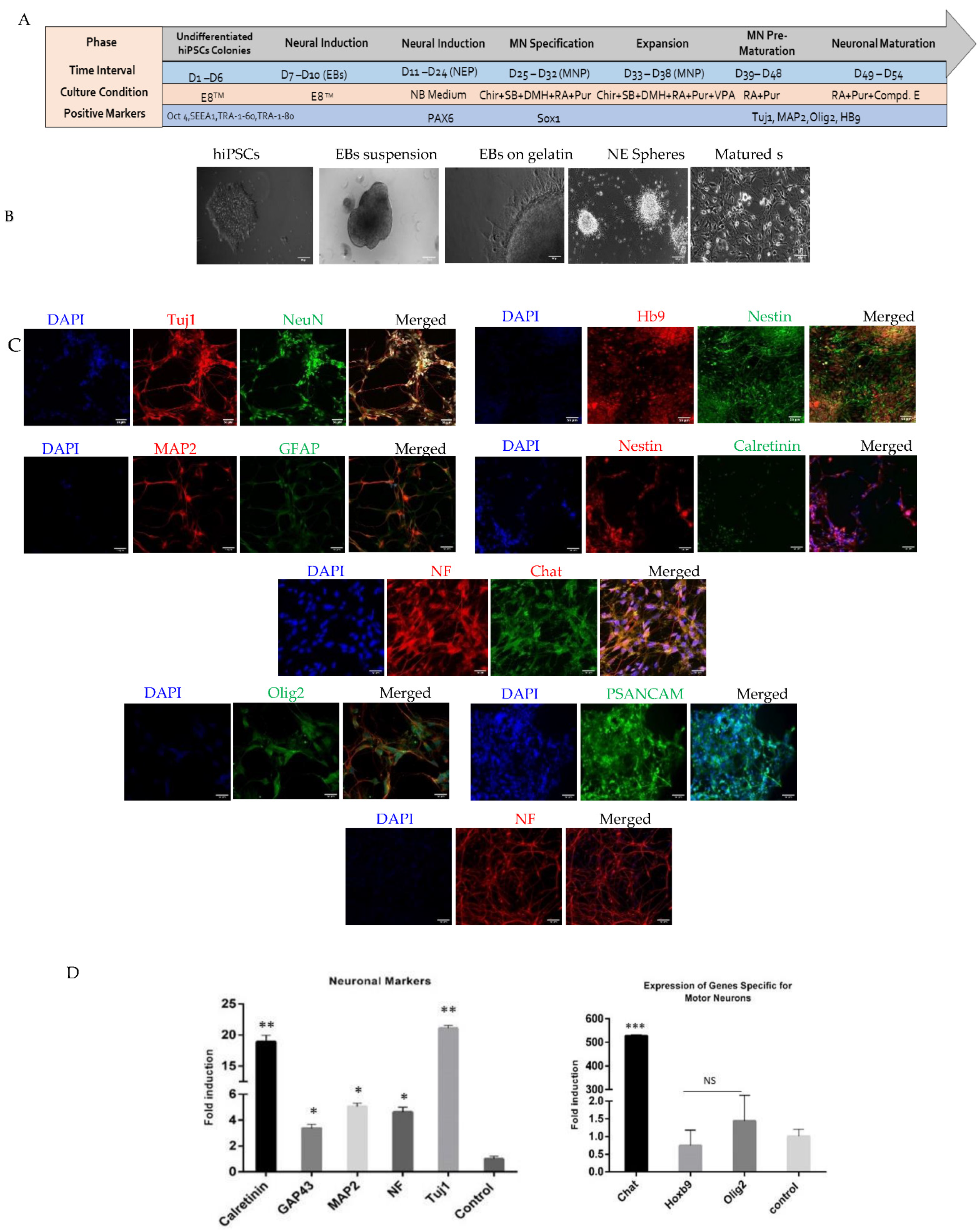
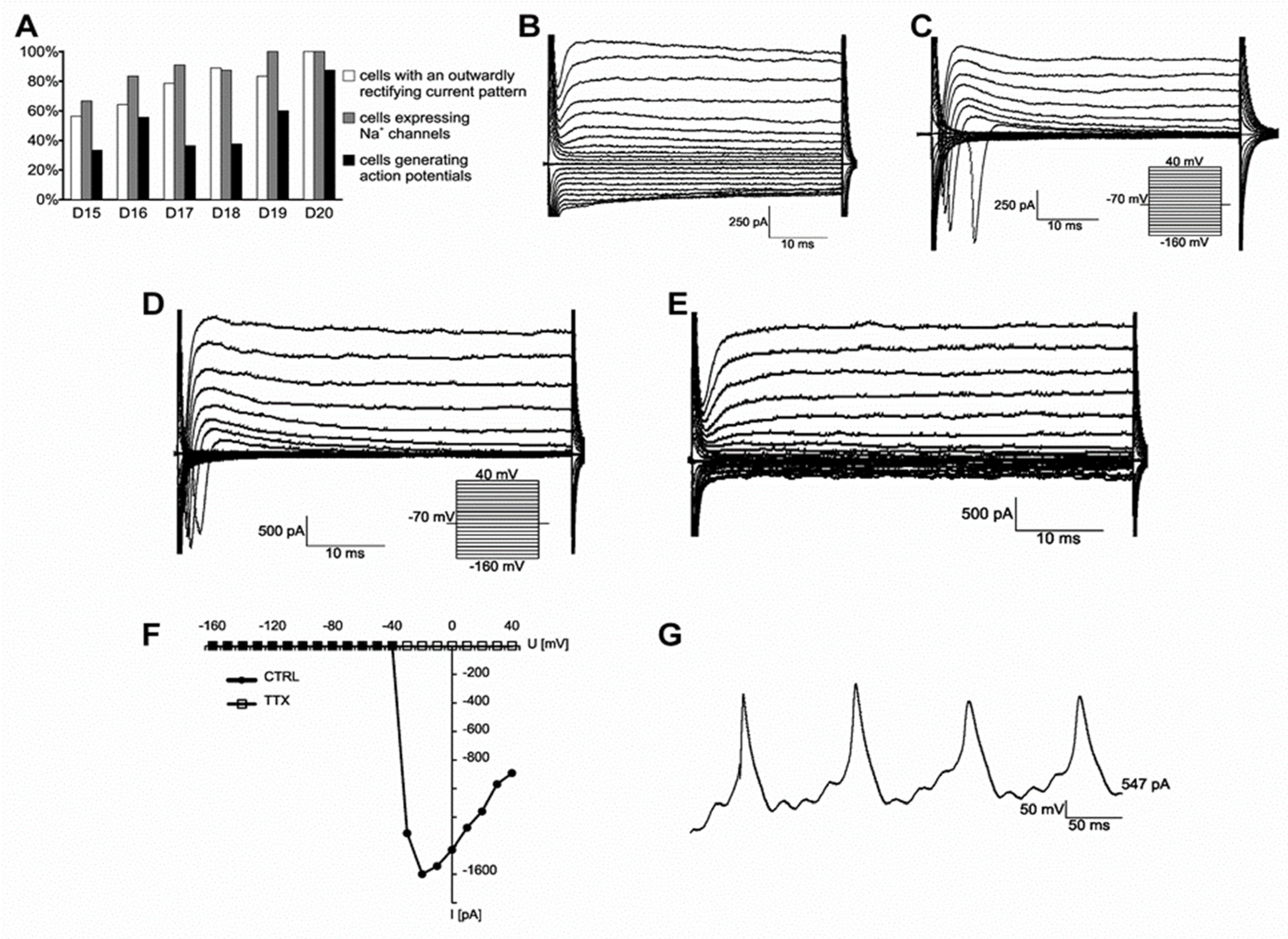
2.1. Generation and Characterization of Motor Neurons
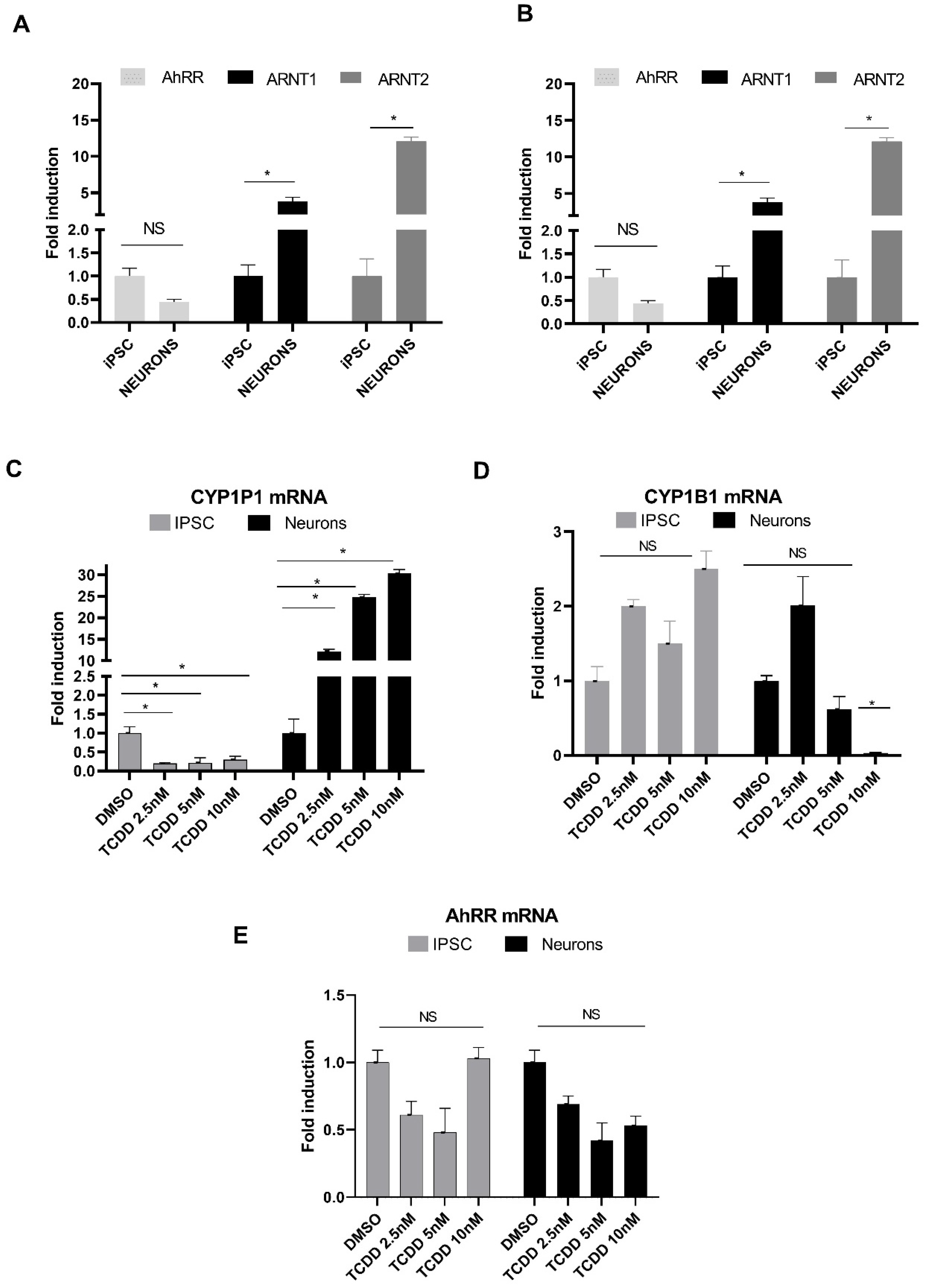
2.2. Monitoring Expression of AhR Signaling Pathway Components in hiPSCs and Neurons
2.3. Response of AhR-Target Genes to Different Ligands in Neurons
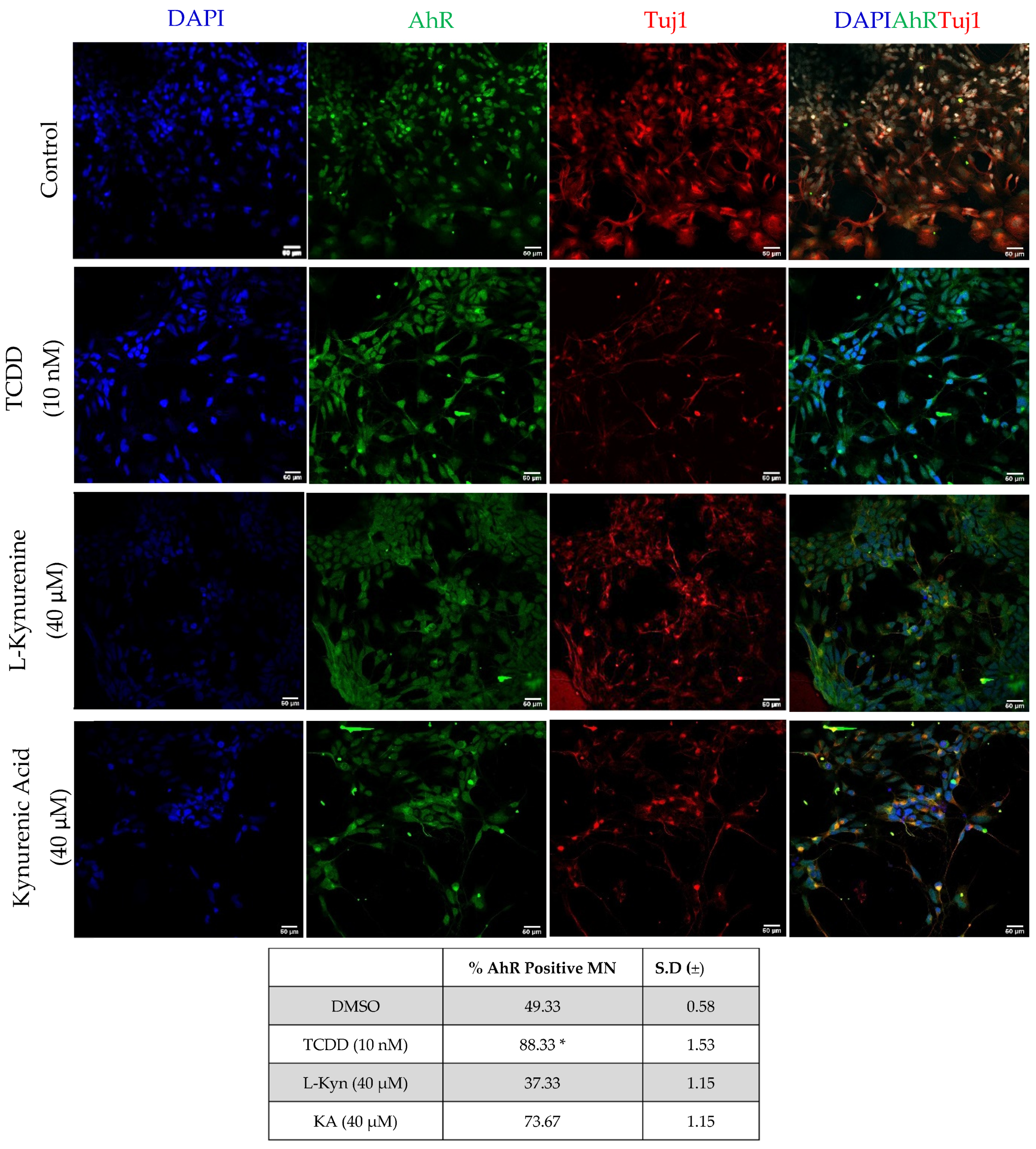
2.4. AhR Protein Expression in Neurons following Exposure to AhR Ligand
2.5. CYP1A1 Expression and Activity in Neurons in Response to AhR Stimulation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Generation of hiPSCs from Human Fibroblasts
4.3. Neural Differentiation from hiPSCs
4.4. RT-qPCR
4.4.1. Immunostaining of Pluripotency and Neural Differentiation Markers
4.4.2. Treatment of iPSCs and Differentiated Neurons
Immunofluorescence
CYP1A1 Enzyme Activity Assay (EROD ASSAY)
4.5. Patch-Clamp Recording
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rachel, H.W.; Christopher, A.B. Rodent genetic models of Ah receptor. Drug Metab. Rew. 2021, 53, 350–374. [Google Scholar] [CrossRef]
- Gassmann, K.; Abel, J.; Bothe, H. Species-specific differential AhR expression protects human neural progenitor cells against developmental neurotoxicity of PAHs. Environ. Health Perspect. 2010, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klocke, C.; Lein, P.J. Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB). Dev. Neurotox. Int. J. Mol. Sci. 2020, 21, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidsen, N.; Lauvås, A.J.; Myhre, O.; Ropsta, E.; Carpi, D.; Gyves, E.M.; Berntsen, H.F.; Dirven, H.; Paulsen, R.E.; Bal-Price, A.; et al. Exposure to human relevant mixtures of halogenated persistent organic pollutants (POPs) alters neurodevelopmental processes in human neural stem cells undergoing differentiation. Reprod. Toxicol. 2021, 100, 17–34. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Balance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef]
