
Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis




Abstract
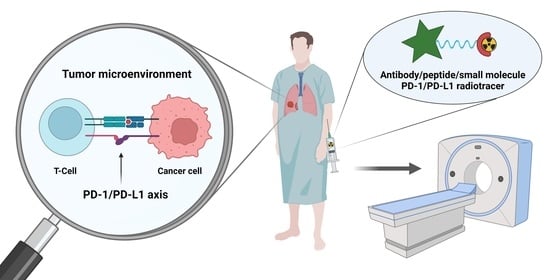
1. Introduction
2. Results

| Target | Class | Substance Name | Imaging Agent | Intended Application | KD/EC50 [nM] | Ref. |
|---|---|---|---|---|---|---|
| PD-1 | mAb | DOTA-anti-mouse-PD-L1 | 64Cu | PET | n.a. | [14] |
| mAb | JS001 | 99mTc | SPECT | n.a. | [15] | |
| mAb | Df-Nivolumab | 89Zr | PET | 3.75 nM | [16,17,18] | |
| mAb | Df-Pembrolizumab | 89Zr | PET | n.a. | [19,20] | |
| Keytruda | 64Cu 89Zr | PET | n.a. | [21,22] | ||
| N-sucDf-Pembrolizumab | 89Zr | PET | n.a. | [23] | ||
| mAb | PD-1-Liposome-DOX | 64Cu IRDye800CW | PET NIRF | n.a. | [24] | |
| mAb | PD-1-IRDye800CW | IRDye800CW | NIRF | n.a. | [25] | |
| PD-1/PD-L1 | mAb | NOTA-α-PD-1 (RMP1-14) NOTA-α-PD-L1 (10F.9G2) | 64Cu | PET | n.a. | [26] |
| mAb Adnectin | Df-Nivolumab BMS-986192 | 89Zr [18F]AlF | PET | n.a. | [18] | |
| PD-L1 | mAb | PD-L1.3.1 | 111In | SPECT | 0.97 nM | [27] |
| mAb | DTPA-anti-PDL1 | 111In | SPECT | 0.6 ± 0.1 nM | [28,29] | |
| mAb | anti-mPD-L1 | 111In | SPECT | 1.1 ± 0.1 nM | [30] | |
| mAb | C4 | 89Zr | PET | 4.2 ± 0.7 nM 1.5 ± 0.34 nM | [31] | |
| mAb | Avelumab | 89Zr | PET | 0.3 nM | [32,33] | |
| mAb | Atezolizumab | Licor 800 | NIRF | 0.43 nM 2 (human) 0.13 nM 1 (mouse) | [34,35] | |
| 111In | SPECT | |||||
| 64Cu | PET | [36] | ||||
| 89Zr | [37,38,39] | |||||
| 99mTc | SPECT | 111.8 ± 17.85 nM | [40] | |||
| mAb | NOTA-MX001 | 64Cu | PET | 5.40 ± 2.30 nM | [41] | |
| mAb | [89Zr]Zr-DFO-anti-PD-L1 mAb | 89Zr | PET | n.a. | [42] | |
| mAb | Df-KN035 | 89Zr | PET | 2.86 ± 0.23 nM | [43,44] | |
| mAb | PD-L1-Mab | 131I | Cherenkov Luminescence | 1.069 nM | [45] | |
| mAb | PD-L1 mAb | 131I | Optical | n.a. | [46] | |
| mAb | NIR-PD-L1-mAb | Licor 800 | NIRF | n.a. | [47] | |
| HCAb | anti-hPD-L1 Nb6 | 124I 125I | PET | 2.19 nM | [48] | |
| NOTA-Nb6 | 64Cu | PET | 3.60 nM | [49] | ||
| Fab Fragment | NOTA-αPD-L1 | 64Cu | PET | 0.72 nM (EC50) | [50] | |
| Nanobody | NOTA-Nb109 | 68Ga | PET | 2.9 nM | [51] | |
| Nanobody | C3 C7 E2 E4 | 99mTc | SPECT | 0.5 nM 17.0 nM 2.1 nM 4.0 nm | [52] | |
| Nanobody | NOTA-(hPD-L1) | 68Ga | PET | 0.8 nM 1.2 nM | [53] | |
| Nanobody | NM-01 | 99mTc | SPECT | n.a. | [54] | |
| Affibody | NOTA-ZPD-L1_1 | 18FAl | PET | 1 nM | [55] | |
| Affibody | NOTA-ZPD-L1_4 | 18FAl 68Ga | PET | 0.07 nM | [56] | |
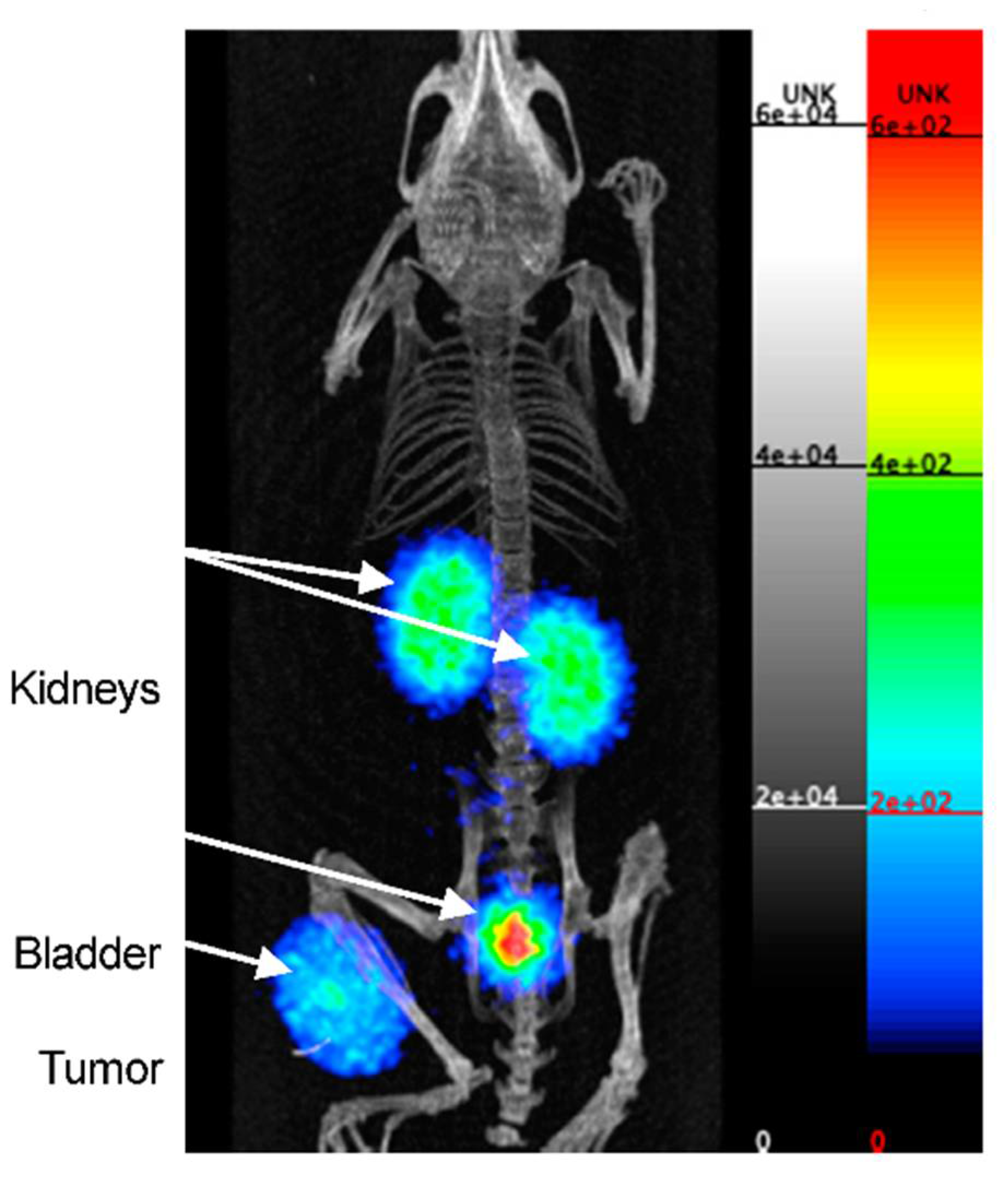
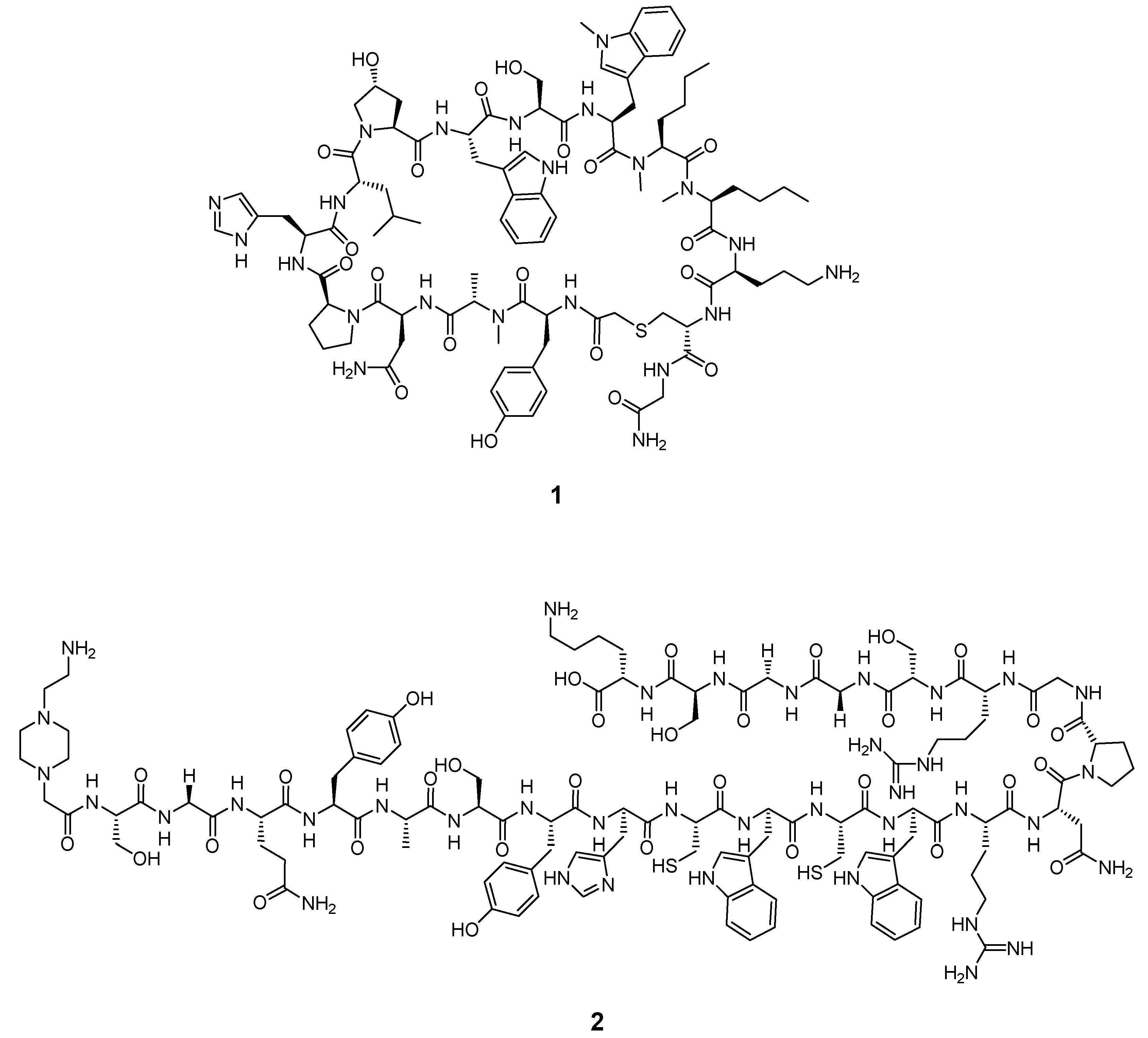
| PD-L1 | Peptide | [64Cu]Cu-WL12 | 64Cu | PET | 2.9 nM | [57] |
| [68Ga]Ga-WL12 | 68Ga | PET | n.a. | [58] | ||
| [18F]FPy-WL12 | 18F | PET | 37.1 nM | [59] | ||
| Peptide | AlF-TPP-1 AlF-PEG-TPP-1 | 18F | PET | 95 nM 2 [60] | [61] | |
| TPP-1 PEG-TPP-1 | 64Cu | |||||
| [18F]AlF-NOTA-IPB-PDL1P | 18F | PET | n.a. | [62] | ||
| [68Ga]Ga-NJMP1 | 68Ga | PET | 25.9 µM | [63] | ||
| Small Molecule | [18F]FLN | 18F | PET | 65.3 nM | [64] | |
| [18F]FLG-1 | 18F | 63.1 nM | [65] | |||
| HAC-PD1 | DOTA-HAC | 64Cu | PET | ~110 pM | [66] | |
| NOTA-HAC NOTA-HACA DOTA-HACA | 64Cu 68Ga | PET | ~110 pM | [67] | ||
| Engineered Protein | FN3hPD-L1 | 64Cu | PET | 1.4 ± 0.3 nM | [68] | |
| Adnectin | BMS-986192 | [18F]AlF | PET | <35 pM | [18,69,70,71] | |
| Nanoparticle | αPDL1-GNP | GNP | CT | n.a. | [72] |
2.1. PD-1-Targeting Radiotracers
Antibodies
2.2. Combined Studies of PD-1 and PD-L1-Targeting Radiotracers
2.3. PD-L1-Targeting Radiotracers
2.3.1. Antibodies
2.3.2. Heavy Chain Antibodies (HCAb)
2.3.3. Fab Fragments
2.3.4. Nanobodies
2.3.5. Affibodies
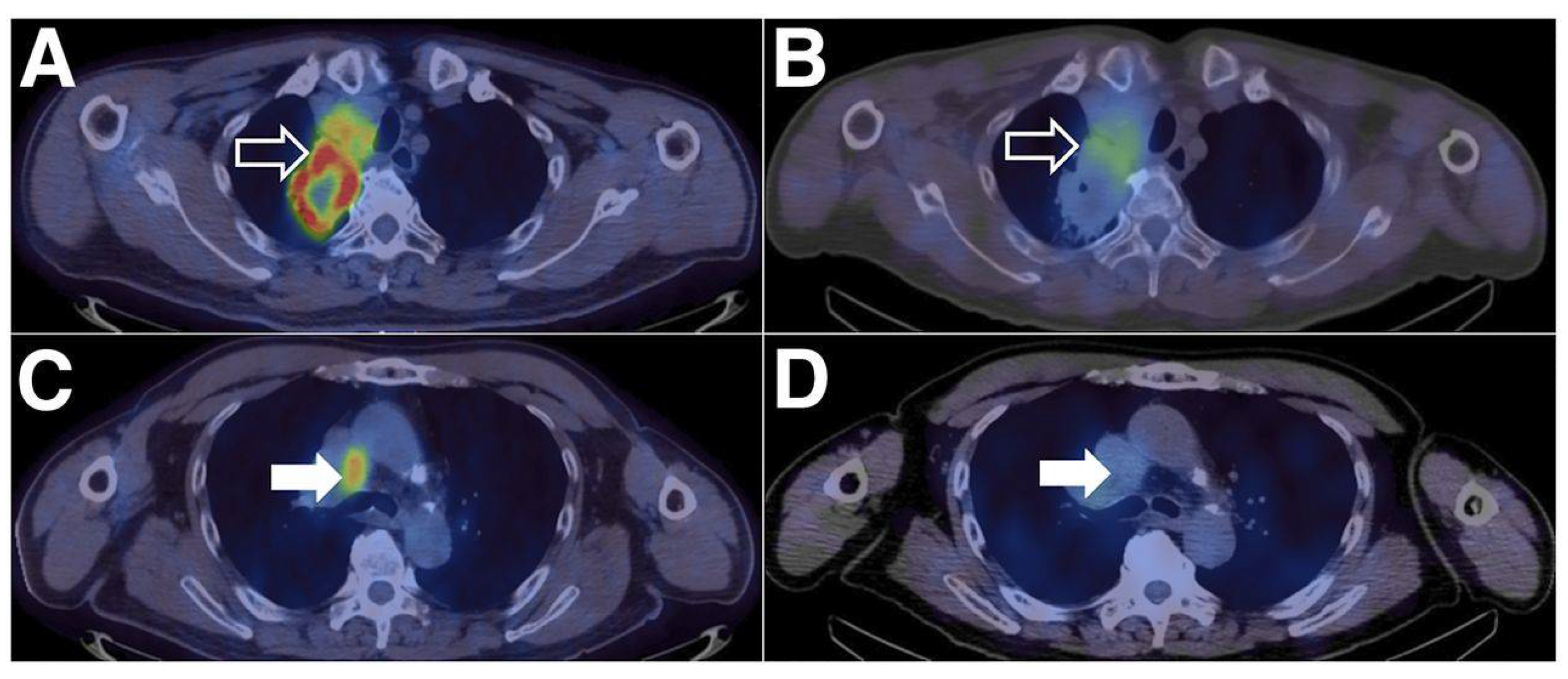
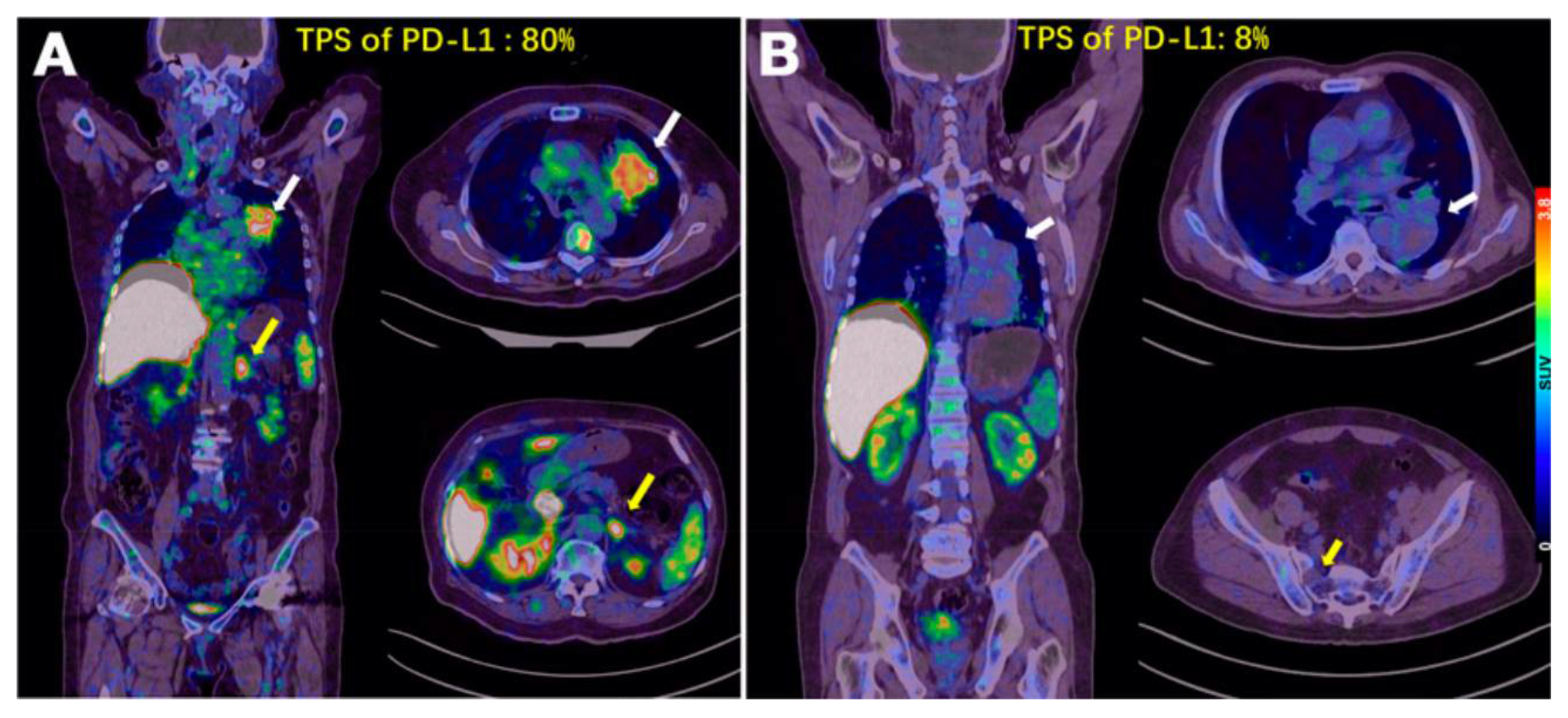
2.3.6. Peptides
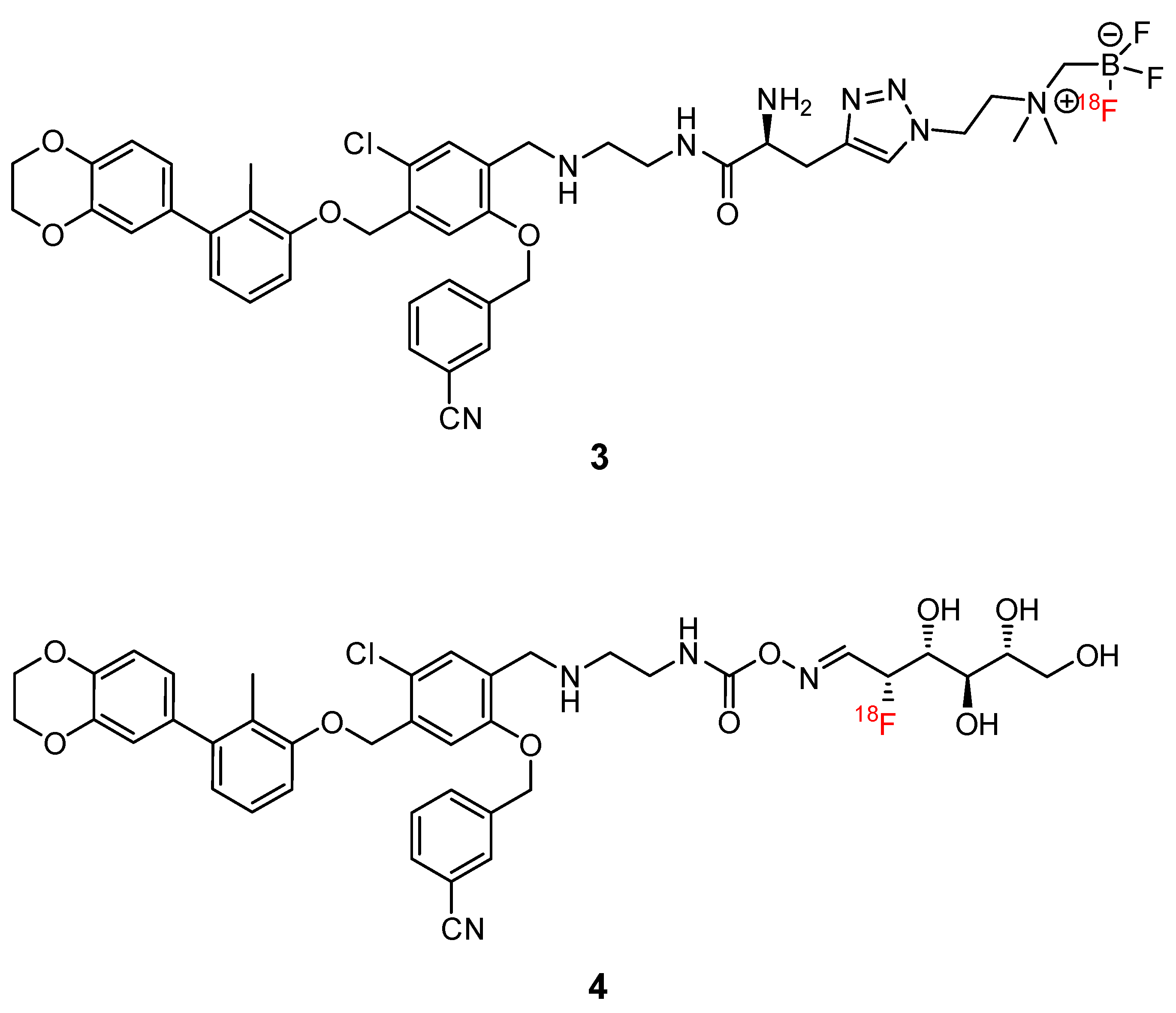
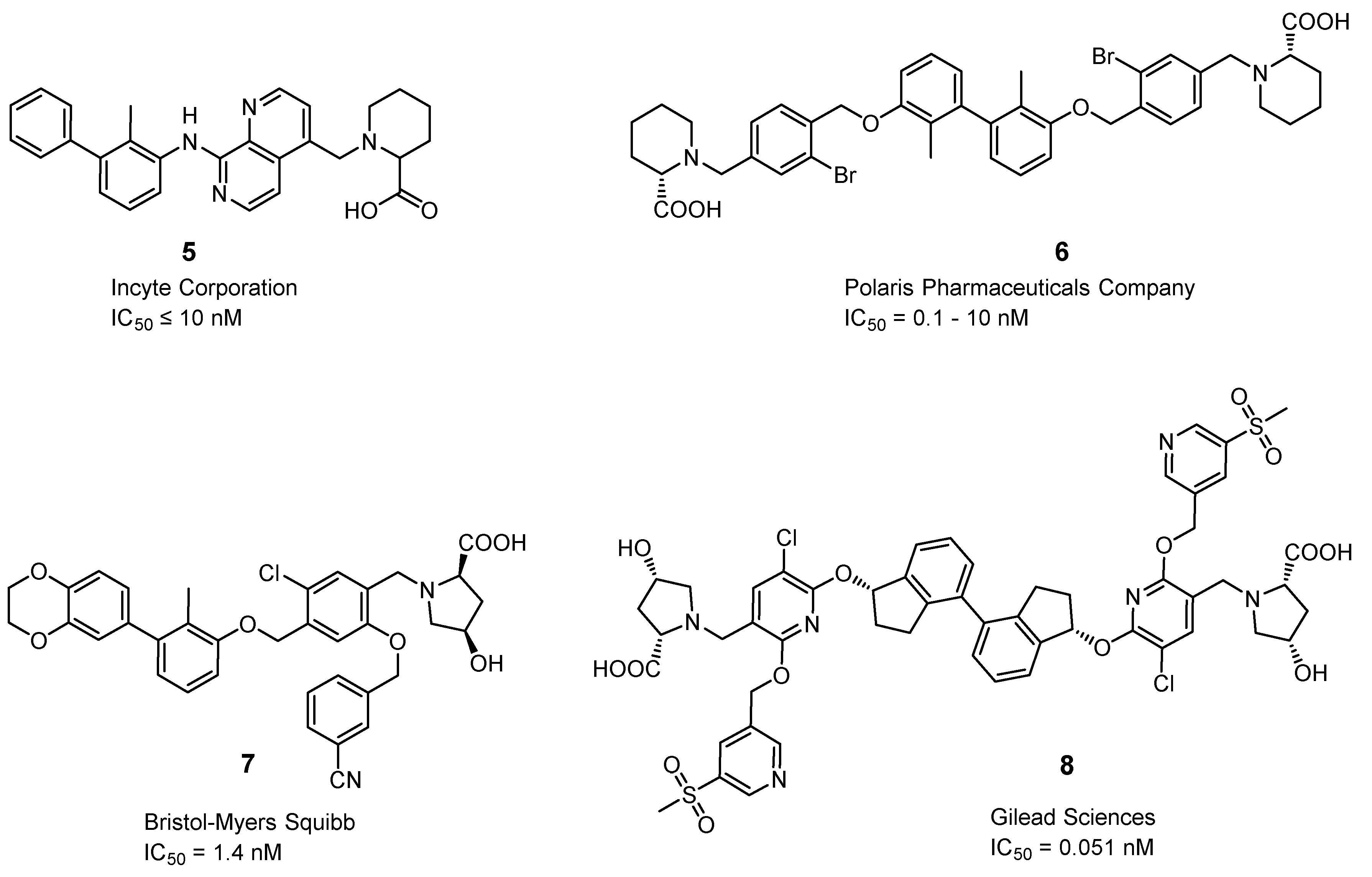
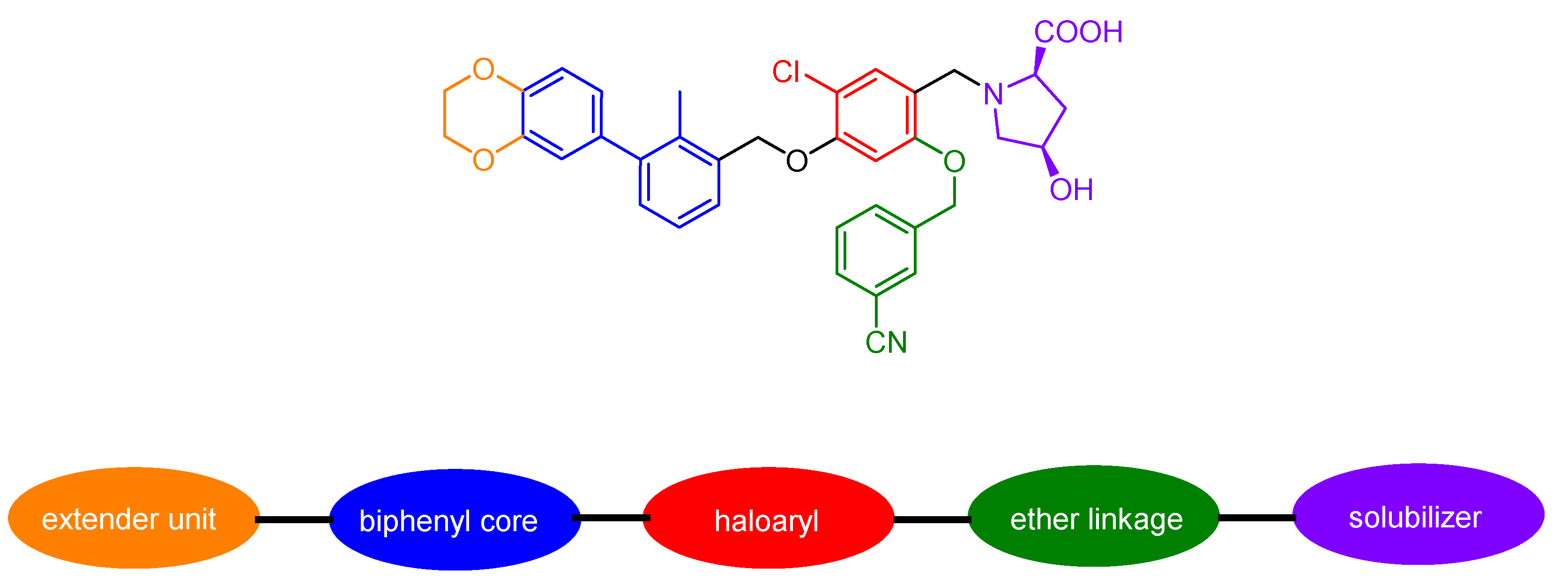
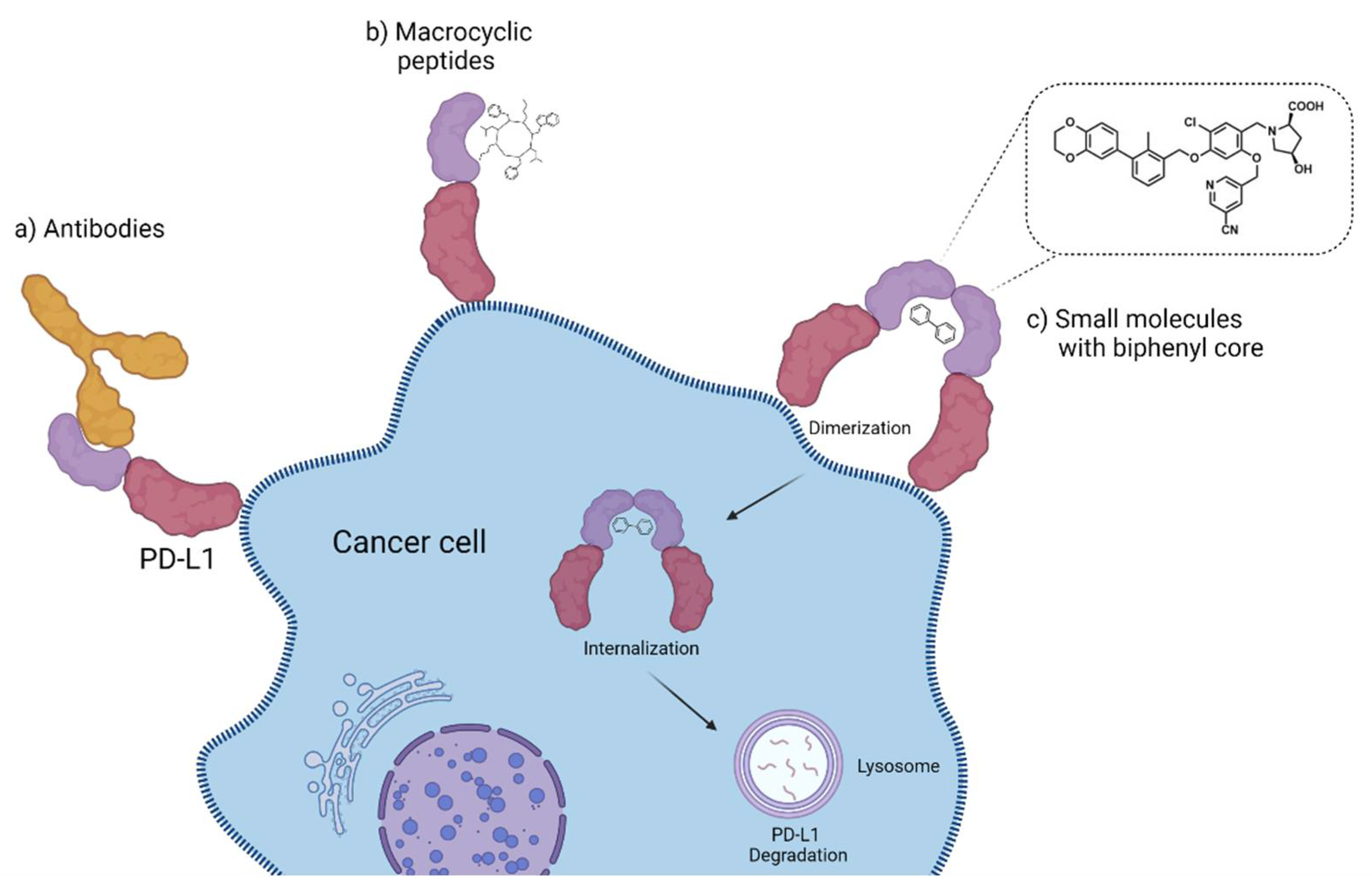
2.3.7. Small Molecules
2.3.8. Miscellaneous
3. Discussion and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef] [PubMed]
