
Mechanistic Analysis of Chemically Diverse Bromodomain-4 Inhibitors Using Balanced QSAR Analysis and Supported by X-ray Resolved Crystal Structures


 and
and
Abstract
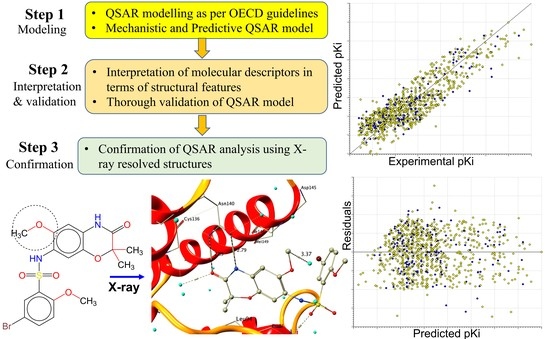
:
1. Introduction
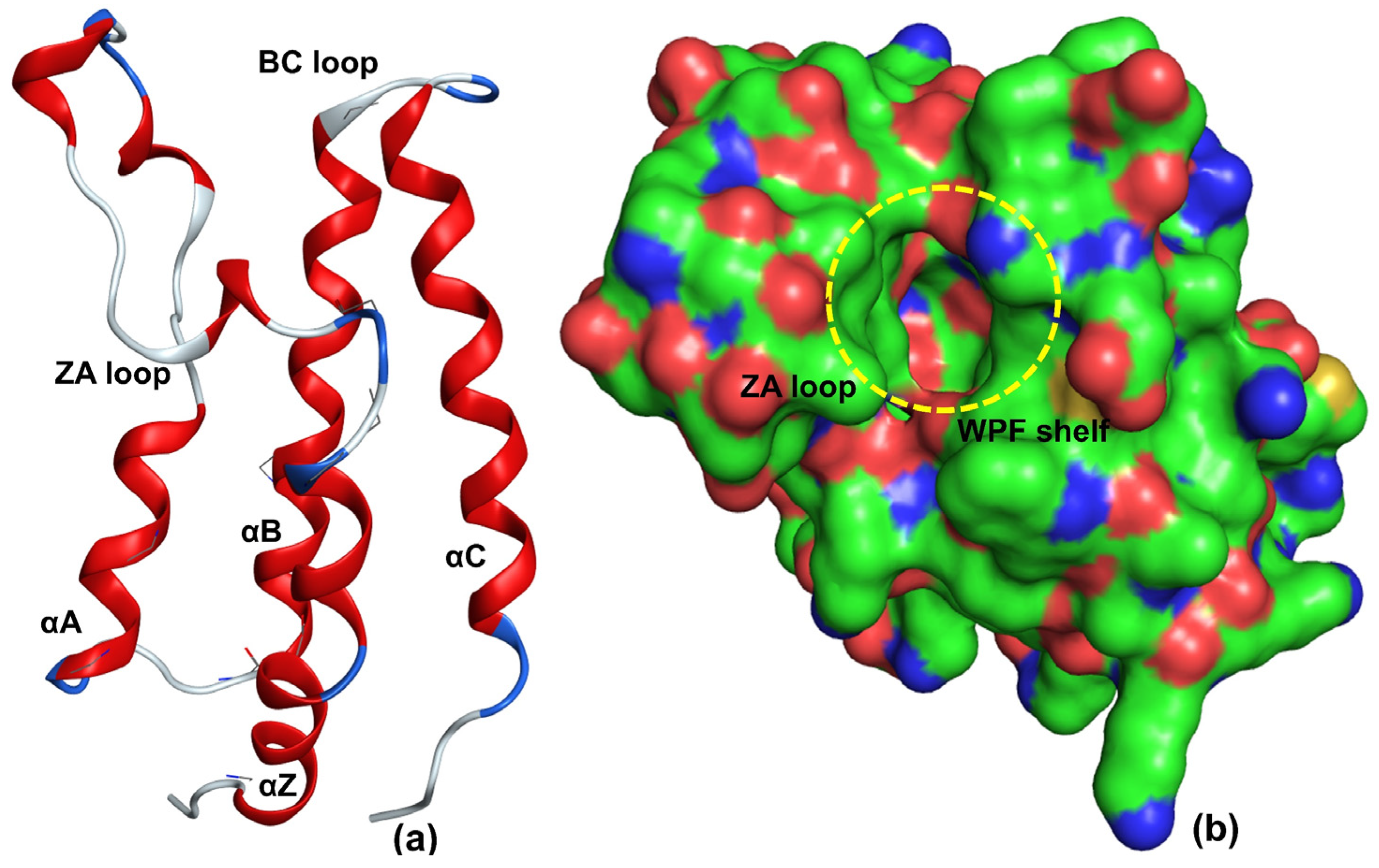
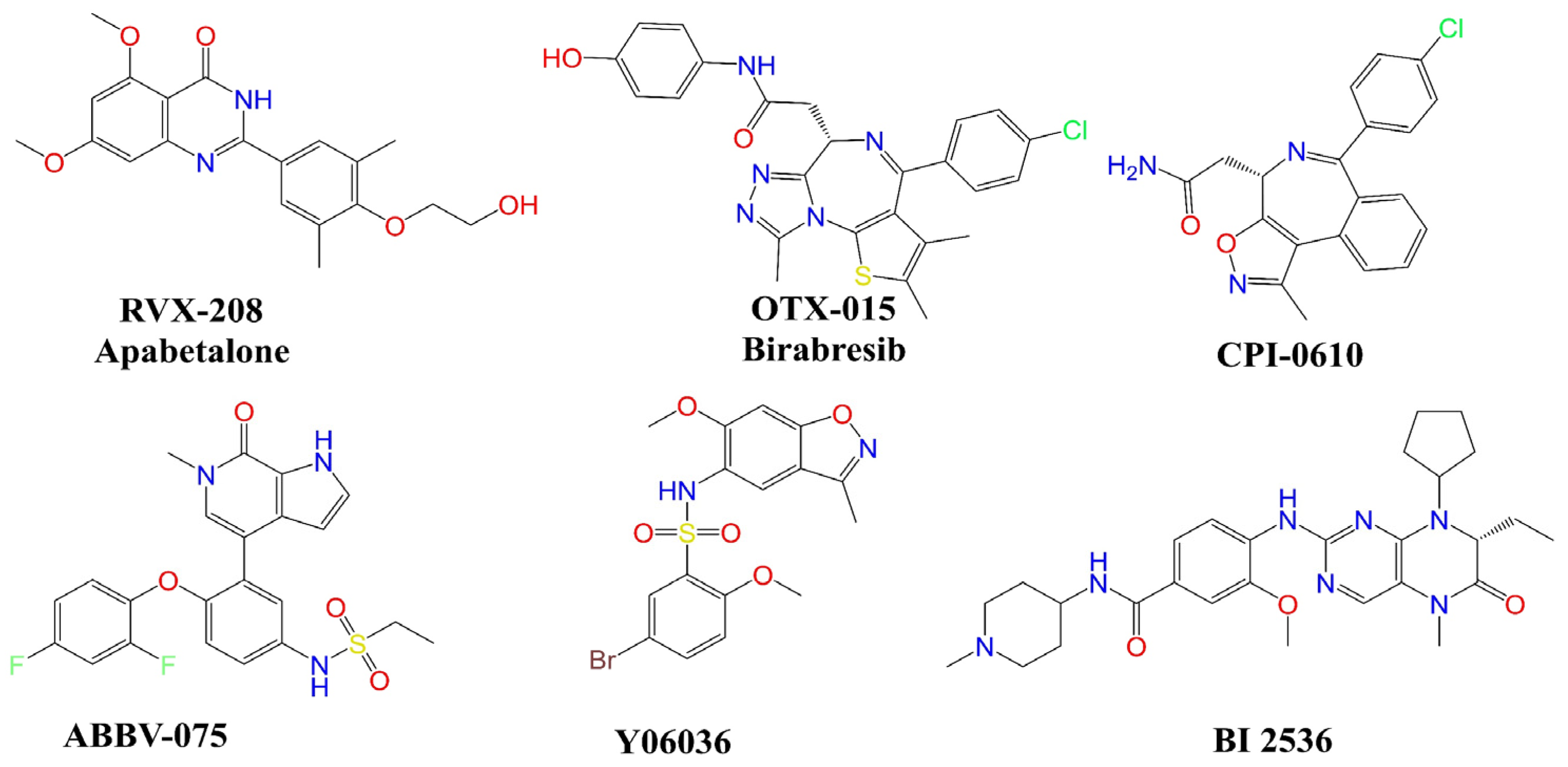
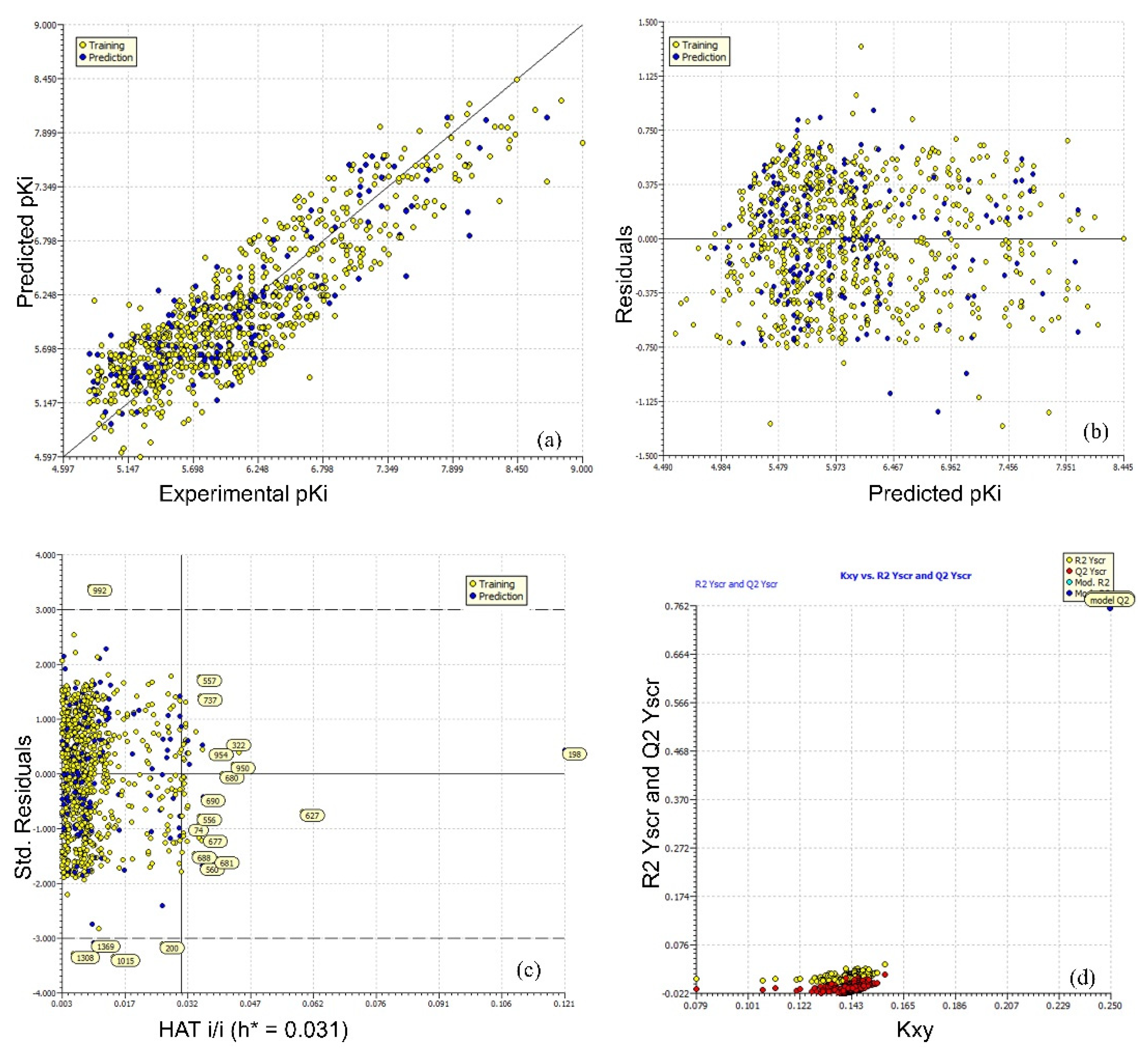
2. Results
3. Discussion
Mechanistic Interpretation of QSAR Model
4. Materials and Methods
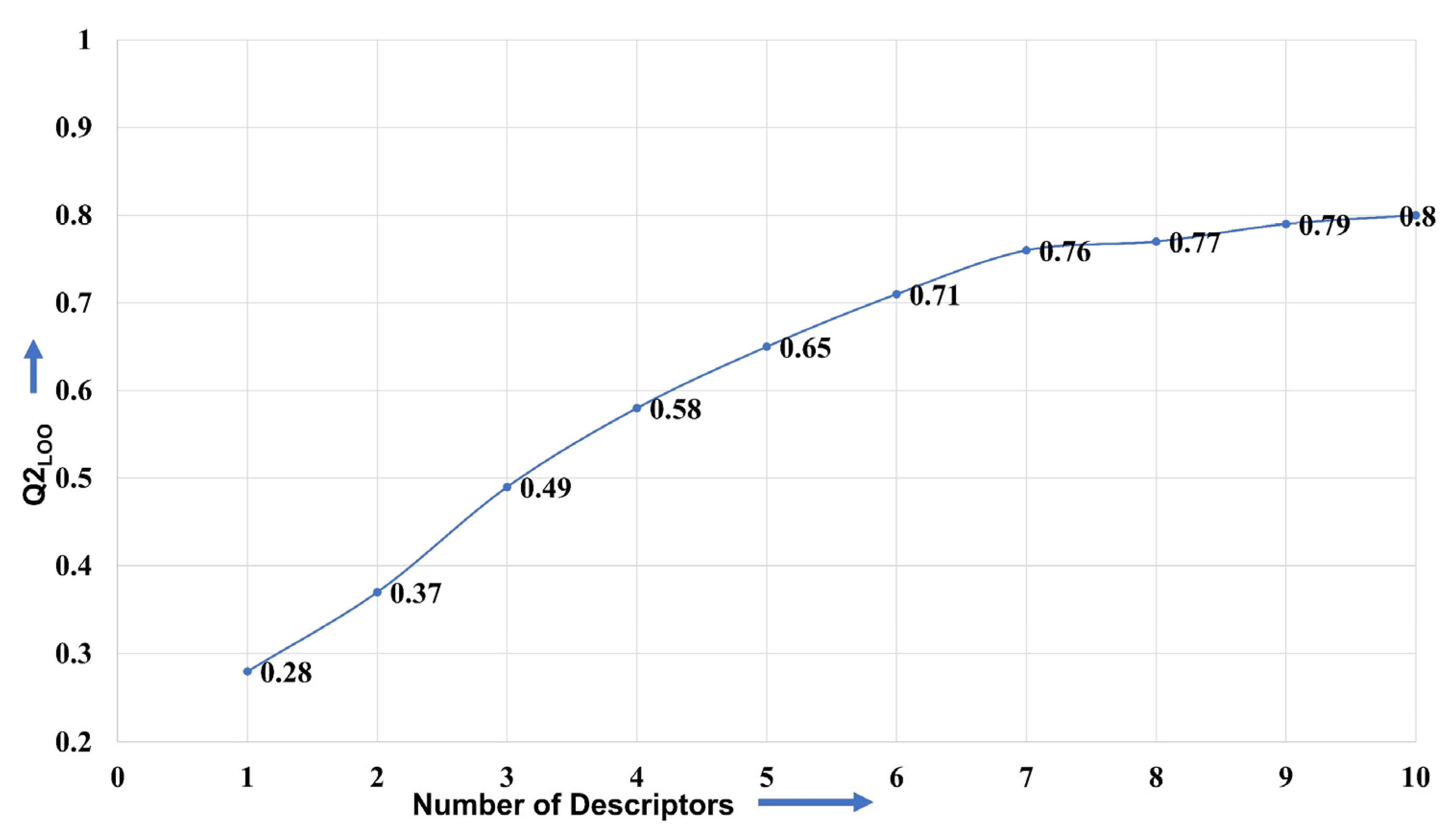
4.1. Splitting the Data Set into Training and External Sets and Subjective Feature Selection (SFS)
4.2. Building Regression Model and Its Validation
4.3. Pharmacophore Model
4.4. Other Experimental Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SMILES | Simplified molecular-input line-entry system |
| GA | Genetic algorithm |
| MLR | Multiple linear regression |
| QSAR | Quantitative structure−activity relationship |
| WHO | World Health Organization |
| OLS | Ordinary least square |
| QSARINS | QSAR Insubria |
| OECD | Organisation for Economic Co-operation and Development |
References
- Boer, R.A.; Meijers, W.C.; Meer, P.; Veldhuisen, D.J. Cancer and heart disease: Associations and relations. Eur. J. Heart Fail. 2019, 21, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidanze, S.D.; Liu, D.; Mantei, R.A.; Hasvold, L.A.; Pratt, J.K.; Sheppard, G.S.; Wang, L.; Holms, J.H.; Dai, Y.; Aguirre, A.; et al. Discovery and optimization of novel constrained pyrrolopyridone BET family inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.E.; Pickett, S.D.; Hirst, J.D. Structural variation of protein–ligand complexes of the first bromodomain of BRD4. Org. Biomol. Chem. 2021, 19, 5632–5641. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, H.; Wang, P.; Li, Y.; Wold, E.A.; Leonard, P.G.; Joseph, S.; Brasier, A.R.; Tian, B.; Zhou, J. Discovery of Orally Bioavailable Chromone Derivatives as Potent and Selective BRD4 Inhibitors: Scaffold Hopping, Optimization, and Pharmacological Evaluation. J. Med. Chem. 2020, 63, 5242–5256. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers 2018, 23, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Guan, Y.; Qin, W.; Zhai, X.; Yu, B.; Liu, H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. MedChemComm 2018, 9, 1779–1802. [Google Scholar] [CrossRef]
- Zhao, Y.; Bai, L.; Liu, L.; McEachern, D.; Stuckey, J.A.; Meagher, J.L.; Yang, C.-Y.; Ran, X.; Zhou, B.; Hu, Y.; et al. Structure-Based Discovery of 4-(6-Methoxy-2-methyl-4-(quinolin-4-yl)-9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethylisoxazole (CD161) as a Potent and Orally Bioavailable BET Bromodomain Inhibitor. J. Med. Chem. 2017, 60, 3887–3901. [Google Scholar] [CrossRef]
- Xing, J.; Lu, W.; Liu, R.; Wang, Y.; Xie, Y.; Zhang, H.; Shi, Z.; Jiang, H.; Liu, Y.-C.; Chen, K.; et al. Machine-Learning-Assisted Approach for Discovering Novel Inhibitors Targeting Bromodomain-Containing Protein 4. J. Chem. Inf. Model. 2017, 57, 1677–1690. [Google Scholar] [CrossRef]
- Kuang, M.; Zhou, J.; Wang, L.; Liu, Z.; Guo, J.; Wu, R. Binding Kinetics versus Affinities in BRD4 Inhibition. J. Chem. Inf. Model. 2015, 55, 1926–1935. [Google Scholar] [CrossRef]