- Bailon-Moscoso, N.; Cevallos-Solorzano, G.; Romero-Benavides, J.C.; Orellana, M.I. Natural compounds as modulators of cell cycle arrest: Application for anticancer chemotherapies. Curr. Genom. 2017, 18, 106–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avilla, M.N.; Malecki, K.M.C.; Hahn, M.E.; Wilson, R.H.; Bradfield, C.A. The Ah Receptor: Adaptive Metabolism, Ligand Diversity, and the Xenokine Model. Chem. Res. Toxicol. 2020, 33, 860–879. [Google Scholar] [CrossRef]
- Gomez-Zepeda, D.; Taghi, M.; Scherrmann, J.M.; Decleves, X.; Menet, M.C. ABC Transporters at the Blood-Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, Y.; Zhu, R.; Xu, L.; Xie, H.Q.; Zhao, B. Rutaecarpine Inhibits U87 Glioblastoma Cell Migration by Activating the Aryl Hydrocarbon Receptor Pathway. Front. Mol. Neurosci. 2021, 14, 765712. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lee, P.S.; Chou, Y.; Hwang, L.L.; Juan, S.H. Mediating effects of aryl-hydrocarbon receptor and RhoA in altering brain vascular integrity: The therapeutic potential of statins. Am. J. Pathol. 2012, 181, 211–221. [Google Scholar] [CrossRef]
- Iqbal, K.; Pierce, S.H.; Kozai, K.; Dhakal, P.; Scott, R.L.; Roby, K.F.; Vyhlidal, C.A.; Soares, M.J. Evaluation of Placentation and the Role of the Aryl Hydrocarbon Receptor Pathway in a Rat Model of Dioxin Exposure. Environ. Health Perspect. 2021, 129, 117001. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza-Ojeda, M.; Apatiga-Vega, E.; Arenas-Huertero, F. Role of aryl hydrocarbon receptor in central nervous system tumors: Biological and therapeutic implications. Oncol. Lett. 2021, 21, 460. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Kado, S.Y.; Bein, K.J.; He, Y.; Pouraryan, A.A.; Urban, A.; Haarmann-Stemmann, T.; Sweeney, C.; Vogel, C.F.A. Aryl Hydrocarbon Receptor Synergizes with TLR/NF-κB- for Induction of IL-22 Through Canonical and Non-Canonical AhR Pathways. Front. Toxicol. 2022, 3, 787360. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Xiao, G.H.; Leingartner, K.; Chu, I.; Musicki, B.; Tsang, B.K. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-omethyltransferase in prepubertal female Sprague-Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 86, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galijatovic, A.; Beaton, D.; Nguyen, N. The human CYP1A1 gene is regulated in a developmental and tissue-specific fashion in transgenic mice. J. Biol. Chem. 2004, 275, 23969–23976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar-Mohamed, A.; Abdelhamid, G.; Amara, I.E.; El-Kadi, A.O. Differential modulation of cytochrome P450 1a1 by arsenite in vivo and in vitro in C57BL/6 mice. Free Radic. Biol. Med. 2013, 58, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Park, H.J.; Kim, S.J.; Kim, H.K. Identification of cytochrome P450 1A1 in human brain. Biochem. Biophys. Res. Commun. 1998, 243, 808–810. [Google Scholar] [CrossRef]
- McMillan, D.M.; Tyndale, R.F. CYP-mediated drug metabolism in the brain impacts drug response. Pharmacol. Ther. 2018, 184, 189–200. [Google Scholar] [CrossRef]
- Consiglio, E.D.; Pistollato, F.; Gyves, E.M.; Bal-Price, A.; Testai, E. Integrating biokinetics and in vitro studies to evaluate developmental neurotoxicity induced by chlorpyrifos in human iPSC-derived neural stem cells undergoing differentiation towards al and glial cells. Reprod. Toxicol. 2020, 98, 174–188. [Google Scholar] [CrossRef]