- Topalian Suzanne, L.; Drake Charles, G.; Pardoll Drew, M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non–Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.C.; Sosman, J.A.; Sznol, M.; Gordon, M.S.; Hollebecque, A.; Hamid, O.; McDermott, D.F.; Delord, J.-P.; Rhee, I.P.; Mokatrin, A.; et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (mRCC). J. Clin. Oncol. 2013, 31, 4505. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Minor, D.R.; D’Angelo, S.P.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Grob, J.J.; et al. LBA3_PR—A Phase 3 Randomized, Open-Label Study of Nivolumab (Anti-Pd-1; Bms-936558; Ono-4538) Versus Investigator’S Choice Chemotherapy (Icc) in Patients with Advanced Melanoma After Prior Anti-Ctla-4 Therapy. Ann. Oncol. 2014, 25, v1. [Google Scholar] [CrossRef]
- Guardascione, M.; Toffoli, G. Immune Checkpoint Inhibitors as Monotherapy or Within a Combinatorial Strategy in Advanced Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 6302. [Google Scholar] [CrossRef]
- Hakozaki, T.; Hosomi, Y.; Kitadai, R.; Kitagawa, S.; Okuma, Y. Efficacy of immune checkpoint inhibitor monotherapy for patients with massive non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2957–2966. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef]
- Willmann, J.K.; van Bruggen, N.; Dinkelborg, L.M.; Gambhir, S.S. Molecular imaging in drug development. Nat. Rev. Drug Discov. 2008, 7, 591–607. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Gambhir, S.S. A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications. Physiol. Rev. 2012, 92, 897–965. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry—Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Mayer, A.T.; Xu, L.; Reeves, R.E.; Gano, J.; Gambhir, S.S. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjugate Chem. 2015, 26, 2062–2069. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, H.; Liu, T.; Xu, X.; Kong, Y.; Yao, S.; Sheng, X.; Yang, Z. Development of 99mTc-Conjugated JS001 Antibody for In Vivo Mapping of PD-1 Distribution in Murine. Bioorg. Med. Chem. Lett. 2019, 29, 2178–2181. [Google Scholar] [CrossRef]
- Cole, E.L.; Kim, J.; Donnelly, D.J.; Smith, R.A.; Cohen, D.; Lafont, V.; Morin, P.E.; Huang, R.Y.C.; Chow, P.L.; Hayes, W.; et al. Radiosynthesis and Preclinical PET Evaluation of 89Zr-Nivolumab (BMS-936558) in Healthy Non-Human Primates. Bioorg. Med. Chem. 2017, 25, 5407–5414. [Google Scholar] [CrossRef]
- England, C.G.; Jiang, D.; Ehlerding, E.B.; Rekoske, B.T.; Ellison, P.A.; Hernandez, R.; Barnhart, T.E.; McNeel, D.G.; Huang, P.; Cai, W. 89Zr-Labeled Nivolumab for Imaging of T-Cell Infiltration in a Humanized Murine Model of Lung Cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 110–120. [Google Scholar] [CrossRef]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; Van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole Body PD-1 and PD-L1 Positron Emission Tomography in Patients with Non-Small-Cell Lung Cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- England, C.G.; Ehlerding, E.B.; Hernandez, R.; Rekoske, B.T.; Graves, S.A.; Sun, H.; Liu, G.; McNeel, D.G.; Barnhart, T.E.; Cai, W. Preclinical Pharmacokinetics and Biodistribution Studies of 89Zr-Labeled Pembrolizumab. J. Nucl. Med. 2017, 58, 162–168. [Google Scholar] [CrossRef]
- van der Veen, E.L.; Giesen, D.; Pot-de Jong, L.; Jorritsma-Smit, A.; De Vries, E.G.E.; Lub-de Hooge, M.N. 89Zr-pembrolizumab biodistribution is influenced by PD-1-mediated uptake in lymphoid organs. J Immunother Cancer 2020, 8, e000938. [Google Scholar] [CrossRef]
