
Bromodomain and Extra-Terminal Protein Inhibitors: Biologic Insights and Therapeutic Potential in Pediatric Brain Tumors


Abstract
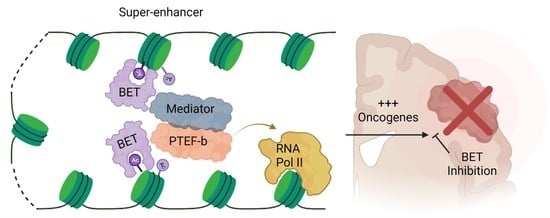
:
1. Introduction
2. BET Protein Structure and Function
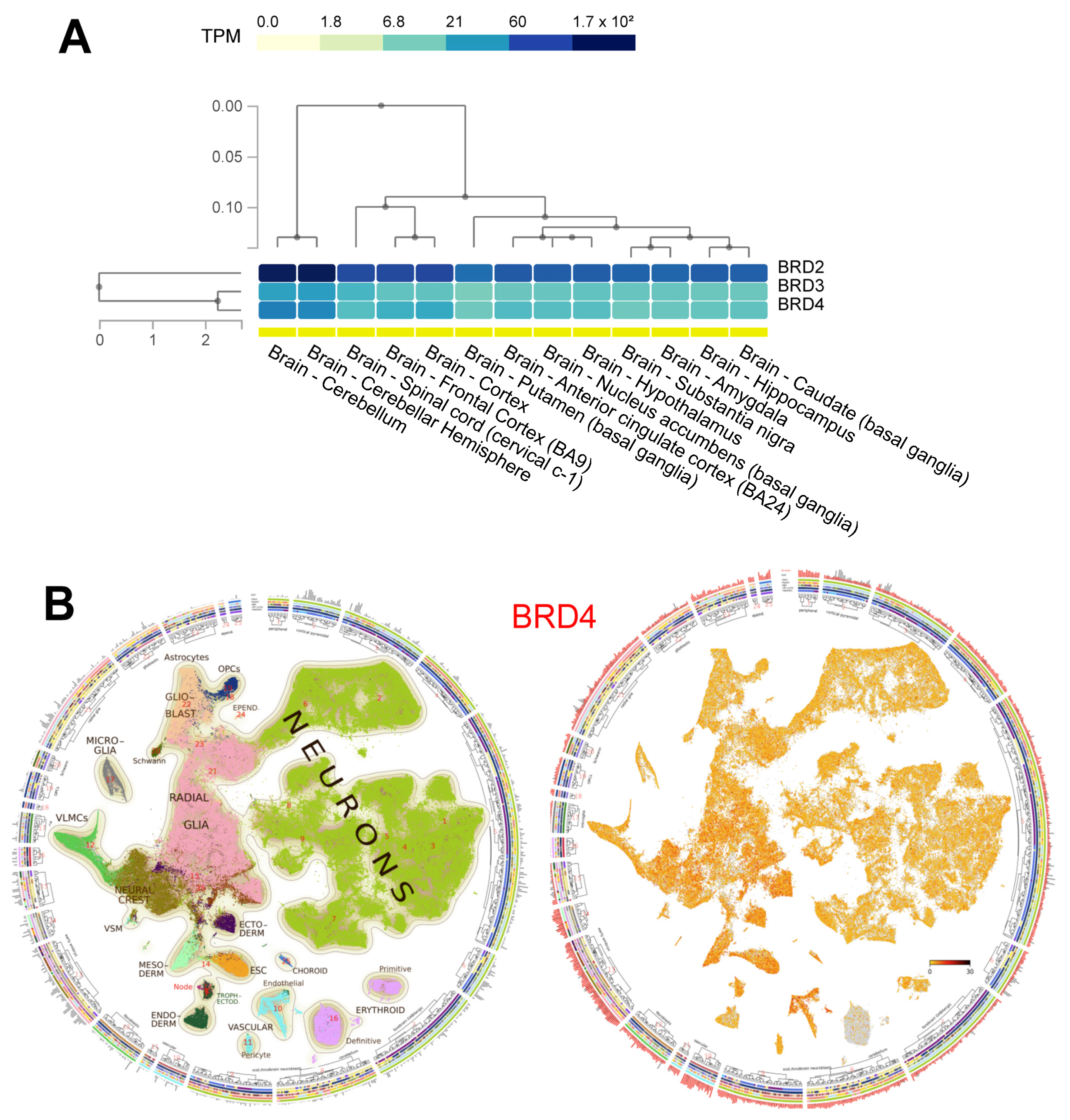
3. BET Inhibitors