- Tripathi, V.K.; Kumar, V.; Singh, A.K. Monocrotophos induces the expression and activity of xenobiotic metabolizing enzymes in pre-sensitized cultured human brain cells. PLoS ONE 2014, 9, e91946. [Google Scholar] [CrossRef]
- Latchney, S.E.; Majewska, A.K. Persistent organic pollutants at the synapse: Shared phenotypes and converging mechanisms of developmental neurotoxicity. Dev. Neuorobiol. 2021, 81, 623–652. [Google Scholar] [CrossRef]
- Wu, M.L.; Li, H.; Wu, D.C. CYP1A1 and CYP1B1 expressions in medulloblastoma cells are AhR independent and have no direct link with resveratrol-induced differentiation and apoptosis. Neurosci. Lett. 2005, 384, 33–37. [Google Scholar] [CrossRef]
- Re, D.B.; Yan, B.; Calderón-Garcidueñas, L.; Andrew, A.S.; Tischbein, M.; Stommel, E.W. A perspective on persistent toxicants in veterans and amyotrophic lateral sclerosis: Identifying exposures determining higher ALS risk. J. Neurol. 2022, 269, 2359–2377. [Google Scholar] [CrossRef] [PubMed]
- Su, F.C.; Goutman, S.A.; Chernyak, S.; Mukherjee, B.; Callaghan, B.C.; Batterman, S.; Feldman, E.L. Association of Environmental Toxins with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.; Warner, M.; Siracusa, C.; Signorini, S.; Brambilla, P.; Mocarelli, P.; Eskenazi, B. Prenatal dioxin exposure and neuropsychological functioning in the Seveso Second Generation Health Study. Int. J. Hyg. Environ. Health 2019, 222, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.P.; Rauh, V.; Whyatt, R.M. Effect of prenatal exposure to airborne polycyclic aromatic hydrocarbons on neurodevelopment in the first 3 years of life among inner-city children. Environ. Health Perspect. 2006, 114, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Gu, A.; Ji, G.; Jiang, T. Contributions of aryl hydrocarbon receptor genetic variants to the risk of glioma and PAH-DNA adducts. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 128, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Chevallier, A.; Mialot, A.; Petit, J.M. Oculomotor deficits in aryl hydrocarbon receptor null mouse. PLoS ONE 2013, 8, e53520. [Google Scholar] [CrossRef] [Green Version]