- Natarajan, A.; Mayer, A.T.; Reeves, R.E.; Nagamine, C.M.; Gambhir, S.S. Development of Novel ImmunoPET Tracers to Image Human PD-1 Checkpoint Expression on Tumor-Infiltrating Lymphocytes in a Humanized Mouse Model. Mol. Imaging Biol. 2017, 19, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Patel, C.B.; Habte, F.; Gambhir, S.S. Dosimetry Prediction for Clinical Translation of 64 Cu-Pembrolizumab ImmunoPET Targeting Human PD-1 Expression. Sci. Rep. 2018, 8, 633. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Y.; Rubins, D.; Bennacef, I.; Holahan, M.; Haley, H.; Purcell, M.; Gantert, L.; Hseih, S.; Judo, M.; et al. PET/CT Imaging of 89Zr-N-sucDf-Pembrolizumab in Healthy Cynomolgus Monkeys. Mol. Imaging Biol. 2020, 23, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liang, X.; Li, Y.; Sun, T.; Jin, Z.; Xue, H.; Tian, J. Nuclear and Fluorescent Labeled PD-1-Liposome-DOX-64Cu/IRDye800CW Allows Improved Breast Tumor Targeted Imaging and Therapy. Mol. Pharmaceutics 2017, 14, 3978–3986. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Sun, T.; Liang, X.; Li, Y.; Jin, Z.; Xue, H.; Wan, Y.; Tian, J. Improved Resection and Prolonged Overall Survival with PD-1-IRDye800CW Fluorescence Probe-Guided Surgery and PD-1 Adjuvant Immunotherapy in 4T1 Mouse Model. Int. J. Nanomed. 2017, 12, 8337–8351. [Google Scholar] [CrossRef]
- Hettich, M.; Braun, F.; Bartholomä, M.D.; Schirmbeck, R.; Niedermann, G. High-Resolution PET Imaging with Therapeutic Antibody-Based PD-1/PD-L1 Checkpoint Tracers. Theranostics 2016, 6, 1629–1640. [Google Scholar] [CrossRef]
- Heskamp, S.; Hobo, W.; Molkenboer-Kuenen, J.D.M.; Olive, D.; Oyen, W.J.G.; Dolstra, H.; Boerman, O.C. Noninvasive Imaging of Tumor PD-L1 Expression Using Radiolabeled Anti–PD-L1 Antibodies. Cancer Res. 2015, 75, 2928–2936. [Google Scholar] [CrossRef]
- Josefsson, A.; Nedrow, J.R.; Park, S.; Banerjee, S.R.; Rittenbach, A.; Jammes, F.; Tsui, B.; Sgouros, G. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2016, 76, 472–479. [Google Scholar] [CrossRef]
- Nedrow, J.R.; Josefsson, A.; Park, S.; Ranka, S.; Roy, S.; Sgouros, G. Imaging of Programmed Cell Death Ligand 1: Impact of Protein Concentration on Distribution of Anti-PD-L1 SPECT Agents in an Immunocompetent Murine Model of Melanoma. J. Nucl. Med. 2017, 58, 1560–1566. [Google Scholar] [CrossRef]
- Heskamp, S.; Wierstra, P.J.; Molkenboer-Kuenen, J.D.M.; Sandker, G.W.; Thordardottir, S.; Cany, J.; Olive, D.; Bussink, J.; Boerman, O.C.; Dolstra, H.; et al. PD-L1 microSPECT/CT Imaging for Longitudinal Monitoring of PD-L1 Expression in Syngeneic and Humanized Mouse Models for Cancer. Cancer Immunol. Res. 2019, 7, 150–161. [Google Scholar] [CrossRef]
- Truillet, C.; Oh, H.L.J.; Yeo, S.P.; Lee, C.-Y.; Huynh, L.T.; Wei, J.; Parker, M.F.L.; Blakely, C.; Sevillano, N.; Wang, Y.-H.; et al. Imaging PD-L1 Expression with ImmunoPET. Bioconjugate Chem. 2018, 29, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ehlerding, E.B.; Jiang, D.; Barnhart, T.E.; Chen, W.; Cao, T.; Engle, J.W.; Cai, W. In Vivo Characterization of PD-L1 Expression in Breast Cancer by Immuno-PET with (89)Zr-Labeled Avelumab. Am. J. Transl. Res. 2020, 12, 1862–1872. [Google Scholar] [PubMed]
- Jagoda, E.M.; Vasalatiy, O.; Basuli, F.; Opina, A.C.L.; Williams, M.R.; Wong, K.; Lane, K.C.; Adler, S.; Ton, A.T.; Szajek, L.P.; et al. Immuno-PET Imaging of the Programmed Cell Death-1 Ligand (PD-L1) Using a Zirconium-89 Labeled Therapeutic Antibody, Avelumab. Mol. Imaging 2019, 18, 1536012119829986. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Lesniak, W.G.; Gabrielson, M.; Lisok, A.; Wharram, B.; Sysa-Shah, P.; Azad, B.B.; Pomper, M.G.; Nimmagadda, S. A Humanized Antibody for Imaging Immune Checkpoint Ligand PD-L1 Expression in Tumors. Oncotarget 2016, 7, 10215–10227. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lesniak, W.G.; Nimmagadda, S. Noninvasive Imaging of Immune Checkpoint Ligand PD-L1 in Tumors and Metastases for Guiding Immunotherapy. Mol. Imaging 2017, 16, 1536012117718459. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Chatterjee, S.; Gabrielson, M.; Lisok, A.; Wharram, B.; Pomper, M.G.; Nimmagadda, S. PD-L1 Detection in Tumors Using [64Cu]Atezolizumab with PET. Bioconjugate Chem. 2016, 27, 2103–2110. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Vento, J.; Mulgaonkar, A.; Woolford, L.; Nham, K.; Christie, A.; Bagrodia, A.; de Leon, A.D.; Hannan, R.; Bowman, I.; McKay, R.M.; et al. PD-L1 Detection Using 89Zr-Atezolizumab Immuno-PET in Renal Cell Carcinoma Tumorgrafts from a Patient with Favorable Nivolumab Response. J. Immunother. Cancer 2019, 7, 144. [Google Scholar] [CrossRef]