- Ember, S.W.J.; Zhu, J.-Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schönbrunn, E. Acetyl-lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pratt, J.K.; Soltwedel, T.; Sheppard, G.S.; Fidanze, S.D.; Liu, D.; Hasvold, L.A.; Mantei, R.A.; Holms, J.H.; McClellan, W.J.; et al. Fragment-Based, Structure-Enabled Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extra-Terminal Domain (BET) Family Bromodomain Inhibitors. J. Med. Chem. 2017, 60, 3828–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.-M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef]
- Sheppard, G.S.; Wang, L.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Pratt, J.K.; Park, C.H.; Longenecker, K.; Qiu, W.; Torrent, M.; et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 2020, 63, 5585–5623. [Google Scholar] [CrossRef]
- Masand, V.H.; Patil, M.K.; El-Sayed, N.N.E.; Zaki, M.E.A.; Almarhoon, Z.; Al-Hussain, S.A. Balanced QSAR analysis to identify the structural requirements of ABBV-075 (Mivebresib) analogues as bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J. Mol. Struct. 2021, 1229, 129597. [Google Scholar] [CrossRef]
- Tahir, A.; Alharthy, R.D.; Naseem, S.; Mahmood, N.; Ahmed, M.; Shahzad, K.; Akhtar, M.N.; Hameed, A.; Sadiq, I.; Nawaz, H.; et al. Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches. Molecules 2018, 23, 1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.-B.; Luo, D.; Feng, Y.; Bian, S.; Zhang, X.; Wang, T.-H. Structural modification of 4, 5-dihydro-[1, 2, 4] triazolo [4, 3-f] pteridine derivatives as BRD4 inhibitors using 2D/3D-QSAR and molecular docking analysis. Mol. Divers. 2021, 25, 1855–1872. [Google Scholar] [CrossRef] [PubMed]