- Moran, T.B.; Brannick, K.E.; Raetzman, L.T. Aryl-hydrocarbon receptor activity modulates prolactin expression in the pituitary. Toxicol. Appl. Pharmacol. 2012, 265, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.L.; Williamson, M.A.; Thompson, B.D.; Dever, D.P.; Gasiewicz, T.A.; Opanashuk, L.A. 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule precursor maturation in the developing mouse cerebellum. Toxicol. Sci. Off. J. Soc. Toxicol. 2008, 103, 125–136. [Google Scholar] [CrossRef]
- Juricek, L.; Coumoul, X. The Aryl Hydrocarbon Receptor and the Nervous System. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef] [Green Version]
- Dever, D.P.; Adham, Z.O.; Thompson, B.; Genestine, M.; Cherry, J.; Olschowka, J.A.; DiCicco-Bloom, E.; Opanashuk, L.A. Aryl hydrocarbon receptor deletion in cerebellar granule precursors impairs neurogenesis. Dev. Neurobiol. 2015, 76, 533–550. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand that Synergistically Induces Interleukin-6 in the Presence of Inflammatory. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 2013, 34, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.; Fabbrocini, G.; Martora, F.; Picone, V.; Morelli, P.; Patruno, C. Role of Aryl Hydrocarbon Receptor Activation in Inflammatory Chronic Skin Diseases. Cells 2021, 10, 3559. [Google Scholar] [CrossRef]
- Imran, S.; Ferretti, P.; Vrzal, R. Different regulation of aryl hydrocarbon receptor-regulated genes in response to dioxin in undifferentiated and ally differentiated human neuroblastoma SHSY5Y cells. Toxicol. Mech. Methods 2015, 25, 689–697. [Google Scholar] [CrossRef]