- Ehlerding, E.B.; Lee, H.J.; Barnhart, T.E.; Jiang, D.; Kang, L.; McNeel, D.G.; Engle, J.W.; Cai, W. Noninvasive Imaging and Quantification of Radiotherapy-Induced PD-L1 Upregulation with 89Zr–Df–Atezolizumab. Bioconjugate Chem. 2019, 30, 1434–1441. [Google Scholar] [CrossRef]
- Qiu, L.; Tan, H.; Lin, Q.; Si, Z.; Mao, W.; Wang, T.; Fu, Z.; Cheng, D.; Shi, H. A Pretargeted Imaging Strategy for Immune Checkpoint Ligand PD-L1 Expression in Tumor Based on Bioorthogonal Diels-Alder Click Chemistry. Mol. Imaging Biol. 2020, 22, 842–853. [Google Scholar] [CrossRef]
- Xu, M.; Han, Y.; Liu, G.; Xu, Y.; Duan, D.; Liu, H.; Du, F.; Luo, P.; Liu, Z. Preclinical Study of a Fully Human Anti-PD-L1 Antibody as a Theranostic Agent for Cancer Immunotherapy. Mol. Pharm. 2018, 15, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Clump, D.A.; Srivastava, R.M.; Sun, L.; Zeng, D.; Diaz-Perez, J.A.; Anderson, C.J.; Edwards, W.B.; Ferris, R.L. Preclinical ImmunoPET/CT Imaging Using Zr-89-Labeled Anti-PD-L1 Monoclonal Antibody for Assessing Radiation-Induced PD-L1 Upregulation in head and neck cancer and melanoma. OncoImmunology 2017, 6, e1329071. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheng, S.; Zou, S.; Zhu, D.; Zhu, T.; Wang, P.; Zhu, X. Immuno-PET Imaging of (89)Zr Labeled Anti-PD-L1 Domain Antibody. Mol. Pharm. 2018, 15, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476. [Google Scholar] [CrossRef]
- Zhao, S.; Pan, W.; Jiang, H.; Zhang, R.; Jiang, H.; Liang, Z.; Hu, H. Cerenkov Luminescence Imaging is an Effective Preclinical Tool for Assessing Colorectal Cancer PD-L1 Levels In Vivo. EJNMMI Res. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Pang, X.; Liu, M.; Wang, R.; Liao, X.; Yan, P.; Zhang, C. Radioimmunoimaging and Targeting Treatment in an Immunocompetent Murine Model of Triple-Negative Breast Cancer Using Radiolabeled Anti–Programmed Death-Ligand 1 Monoclonal Antibody. J. Label. Compd. Radiopharm. 2018, 61, 826–836. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, H.; Zhang, R.; Jiang, H.; Xu, H.; Pan, W.; Gao, X.; Sun, Z. Near-Infrared Fluorescence-Labeled Anti-PD-L1-mAb for Tumor Imaging in Human Colorectal Cancer Xenografted Mice. J. Cell. Biochem. 2019, 120, 10239–10247. [Google Scholar] [CrossRef]
- Huang, H.-F.; Zhu, H.; Li, G.-H.; Xie, Q.; Yang, X.-T.; Xu, X.-X.; Tian, X.-B.; Wan, Y.-K.; Yang, Z. Construction of Anti-hPD-L1 HCAb Nb6 and in Situ 124I Labeling for Noninvasive Detection of PD-L1 Expression in Human Bone Sarcoma. Bioconjugate Chem. 2019, 30, 2614–2623. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, M.; Li, G.; Liu, T.; Wan, Y.; Liu, Z.; Zhu, H.; Yang, Z. Evaluation of 64Cu Radiolabeled Anti-hPD-L1 Nb6 for Positron Emission Tomography Imaging in Lung Cancer Tumor Mice Model. Bioorg. Med. Chem. Lett. 2020, 30, 126915. [Google Scholar] [CrossRef]
- Wissler, H.L.; Ehlerding, E.B.; Lyu, Z.; Zhao, Y.; Zhang, S.; Eshraghi, A.; Buuh, Z.Y.; McGuth, J.C.; Guan, Y.; Engle, J.W.; et al. Site-Specific Immuno-PET Tracer to Image PD-L1. Mol. Pharm. 2019, 16, 2028–2036. [Google Scholar] [CrossRef]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J. Nucl. Med. 2020, 61, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N. Non-Invasive Assessment of Murine PD-L1 Levels in Syngeneic Tumor Models by Nuclear Imaging with Nanobody Rracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef] [PubMed]
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a 99mTc-Labeled Anti–Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non–Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- González Trotter, D.E.; Meng, X.; McQuade, P.; Rubins, D.; Klimas, M.; Zeng, Z.; Connolly, B.M.; Miller, P.J.; O’Malley, S.S.; Lin, S.-A.; et al. In Vivo Imaging of the Programmed Death Ligand 1 by 18F PET. J. Nucl. Med. 2017, 58, 1852–1857. [Google Scholar] [CrossRef]