- Babatunde Samuel Obadawo, O.E.O. Mayowa Monday Anifowose, Kehinde Henry Fagbohungbe, Justinah Solayide Amoko, QSAR modeling of novel substituted 4-Phenylisoquinolinones as potent BET bromodomain (BRD4-BD1) inhibitors. Biomed. Lett. 2019, 5, 69–78. [Google Scholar]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Lewaa, I. QSAR and Pharmacophore Modeling of Nitrogen Heterocycles as Potent Human N-Myristoyltransferase (Hs-NMT) Inhibitors. Molecules 2021, 26, 1834. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Modeling. Int. J. Quant. Struct. Prop. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Polishchuk, P. Interpretation of Quantitative Structure–Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model. 2017, 57, 2618–2639. [Google Scholar] [CrossRef]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “Two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef]
- Krstajic, D.; Buturovic, L.J.; Leahy, D.E.; Thomas, S. Cross-validation pitfalls when selecting and assessing regression and classification models. J. Cheminform. 2014, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P. External Evaluation of QSAR Models, in Addition to Cross-Validation Verification of Predictive Capability on Totally New Chemicals. Mol. Inf. 2014, 33, 311–314. [Google Scholar] [CrossRef]
- Gütlein, M.; Helma, C.; Karwath, A.; Kramer, S. A Large-Scale Empirical Evaluation of Cross-Validation and External Test Set Validation in (Q)SAR. Mol. Inf. 2013, 32, 516–528. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the definition of the Q2 parameter for QSAR validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef]