- Hu, B.Y.; Zhang, S.C. Differentiation of spinal motor s from pluripotent human stem cells. Nat. Protoc. 2009, 4, 1295–1304. [Google Scholar] [CrossRef]
- Semkova, V.; Haupt, S.; Segschneider, M.; Bell, C.; Ingelman-Sundberg, M.; Hajo, M.; Weykopf, B.; Muthukottiappan, P.; Till, A.; Brüstle, O. Dynamics of Metabolic Pathways and Stress Response Patterns during Human Neural Stem Cell Proliferation and Differentiation. Cells 2022, 11, 1388. [Google Scholar] [CrossRef]
- Walczak, K.; Langner, E.; Makuch-Kocka, A.; Szelest, M.; Szalast, K.; Marciniak, S.; Tomasz, P. Effect of Tryptophan-Derived AhR Ligands, Kynurenine, Kynurenic Acid and FICZ, on Proliferation, Cell Cycle Regulation and Cell Death of Melanoma Cells—In Vitro Studies. Int. J. Mol. Sci. 2020, 21, 7946. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wahlang, B.; Thapa, M.; Head, K.Z.; Hardesty, J.E.; Srivastava, S.; Merchant, M.L.; Rai, S.N.; Prough, R.A.; Cave, M.C. Proteomics and metabolic phenotyping define principal roles for the aryl hydrocarbon receptor in mouse liver. Acta Pharm. Sin. B 2021, 11, 3806–3819. [Google Scholar] [CrossRef] [PubMed]
- Teino, I.; Matvere, A.; Pook, M.; Varik, I.; Pajusaar, L.; Uudeküll, K.; Vaher, H.; Trei, A.; Kristjuhan, A.; Org, T.; et al. Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation. Int. J. Mol. Sci. 2020, 21, 9052. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.; Bhakti, V.L.; Peet, D.J.; Whitelaw, M.L. Reciprocal regulation of the basic helix-loop-helix/Per-Arnt-Sim partner proteins, Arnt and Arnt2, during al differentiation. Nucle Acids Res. 2013, 41, 5626–5638. [Google Scholar] [CrossRef]