- Rubins, D.J.; Meng, X.; McQuade, P.; Klimas, M.; Getty, K.; Lin, S.-A.; Connolly, B.M.; O’Malley, S.S.; Haley, H.; Purcell, M.; et al. In Vivo Evaluation and Dosimetry Estimate for a High Affinity Affibody PET Tracer Targeting PD-L1. Mol. Imaging Biol. 2020, 23, 241–249. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lesniak, W.G.; Miller, M.S.; Lisok, A.; Sikorska, E.; Wharram, B.; Kumar, D.; Gabrielson, M.; Pomper, M.G.; Gabelli, S.B.; et al. Rapid PD-L1 Detection in Tumors with PET Using a Highly Specific Peptide. Biochem. Biophys. Res. Commun. 2017, 483, 258–263. [Google Scholar] [CrossRef]
- De Silva, R.A.; Kumar, D.; Lisok, A.; Chatterjee, S.; Wharram, B.; Venkateswara Rao, K.; Mease, R.; Dannals, R.F.; Pomper, M.G.; Nimmagadda, S. Peptide-Based 68Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol. Pharm. 2018, 15, 3946–3952. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Mease, R.C.; Chatterjee, S.; Kumar, D.; Lisok, A.; Wharram, B.; Kalagadda, V.R.; Emens, L.A.; Pomper, M.G.; Nimmagadda, S. Development of [18F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol. Imaging 2019, 18, 1536012119852189. [Google Scholar] [CrossRef]
- Li, C.; Zhang, N.; Zhou, J.; Ding, C.; Jin, Y.; Cui, X.; Pu, K.; Zhu, Y. Peptide Blocking of PD-1/PD-L1 Interaction for Cancer Immunotherapy. Cancer Immunol. Res. 2018, 6, 178. [Google Scholar] [CrossRef]
- Kuan, H.; Masayuki, H.; Xie, L.; Zhang, Y.; Kotaro, N.; Hisashi, S.; Zhang, M.-R. Developing Native Peptide-Based Radiotracers for PD-L1 PET Imaging and Improving Imaging Contrast by Pegylation. Chem. Commun. 2019, 55, 4162–4165. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Han, Y.; Hu, K.; Huang, S.; Wang, M.; Zhou, K.; Fu, L.; Chen, H.; Tang, G. Synthesis and biological evaluation of Al[18F]-NOTA-IPB-PDL1P as a molecular probe for PET imaging of PD-L1 positive tumors. Bioorganic Chem. 2022, 122, 105682. [Google Scholar] [CrossRef] [PubMed]
- Jouini, N.; Cardinale, J.; Mindt, T.L. Evaluation of a Radiolabeled Macrocyclic Peptide as Potential PET Imaging Probe for PD-L1. ChemMedChem 2022, n/a, e202200091. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Lv, G.; Chen, Y.; Qiu, L.; Xie, M.; Lin, J. One-Step Radiosynthesis and Initial Evaluation of a Small Molecule PET Tracer for PD-L1 Imaging. Bioorganic Med. Chem. Lett. 2020, 30, 127572. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Miao, Y.; Chen, Y.; Lu, C.; Wang, X.; Xie, M.; Qiu, L.; Lin, J. Promising potential of a 18F-labelled small-molecular radiotracer to evaluate PD-L1 expression in tumors by PET imaging. Bioorganic Chem. 2021, 115, 105294. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.L.; Gordon, S.R.; Mayer, A.T.; McCracken, M.N.; Natarajan, A.; Ring, N.G.; Kimura, R.; Tsai, J.M.; Manglik, A.; Kruse, A.C.; et al. Engineering High-Affinity PD-1 Variants for Optimized Immunotherapy and Immuno-PET Imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E6506–E6514. [Google Scholar] [CrossRef]
- Mayer, A.T.; Natarajan, A.; Gordon, S.R.; Maute, R.L.; McCracken, M.N.; Ring, A.M.; Weissman, I.L.; Gambhir, S.S. Practical Immuno-PET Radiotracer Design Considerations for Human Immune Checkpoint Imaging. J. Nucl. Med. 2017, 58, 538–546. [Google Scholar] [CrossRef]
- Natarajan, A.; Patel, C.B.; Ramakrishnan, S.; Panesar, P.S.; Long, S.R.; Gambhir, S.S. A Novel Engineered Small Protein for Positron Emission Tomography Imaging of Human Programmed Death Ligand-1: Validation in Mouse Models and Human Cancer Tissues. Clin. Cancer Res. 2019, 25, 1774–1785. [Google Scholar] [CrossRef]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovšek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and Biologic Evaluation of a Novel 18F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef]