- Rao, R.B.; Fung, G.; Rosales, R. On the Dangers of Cross-Validation. An Experimental Evaluation; SIAM: Philadelphia, PA, USA, 2008; pp. 588–596. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P.; Giani, E.; Papa, E. Statistical external validation and consensus modeling: A QSPR case study for Koc prediction. J. Mol. Graph. Model. 2007, 25, 755–766. [Google Scholar] [CrossRef]
- Hawkins, D.M.; Basak, S.C.; Mills, D. Assessing model fit by cross-validation. J. Chem. Inf. Comput. Sci. 2003, 43, 579–586. [Google Scholar] [CrossRef]
- Masand, V.H.; Mahajan, D.T.; Nazeruddin, G.M.; Hadda, T.B.; Rastija, V.; Alfeefy, A.M. Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 2015, 24, 1241–1264. [Google Scholar] [CrossRef]
- Zaki, M.E.A.; Al-Hussain, S.A.; Bukhari, S.N.A.; Masand, V.H.; Rathore, M.M.; Thakur, S.D.; Patil, V.M. Exploring the Prominent and Concealed Inhibitory Features for Cytoplasmic Isoforms of Hsp90 Using QSAR Analysis. Pharmaceuticals 2022, 15, 303. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability Domain: A Step Toward Confident Predictions and Decidability for QSAR Modeling. In Computational Toxicology; Humana Press: New York, NY, USA, 2018; pp. 141–169. [Google Scholar]
- Gramatica, P.; Kovarich, S.; Roy, P.P. Reply to the comment of S. Rayne on “QSAR model reproducibility and applicability: A case study of rate constants of hydroxyl radical reaction models applied to polybrominated diphenyl ethers and (benzo-)triazoles”. J. Comput. Chem. 2013, 34, 1796. [Google Scholar] [CrossRef]
- Roy, P.P.; Kovarich, S.; Gramatica, P. QSAR model reproducibility and applicability: A case study of rate constants of hydroxyl radical reaction models applied to polybrominated diphenyl ethers and (benzo-)triazoles. J. Comput. Chem. 2011, 32, 2386–2396. [Google Scholar] [CrossRef]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability Domain for QSAR Models. Int. J. Quant. Struct. -Prop. Relatsh. 2016, 1, 45–63. [Google Scholar] [CrossRef]