- Sadowska, A.; Paukszto, L.; Nynca, A.; Szczerbal, I.; Orlowska, K.; Swigonska, S.; Ruszkowska, M.; Molcan, T.; Jastrzebski, J.P.; Panasiewicz, G.; et al. Transcript variations, phylogenetic tree and chromosomal localization of porcine aryl hydrocarbon receptor (AhR) and AhR nuclear translocator (ARNT) genes. J. Genet. 2017, 96, 75–85. [Google Scholar] [CrossRef]
- Xie, H.Q.; Xu, H.M.; Fu, H.L.; Hu, Q.; Tian, W.J.; Pei, X.H.; Zhao, B. AhR-mediated effects of dioxin on al acetylcholinesterase expression in vitro. Environ. Health Perspect. 2013, 121, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Lidin, E.; Sköld, M.K.; Angéria, M.; Davidsson, J.; Risling, M. Hippocampal Expression of Cytochrome P450 1B1 in Penetrating Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 722. [Google Scholar] [CrossRef]
- Farin, F.M.; Omiecinski, C.J. Regiospecific expression of cytochrome P-450s and microsomal epoxide hydrolase in human brain tissue. J. Toxicol. Environ. Health 1993, 40, 317–335. [Google Scholar] [CrossRef]
- D’Uva, G.; Baci, D.; Albini, A.; Noonan, D.M. Cancer chemoprevention revisited: Cytochrome P450 family 1B1 as a target in the tumor and the microenvironment. Cancer Treat. Rev. 2018, 63, 1–18. [Google Scholar] [CrossRef]
- Jacob, A.; Hartz, A.M.; Potin, S. Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1by TCDD and diesel exhaust particles in rat brain microvessels. Fluids Barriers CNS 2011, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Wincent, E.; Bengtsson, J.; Bardbori, A.M.; Alsberg, T.; Luecke, S.; Rannug, U.; Rannug, A. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Biol. Sci 2012, 9, 4479–4484. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Yoshimochi, K.; Li, J.; Takekita, K.; Shimotsuma, M.; Li, L.; Qu, X.; Zhang, J.; Sawa, Y.; Liu, L.; et al. Development and evaluation of a novel xeno-free culture medium for human-induced pluripotent stem cells. Stem Cell Res. Ther. 