- Huisman, M.C.; Niemeijer, A.-L.N.; Windhorst, A.D.; Schuit, R.C.; Leung, D.; Hayes, W.; Poot, A.; Bahce, I.; Radonic, T.; Oprea-Lager, D.E.; et al. Quantification of PD-L1 Expression with 18F-BMS-986192 PET/CT in Patients with Advanced-Stage Non–Small Cell Lung Cancer. J. Nucl. Med. 2020, 61, 1455–1460. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Van Der Veen, E.L.; Kol, A.; Antunes, I.F.; De Vries, E.F.J.; Hospers, G.A.P.; De Vries, E.G.E.; De Jong, S.; Lub-De Hooge, M.N. Molecular Imaging of PD-L1 Expression and Dynamics with the Adnectin-Based PET Tracer 18F-BMS-986192. J. Nucl. Med. 2020, 61, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Meir, R.; Shamalov, K.; Sadan, T.; Motiei, M.; Yaari, G.; Cohen, C.J.; Popovtzer, R. Fast Image-Guided Stratification Using Anti-Programmed Death Ligand 1 Gold Nanoparticles for Cancer Immunotherapy. ACS Nano 2017, 11, 11127–11134. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, P.P.; Kist de Ruijter, L.; Lub-de Hooge, M.N.; Brouwers, A.H.; van der Wekken, A.J.; Oosting, S.F.; Fehrmann, R.S.N.; de Groot, D.J.A.; de Vries, E.G.E. Molecular Imaging Biomarkers for Immune Checkpoint Inhibitor Therapy. Theranostics 2020, 10, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Dolan, D.E.; Gupta, S. PD-1 Pathway Inhibitors: Changing the Landscape of Cancer Immunotherapy. Cancer Control. 2014, 21, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Sun, X.; Niu, W.; Weisman, G.R.; Wong, E.H.; Rheingold, A.L.; Anderson, C.J. Comparative In Vivo Stability of Copper-64-Labeled Cross-Bridged and Conventional Tetraazamacrocyclic Complexes. J. Med. Chem. 2004, 47, 1465–1474. [Google Scholar] [CrossRef]
- Zakaly, H.M.H.; Mostafa, M.Y.A.; Zhukovsky, M. Dosimetry Assessment of Injected 89Zr-Labeled Monoclonal Antibodies in Humans. Radiat. Res. 2019, 191, 466–474. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R. Acquired Resistance to Fractionated Radiotherapy Can Be overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and Anti–PD-L1 Treatment Synergistically Promote Antitumor Immunity in Mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Gulley, J.L. Avelumab demonstrates promise in advanced NSCLC. Oncotarget 2017, 8, 102767–102768. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.-l. MPDL3280A (Anti-PD-L1) Treatment Leads to Clinical Activity in Metastatic Bladder Cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Cheng, J.; Yang, T.; Li, Y.; Zhu, B. EGFR-TKI Down-Regulates PD-L1 in EGFR Mutant NSCLC ThroughIinhibiting NF-κB. Biochem. Biophys. Res. Commun. 2015, 463, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; De Haard, H.J. Properties, Production, and Applications of Camelid Single-Domain Antibody Fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Wernery, U.; Muyldermans, S.; Nguyen, V.K. Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev. Comp. Immunol. 2003, 27, 87–103. [Google Scholar] [CrossRef]
- Tanha, J.; Dubuc, G.; Hirama, T.; Narang, S.A.; MacKenzie, C.R. Selection by Phage Display of Llama Conventional VH Fragments with Heavy Chain Antibody VHH Properties. J. Immunol. Methods 2002, 263, 97–109. [Google Scholar] [CrossRef]
- Fu, R.; Carroll, L.; Yahioglu, G.; Aboagye, E.O.; Miller, P.W. Antibody Fragment and Affibody ImmunoPET Imaging Agents: Radiolabelling Strategies and Applications. ChemMedChem 2018, 13, 2466–2478. [Google Scholar] [CrossRef]
- Hudson, P.J.; Souriau, C. Engineered Antibodies. Nat. Med. 2003, 9, 129–134. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody Molecules: Engineered Proteins for Therapeutic, Diagnostic and Biotechnological Applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, Y.; Liu, T.; Li, Z.; Zhang, X.; Chen, X. Peptide-Based Imaging Agents for Cancer Detection. Adv. Drug Deliv. Rev. 2017, 110, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.-M.; Hu, X.-Q.; Liu, X.-X.; Ruan, B.-F.; Xu, J.; Liao, C. From Monoclonal Antibodies to Small Molecules: The Development of Inhibitors Targeting the PD-1/PD-L1 Pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Dömling, A. Inhibitors of Programmed Cell Death 1 (PD-1): A Patent Review (2010-2015). Expert Opin. Ther. Pat. 2016, 26, 973–977. [Google Scholar] [CrossRef]