- Tropsha, A.; Golbraikh, A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Zhang, Y.; Li, J.; Xue, X.; Wang, C.; Song, M.; Zhang, C.; Wang, R.; Li, C.; Wu, C.; et al. Y08060: A Selective BET Inhibitor for Treatment of Prostate Cancer. ACS Med. Chem. Lett. 2018, 9, 262–267. [Google Scholar] [CrossRef]
- McDaniel, K.F.; Wang, L.; Soltwedel, T.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Mantei, R.A.; Pratt, J.K.; Sheppard, G.S.; Bui, M.H.; et al. Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. J. Med. Chem. 2017, 60, 8369–8384. [Google Scholar] [CrossRef]
- Dearden, J.C.; Cronin, M.T.; Kaiser, K.L. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSAR Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Fan, X. Why QSAR fails: An empirical evaluation using conventional computational approach. Mol. Pharm. 2011, 8, 600–608. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Golbraikh, A.; Muratov, E.; Fourches, D.; Tropsha, A. Data set modelability by QSAR. J. Chem. Inf. Model. 2014, 54, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Sabnani, M.K.; Samad, A. Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity. Int. J. Mol. Sci. 2021, 22, 8352. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Tanrikulu, Y.; Nietert, M.; Scheffer, U.; Proschak, E.; Grabowski, K.; Schneider, P.; Weidlich, M.; Karas, M.; Gobel, M.; Schneider, G. Scaffold hopping by “fuzzy” pharmacophores and its application to RNA targets. Chembiochem 2007, 8, 1932–1936. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Description | Software Used for Calculation |