2022, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Kriska, J.; Janeckova, L.; Kirdajova, D.; Honsa, P.; Knotek, T.; Dzamba, D.; Kolenicova, D.; Butenko, O.; Vojtechova, M.; Capek, M.; et al. Wnt/β-Catenin Promotes Differentiation of Ischemia-Activated Adult Neural Stem/Progenitor Cells to al Precursors. Front. Neurosci. 2021, 15, 628983. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, S.; Forostyak, O.; Kwok, J.C.F.; Romanyuk, N.; Rehorova, M.; Kriska, J.; Dayanithi, G.; Raha-Chowdhury, R.; Jendelova, P.; Anderova, M.; et al. Transplantation of Neural Precursors Derived from Induced Pluripotent Cells Preserve Perial Nets and Stimulate Neural Plasticity in ALS Rats. Int. J. Mol. Sci. 2020, 21, 9593. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, S.J.; Vagaska, B.; Kriska, J.; Anderova, M.; Bortolozzi, M.; Gerosa, G.; Ferretti, P.; Vrzal, R. Aryl Hydrocarbon Receptor (AhR)-Mediated Signaling in iPSC-Derived Human Motor Neurons. Pharmaceuticals 2022, 15, 828. https://doi.org/10.3390/ph15070828
Imran SJ, Vagaska B, Kriska J, Anderova M, Bortolozzi M, Gerosa G, Ferretti P, Vrzal R. Aryl Hydrocarbon Receptor (AhR)-Mediated Signaling in iPSC-Derived Human Motor Neurons. Pharmaceuticals. 2022; 15(7):828. https://doi.org/10.3390/ph15070828
Chicago/Turabian StyleImran, Saima Jalil, Barbora Vagaska, Jan Kriska, Miroslava Anderova, Mario Bortolozzi, Gino Gerosa, Patrizia Ferretti, and Radim Vrzal. 2022. "Aryl Hydrocarbon Receptor (AhR)-Mediated Signaling in iPSC-Derived Human Motor Neurons" Pharmaceuticals 15, no. 7: 828. https://doi.org/10.3390/ph15070828
APA StyleImran, S. J., Vagaska, B., Kriska, J., Anderova, M., Bortolozzi, M., Gerosa, G., Ferretti, P., & Vrzal, R. (2022). Aryl Hydrocarbon Receptor (AhR)-Mediated Signaling in iPSC-Derived Human Motor Neurons. Pharmaceuticals, 15(7), 828. https://doi.org/10.3390/ph15070828