- Miller, M.M.; Mapelli, C.; Allen, M.P.; Bowsher, M.; Boy, K.; Gillis, E.P.; Langley, D.R.; Mull, E.; Poirier, M.A.; Sanghvi, N.; et al. Macrocyclic Inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD-L1 Protein/Protein Interactions. U.S. Patent 9308236-B2, 2016. [Google Scholar]
- Zhou, X.; Jiang, J.; Yang, X.; Liu, T.; Ding, J.; Nimmagadda, S.; Pomper, M.G.; Zhu, H.; Zhao, J.; Yang, Z.; et al. First-in-human evaluation of a PD-L1-binding peptide radiotracer in non-small cell lung cancer patients with PET. J. Nucl. Med. 2022, 63, 536–542. [Google Scholar] [CrossRef]
- Donnelly, D.J. Small Molecule PET Tracers in Drug Discovery. Semin. Nucl. Med. 2017, 47, 454–460. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Daniels, D.L.; Riching, K.M.; Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discov. Today Technol. 2019, 31, 61–68. [Google Scholar] [CrossRef]
- Yang, J.; Hu, L. Immunomodulators Targeting the PD-1/PD-L1 Protein-Protein Interaction: From Antibodies to Small Molecules. Med. Res. Rev. 2019, 39, 265–301. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Yang, Y.; Lai, F. Method for Preparing 2-Hydroxyl-4-(2, 3-Disubstituted Benzyloxy)-5-Substituted Benzaldehyde Derivatives. U.S. Patent EP 3 459 925 A1, 2019. [Google Scholar]
- Yeung, K.-S.; Connolly, T.P.; Frennesson, D.B.; Grant-Young, K.A.; Hewawasam, P.; Langley, D.R.; Meng, Z.; Mull, E.; Parcella, K.E.; Saulnier, M.G.; et al. Compounds Useful as Immunomodulators. WO Patent 2017/066227 A1, 2017. [Google Scholar]
- Aktoudianakis, E.; Appleby, T.; Cho, A.; Du, Z.; Graupe, M.; Aktoudianakis, E.; Appleby, T.; Cho, A.; Du, Z.; Graupe, M.; et al. PD-1/PD-L1 Inhibitors. WO Patent 2018/195321 Al, 2018. [Google Scholar]
- Ankri, R.; Ashkenazy, A.; Milstein, Y.; Brami, Y.; Olshinka, A.; Goldenberg-Cohen, N.; Popovtzer, A.; Fixler, D.; Hirshberg, A. Gold Nanorods Based Air Scanning Electron Microscopy and Diffusion Reflection Imaging for Mapping Tumor Margins in Squamous Cell Carcinoma. ACS Nano 2016, 10, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Popovtzer, A.; Mizrachi, A.; Motiei, M.; Bragilovski, D.; Lubimov, L.; Levi, M.; Hilly, O.; Ben-Aharon, I.; Popovtzer, R. Actively Targeted Gold Nanoparticles as Novel Radiosensitizer Agents: An In Vivo Head and Neck Cancer Model. Nanoscale 2016, 8, 2678–2685. [Google Scholar] [CrossRef] [PubMed]
- Dreifuss, T.; Betzer, O.; Shilo, M.; Popovtzer, A.; Motiei, M.; Popovtzer, R. A Challenge for Theranostics: Is the Optimal Particle for Therapy Also Optimal for Diagnostics? Nanoscale 2015, 7, 15175–15184. [Google Scholar] [CrossRef] [PubMed]
- Lipovšek, D. Adnectins: Engineered Target-Binding Protein Therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The Fibronectin Type III Domain as a Scaffold for Novel Binding Proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Pysz, M.A.; Gambhir, S.S.; Willmann, J.K. Molecular imaging: Current status and emerging strategies. Clin. Radiol. 2010, 65, 500–516. [Google Scholar] [CrossRef]
- Skotland, T. Molecular imaging: Challenges of bringing imaging of intracellular targets into common clinical use. Contrast Media Mol. Imaging 2012, 7, 1–6. [Google Scholar] [CrossRef]
- Nagaya, T.; Nakamura, Y.A.; Choyke, P.L.; Kobayashi, H. Fluorescence-Guided Surgery. Front. Oncol. 2017, 7, 314. [Google Scholar] [CrossRef]
- Ariztia, J.; Solmont, K.; Moïse, N.P.; Specklin, S.; Heck, M.P.; Lamandé-Langle, S.; Kuhnast, B. PET/Fluorescence Imaging: An Overview of the Chemical Strategies to Build Dual Imaging Tools. Bioconjugate Chem. 2022, 33, 24–52. [Google Scholar] [CrossRef]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Blaszkiewicz, U.; Pustula, M.; Butera, R.; Domling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Liu, Z.; Chen, Y.; Feng, F.; Sun, H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur. J. Med. Chem. 2019, 161, 378–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, X.; Guo, C.; Zhang, K.; Xu, J.; Li, Z.; Jiang, S. Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. 2019, 62, 1715–1730. [Google Scholar] [CrossRef] [PubMed]