|---|---|---|
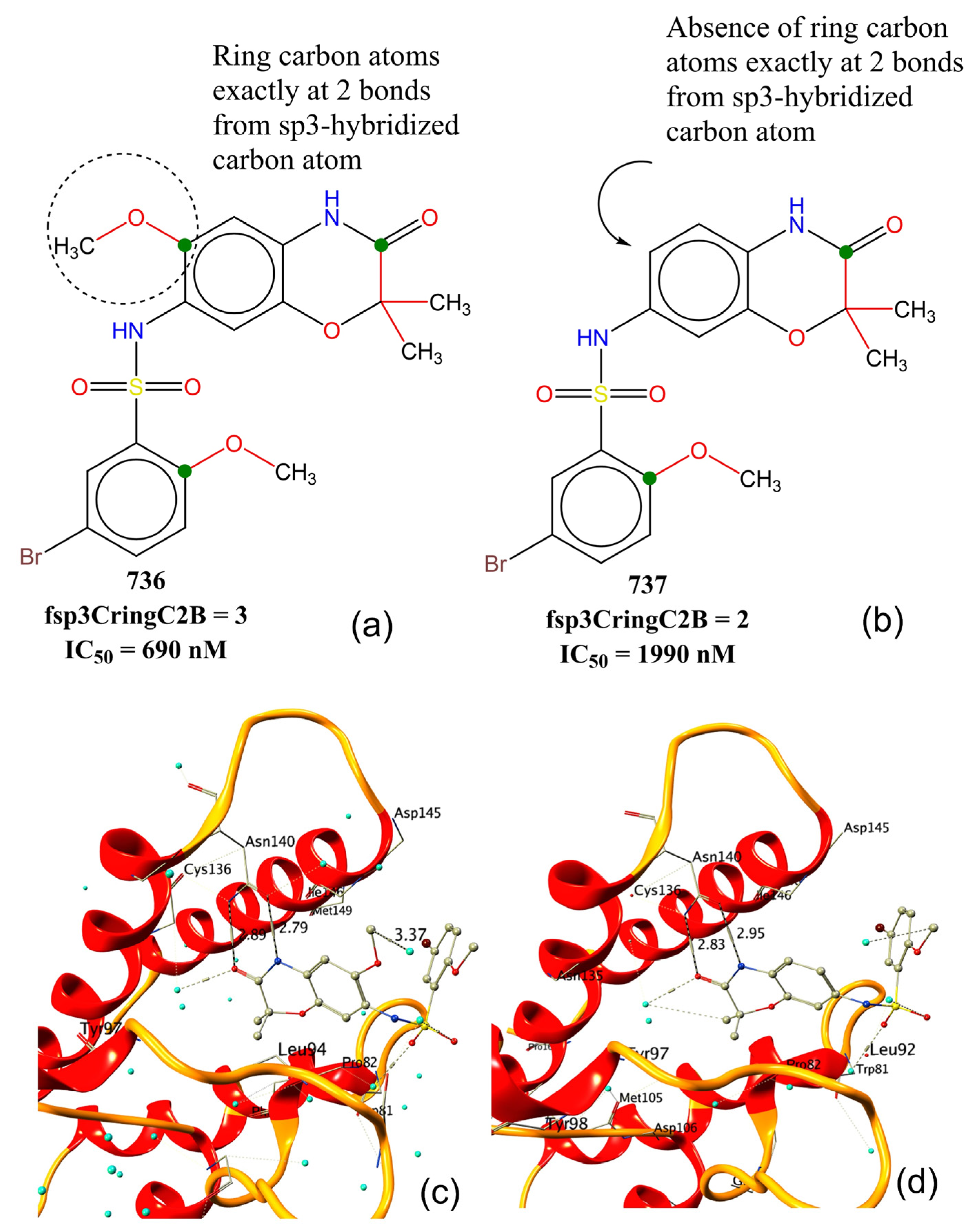
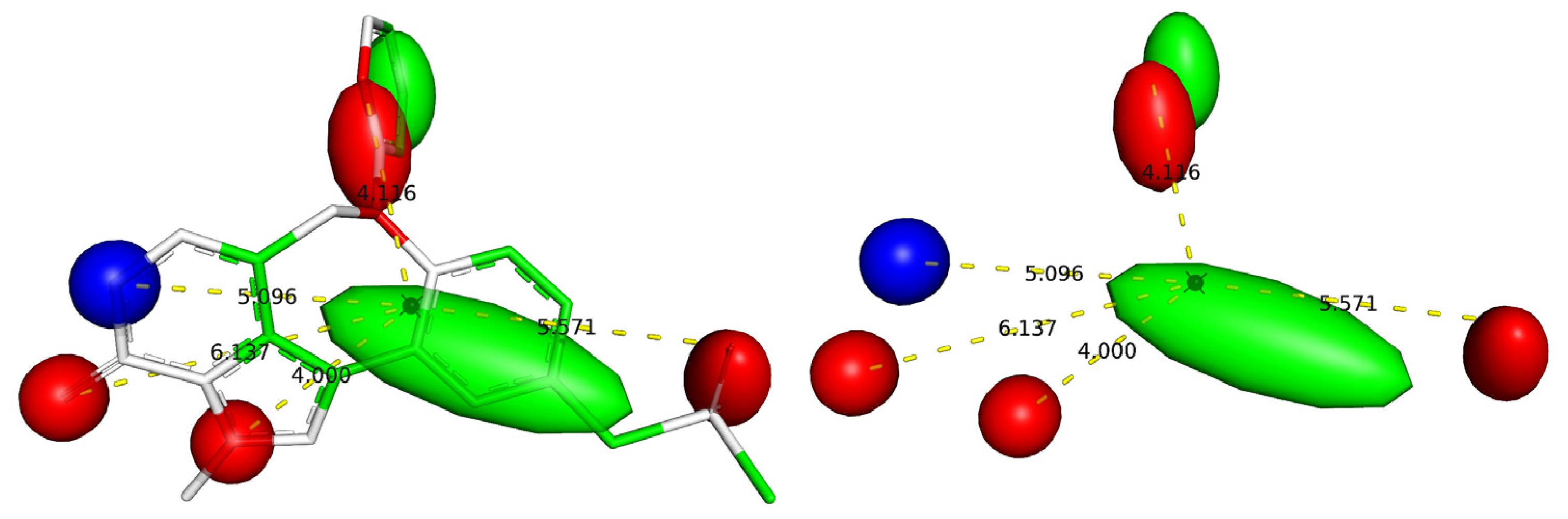
| fsp3CringC2B | Frequency of occurrence of ring carbon atoms exactly at 2 bonds from sp3-hybridised carbon atoms | PyDescriptor [45] |
| com_C_4A | Total number of carbon atoms within 4 Å from centre of mass (com) of molecule | PyDescriptor |
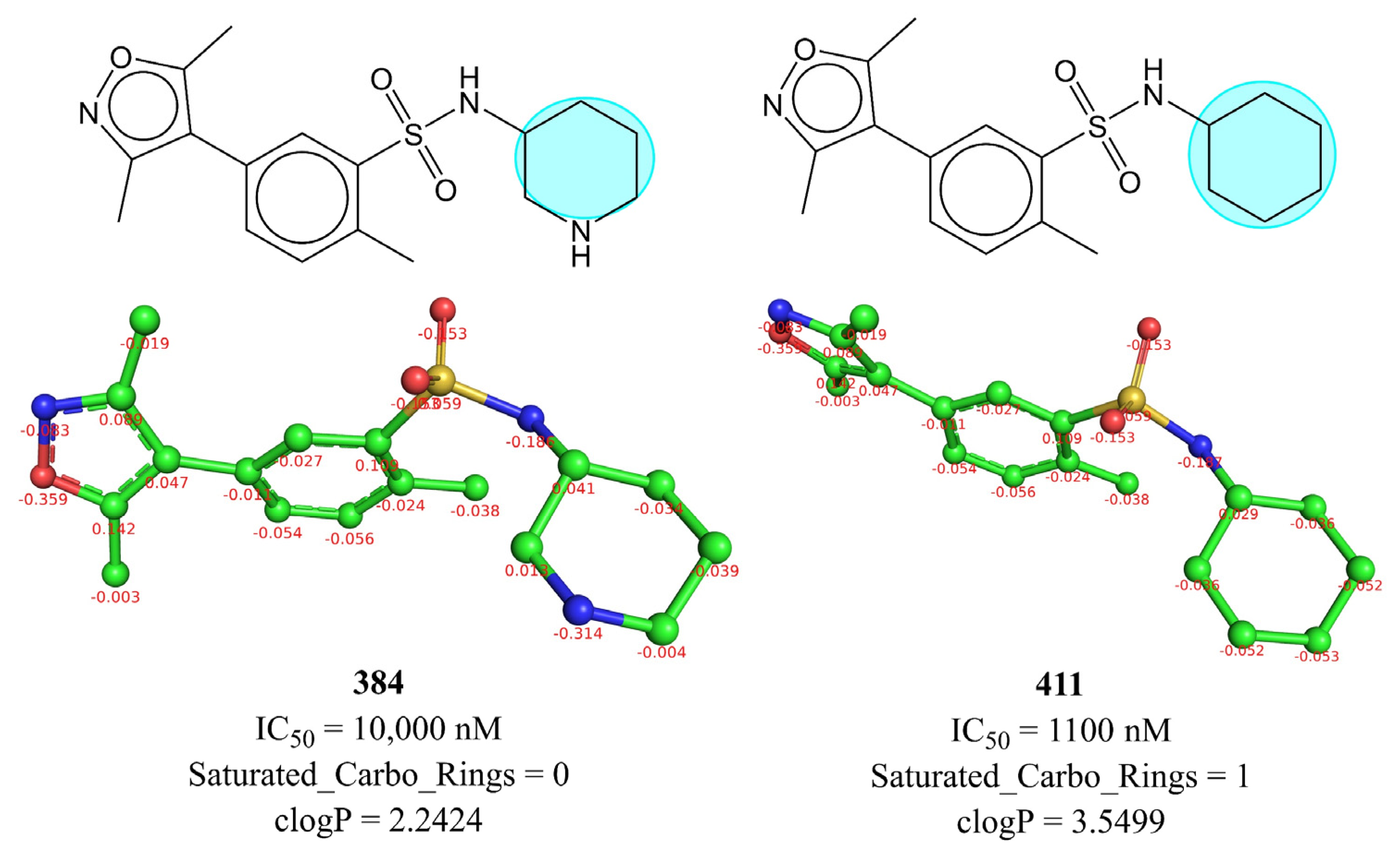
| Saturated_Carbo_Rings | Total number of saturated rings containing carbon atoms only | DataWarrior [46] |
| fsulfonSaroC8B | Frequency of occurrence of aromatic carbon atoms exactly at 8 bonds from sulphur atoms of Sulfone group | PyDescriptor |
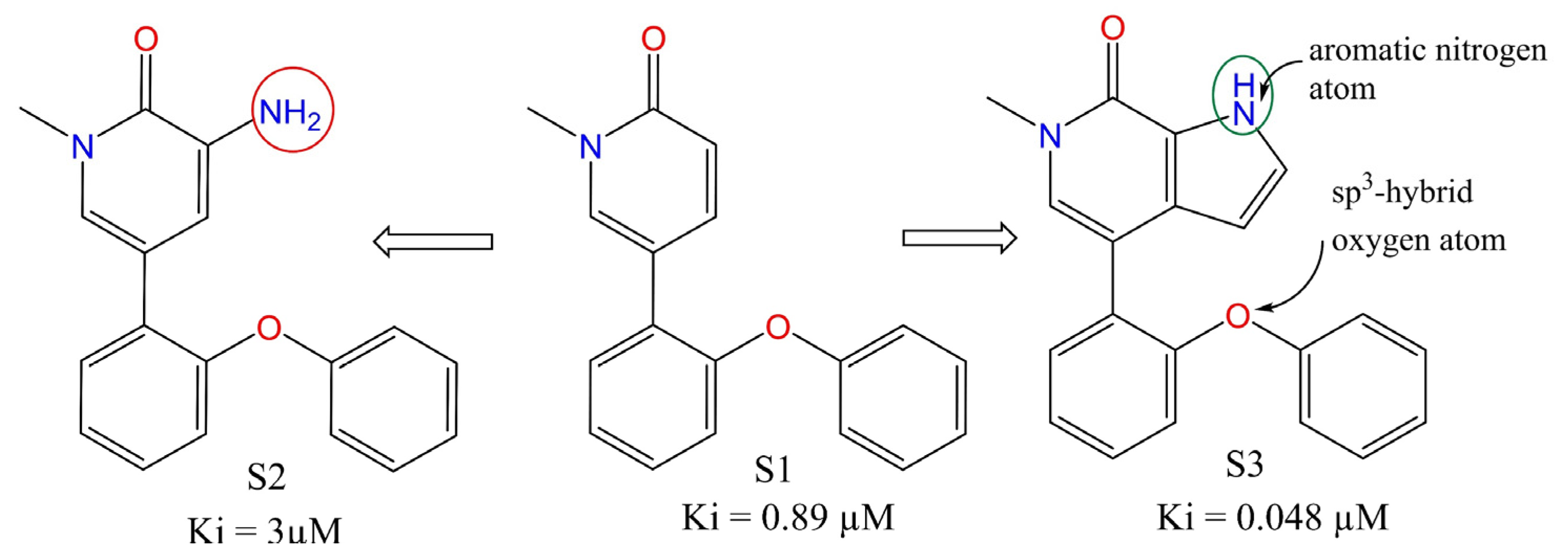
| fsp3OaroN6B | Frequency of occurrence of aromatic nitrogen atoms exactly at 6 bonds from sp3-hybridised oxygen atoms | PyDescriptor |
| flipoacc3B | Frequency of occurrence of H-bond acceptor atoms exactly at 3 bonds from lipophilic atoms | PyDescriptor |
| fplaNN4B | Frequency of occurrence of nitrogen atoms exactly at 4 bonds from planer nitrogen atoms | PyDescriptor |
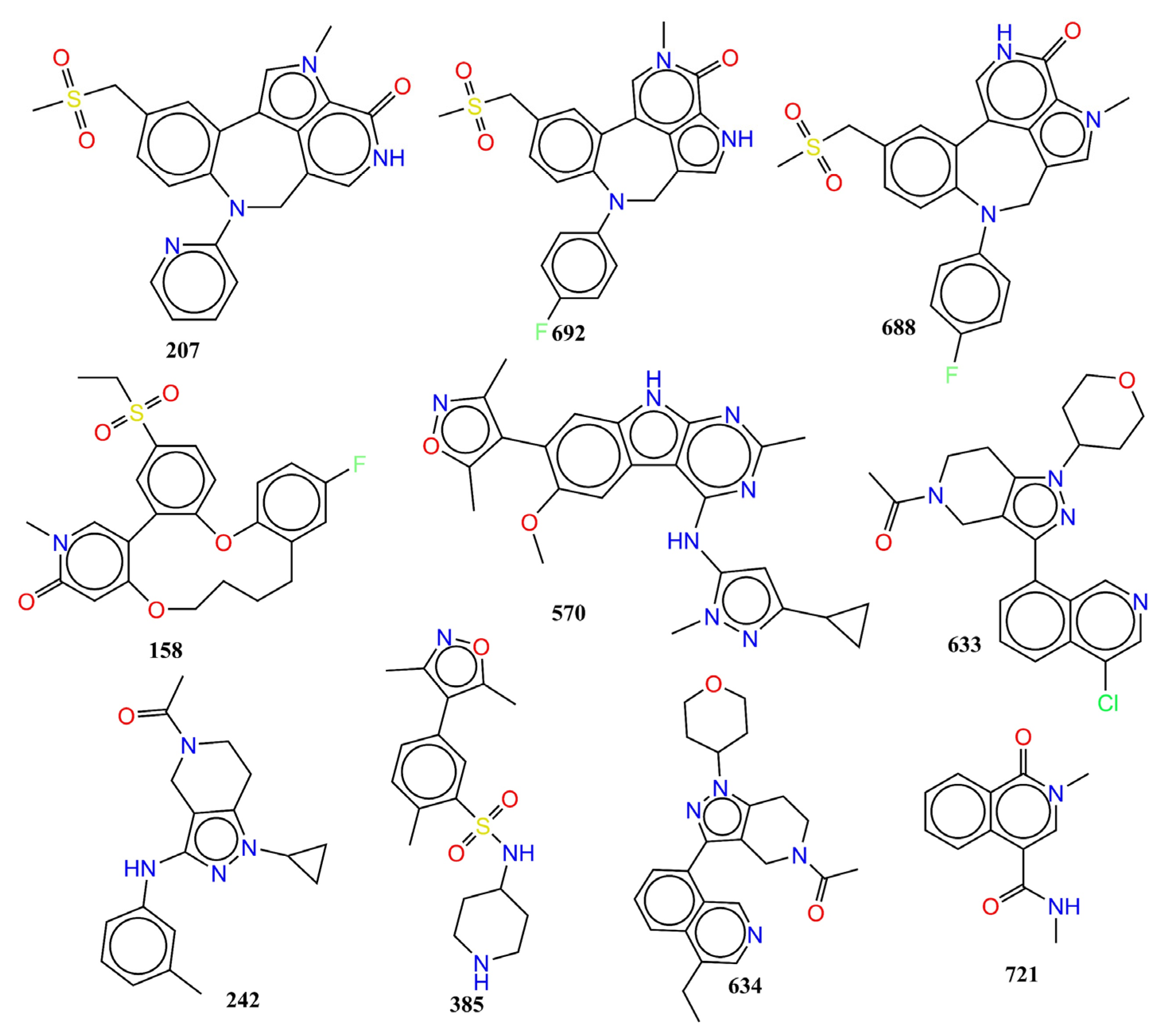
| SN | Ligand SMILES | IC50 (nM) | pIC50 (M) |
|---|---|---|---|
| 207 | Cn1cc2-c3cc(CS(C)(=O)=O)ccc3N(Cc3c[nH]c(=O)c1c23)c1ccccn1 | 1 | 9 |
| 692 | Cn1cc2-c3cc(CS(C)(=O)=O)ccc3N(Cc3c[nH]c(c23)c1=O)c1ccc(F)cc1 | 1.5 | 8.824 |
| 158 | CCS(=O)(=O)c1ccc2Oc3ccc(F)cc3CCCCOc3cc(=O)n(C)cc3-c2c1 | 2 | 8.699 |
| 570 | COc1cc2c(cc1-c1c(C)noc1C)[nH]c1nc(C)nc(Nc3cc(nn3C)C3CC3)c21 | 2 | 8.699 |
| 688 | Cn1cc2CN(c3ccc(F)cc3)c3ccc(CS(C)(=O)=O)cc3-c3c[nH]c(=O)c1c23 | 2.5 | 8.602 |
| 633 | CC(=O)N1CCc2c(C1)c(nn2C1CCOCC1)-c1cccc2c(Cl)cncc12 | 14,000 | 4.854 |
| 242 | CC(=O)N1CCc2c(C1)c(Nc1cccc(C)c1)nn2C1CC1 | 15,000 | 4.824 |
| 385 | Cc1noc(C)c1-c1ccc(C)c(c1)S(=O)(=O)NC1CCNCC1 | 15,000 | 4.824 |
| 634 | CCc1cncc2c(cccc12)-c1nn(C2CCOCC2)c2CCN(Cc12)C(C)=O | 15,000 | 4.824 |
| 721 | CNC(=O)c1cn(C)c(=O)c2ccccc12 | 15,000 | 4.824 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.E.A.; Al-Hussain, S.A.; Al-Mutairi, A.A.; Masand, V.H.; Samad, A.; Jawarkar, R.D. Mechanistic Analysis of Chemically Diverse Bromodomain-4 Inhibitors Using Balanced QSAR Analysis and Supported by X-ray Resolved Crystal Structures. Pharmaceuticals 2022, 15, 745. https://doi.org/10.3390/ph15060745
Zaki MEA, Al-Hussain SA, Al-Mutairi AA, Masand VH, Samad A, Jawarkar RD. Mechanistic Analysis of Chemically Diverse Bromodomain-4 Inhibitors Using Balanced QSAR Analysis and Supported by X-ray Resolved Crystal Structures. Pharmaceuticals. 2022; 15(6):745. https://doi.org/10.3390/ph15060745
Chicago/Turabian StyleZaki, Magdi E. A., Sami A. Al-Hussain, Aamal A. Al-Mutairi, Vijay H. Masand, Abdul Samad, and Rahul D. Jawarkar. 2022. "Mechanistic Analysis of Chemically Diverse Bromodomain-4 Inhibitors Using Balanced QSAR Analysis and Supported by X-ray Resolved Crystal Structures" Pharmaceuticals 15, no. 6: 745. https://doi.org/10.3390/ph15060745
APA StyleZaki, M. E. A., Al-Hussain, S. A., Al-Mutairi, A. A., Masand, V. H., Samad, A., & Jawarkar, R. D. (2022). Mechanistic Analysis of Chemically Diverse Bromodomain-4 Inhibitors Using Balanced QSAR Analysis and Supported by X-ray Resolved Crystal Structures. Pharmaceuticals, 15(6), 745. https://doi.org/10.3390/ph15060745