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, W. Heterocyclic Compounds as Immunomodulators. U.S. Patent 20170107216 A1, 2017. [Google Scholar]
- Chupak, L.S.Z.; Xiao, F. International Publication Date. WO Patent 2015/034820 A1, 12 March 2015. [Google Scholar]
- Yeung, K.-S.; Connolly, T.P.; Frennesson, D.B.; Grant-Young, K.A.; Hewawasam, P.; Langley, D.R.; Meng, Z.; Mull, E.; Parcella, K.E.; Saulnier, M.G.; et al. Compounds Useful as Immunomodulators. U.S. Patent 2017/0107202 A1, 2017. [Google Scholar]
- Vallabhajosula, S. Chemistry of Radiohalogens (F, Br, and I). In Molecular Imaging: Radiopharmaceuticals for PET and SPECT; Springer: Berlin/Heidelberg, 2009; pp. 151–166. [Google Scholar]
- Dubost, E.; McErlain, H.; Babin, V.; Sutherland, A.; Cailly, T. Recent Advances in Synthetic Methods for Radioiodination. J. Org. Chem. 2020, 85, 8300–8310. [Google Scholar] [CrossRef] [PubMed]
- Ferris, T.; Carroll, L.; Jenner, S.; Aboagye, E.O. Use of radioiodine in nuclear medicine—A brief overview. J. Label. Compd. Radiopharm. 2021, 64, 92–108. [Google Scholar] [CrossRef]
- Cavina, L.; van der Born, D.; Klaren, P.H.M.; Feiters, M.C.; Boerman, O.C.; Rutjes, F.P.J.T. Design of Radioiodinated Pharmaceuticals: Structural Features Affecting Metabolic Stability towards in Vivo Deiodination. Eur. J. Org. Chem. 2017, 2017, 3387–3414. [Google Scholar] [CrossRef]
- Feng, Z.; Yang, Y. Iodine Isotope-Labeled Benzyl Phenyl Ether Derivative, and Preparation Method and Medicine Thereof. Composition and Use. CN Patent 111662270 A, 2020. [Google Scholar]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef]
- Altmann, A.; Haberkorn, U.; Siveke, J. The Latest Developments in Imaging of Fibroblast Activation Protein. J. Nucl. Med. 2021, 62, 160–167. [Google Scholar] [CrossRef]
- Stadlbauer, S.; Krutzek, F.; Kopka, K. 3-((3-([1,1‘-Biphenyl]-3-ylmethoxy)phenoxy)methyl)benzonitrile Derivatives and the Use Thereof. EP Patent 21212444, 2021. [Google Scholar]
- Surmiak, E.; Magiera-Mularz, K.; Musielak, B.; Muszak, D.; Kocik-Krol, J.; Kitel, R.; Plewka, J.; Holak, T.A.; Skalniak, L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 11797. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S.; et al. Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-F.; Li, Z.; Ma, D.; Yu, Q. Small-molecule PD-L1 inhibitor BMS1166 abrogates the function of PD-L1 by blocking its ER export. Oncoimmunology 2020, 9, 1831153. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. ImmunoTherapy Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
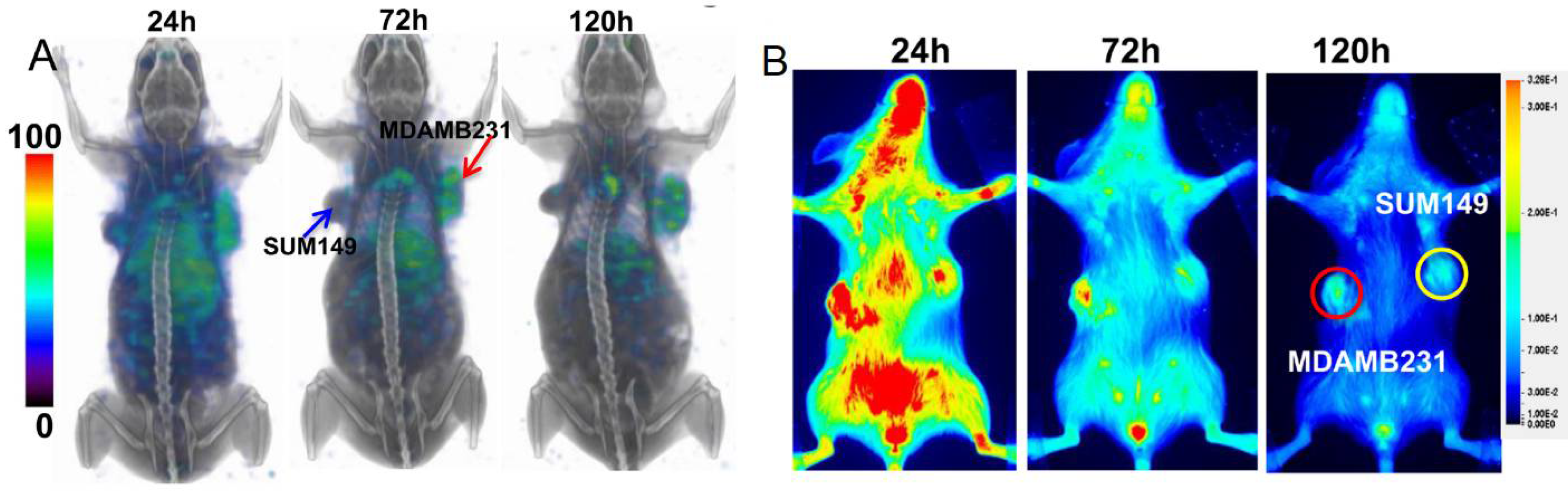{kind=link}
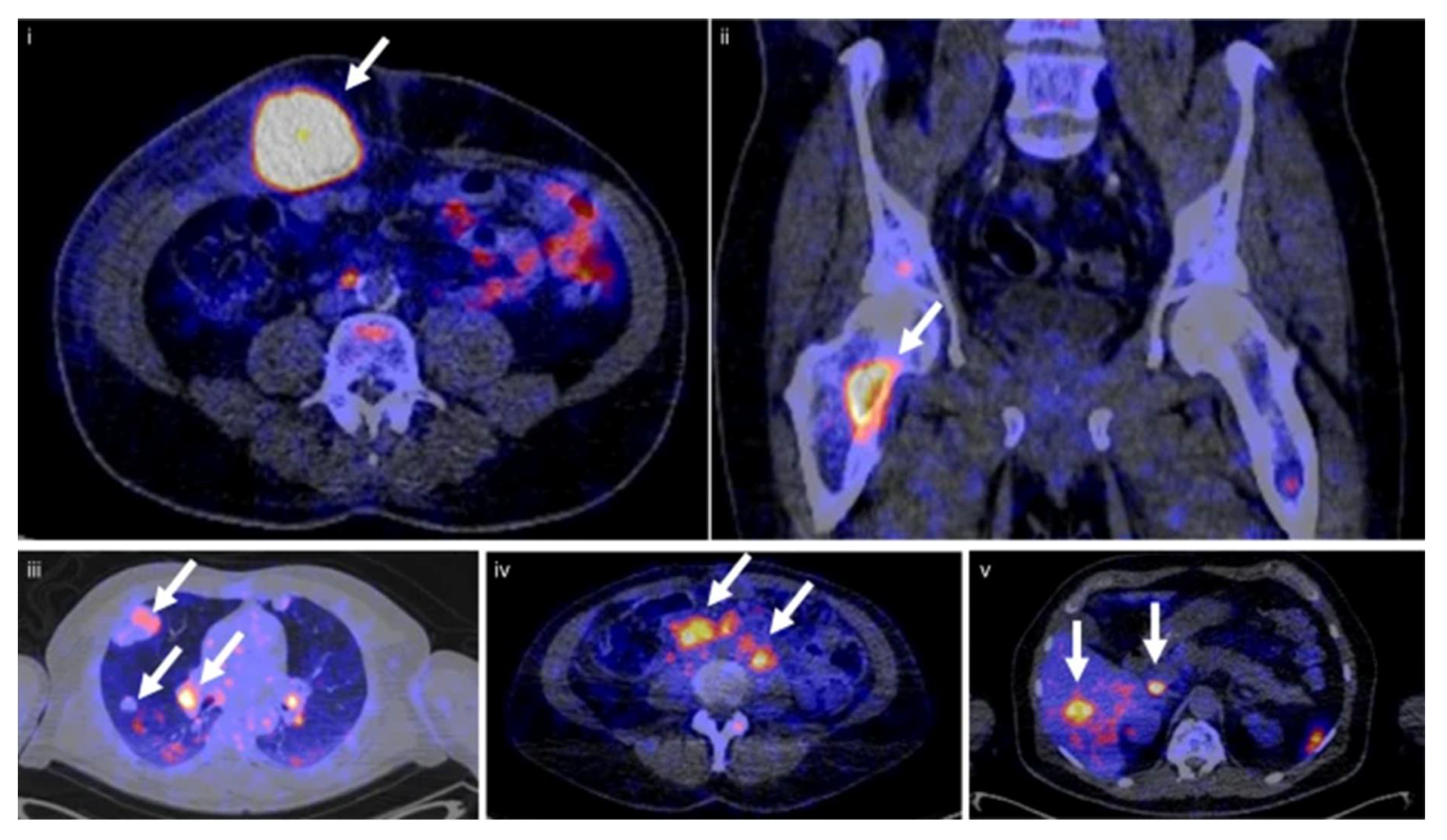{kind=link}
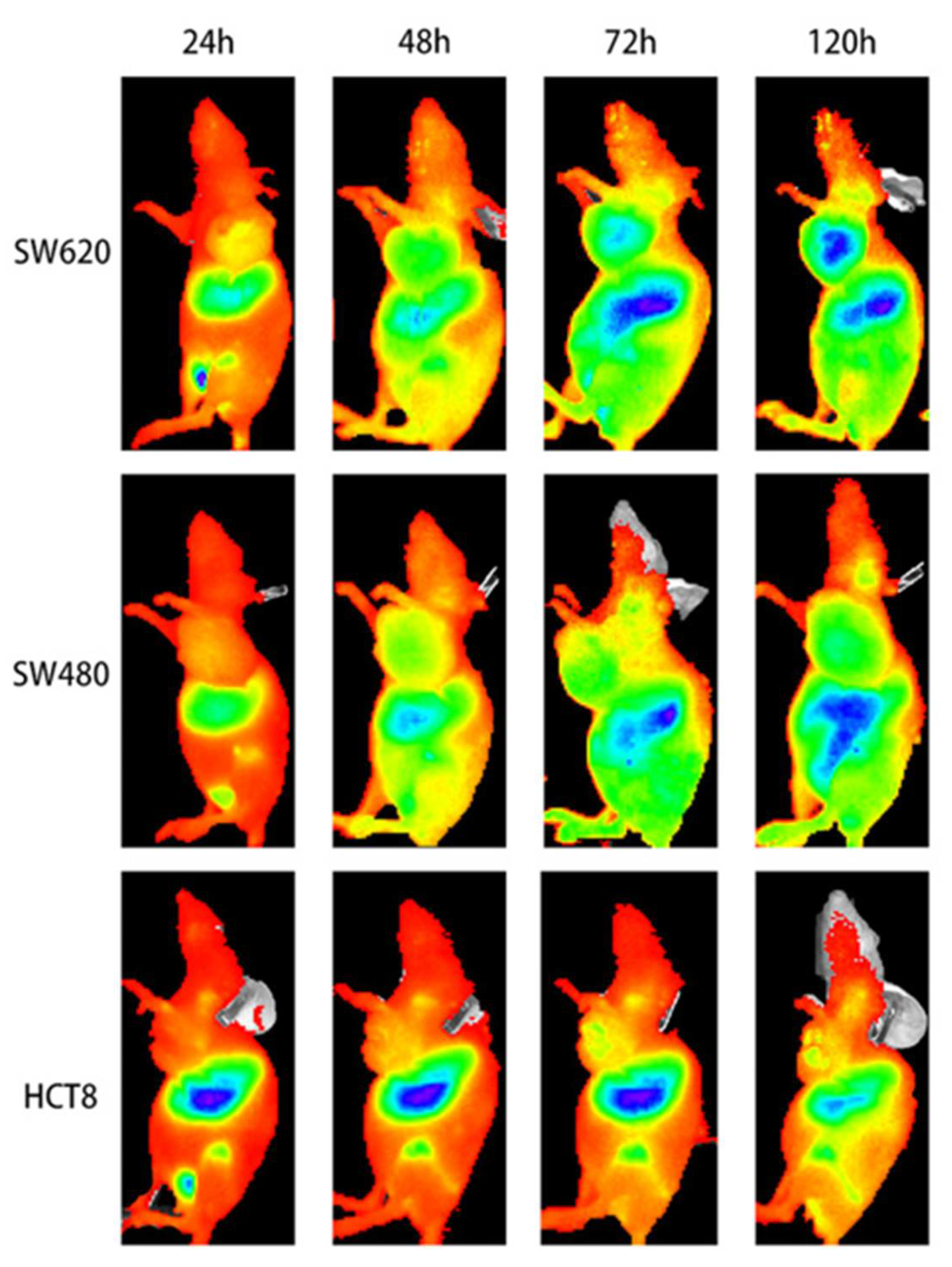{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





| [64Cu]Cu-WL12 | [68Ga]Ga-WL12 | [18F]FPy-WL12 | |
|---|---|---|---|
| Molar Activity | 28.5 ± 1.65 GBq/µmol | 128 GBq/μmol. | 105 ± 54 Ci/mmol |
| IC50 [nM] | 2.9 | n.a. | 37.1 |
| %ID/g at 60 min p.i. | 14.9 ± 0.8 (tumor) 34.4 ± 3.1 (kidney) 24.2 ± 2.5 (liver) | 11.56 ± 3.18 (tumor) 64.7 ± 12.1 (kidney) 15.1 ± 7.6 (liver) | 7.16 ± 1.67 (tumor) ~12 (kidney) ~32 (liver) |
| Tumor-to-Muscle Ratio, 2 h p.i. Tumor-to-Blood Ratio, 1 h p.i. | 25.6 ± 1.9 | 59.79 ± 16.47 | ~18 |
| 4.7 ± 1.2 | 7.56 ± 16.47 | ~4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krutzek, F.; Kopka, K.; Stadlbauer, S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals 2022, 15, 747. https://doi.org/10.3390/ph15060747
Krutzek F, Kopka K, Stadlbauer S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals. 2022; 15(6):747. https://doi.org/10.3390/ph15060747
Chicago/Turabian StyleKrutzek, Fabian, Klaus Kopka, and Sven Stadlbauer. 2022. "Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis" Pharmaceuticals 15, no. 6: 747. https://doi.org/10.3390/ph15060747
APA StyleKrutzek, F., Kopka, K., & Stadlbauer, S. (2022). Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals, 15(6), 747. https://doi.org/10.3390/ph15060747