4. BET Inhibitors in Pediatric Brain Tumor Models
4.1. Medulloblastoma
4.2. Diffuse Intrinsic Pontine Glioma
4.3. Ependymoma
4.4. Embryonal Tumor with Multilayer Rosettes (ETMR)
4.5. Atypical Teratoid/Rhabdoid Tumor (ATRT)

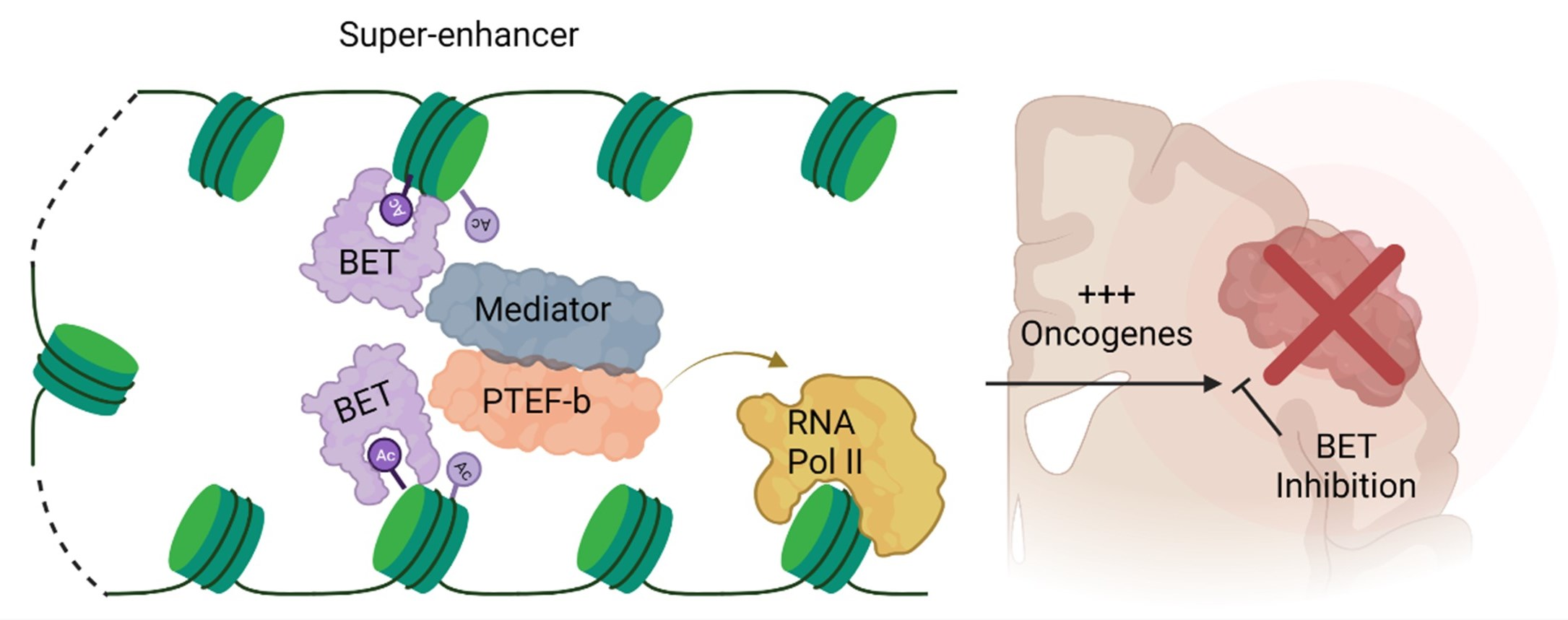{kind=link}
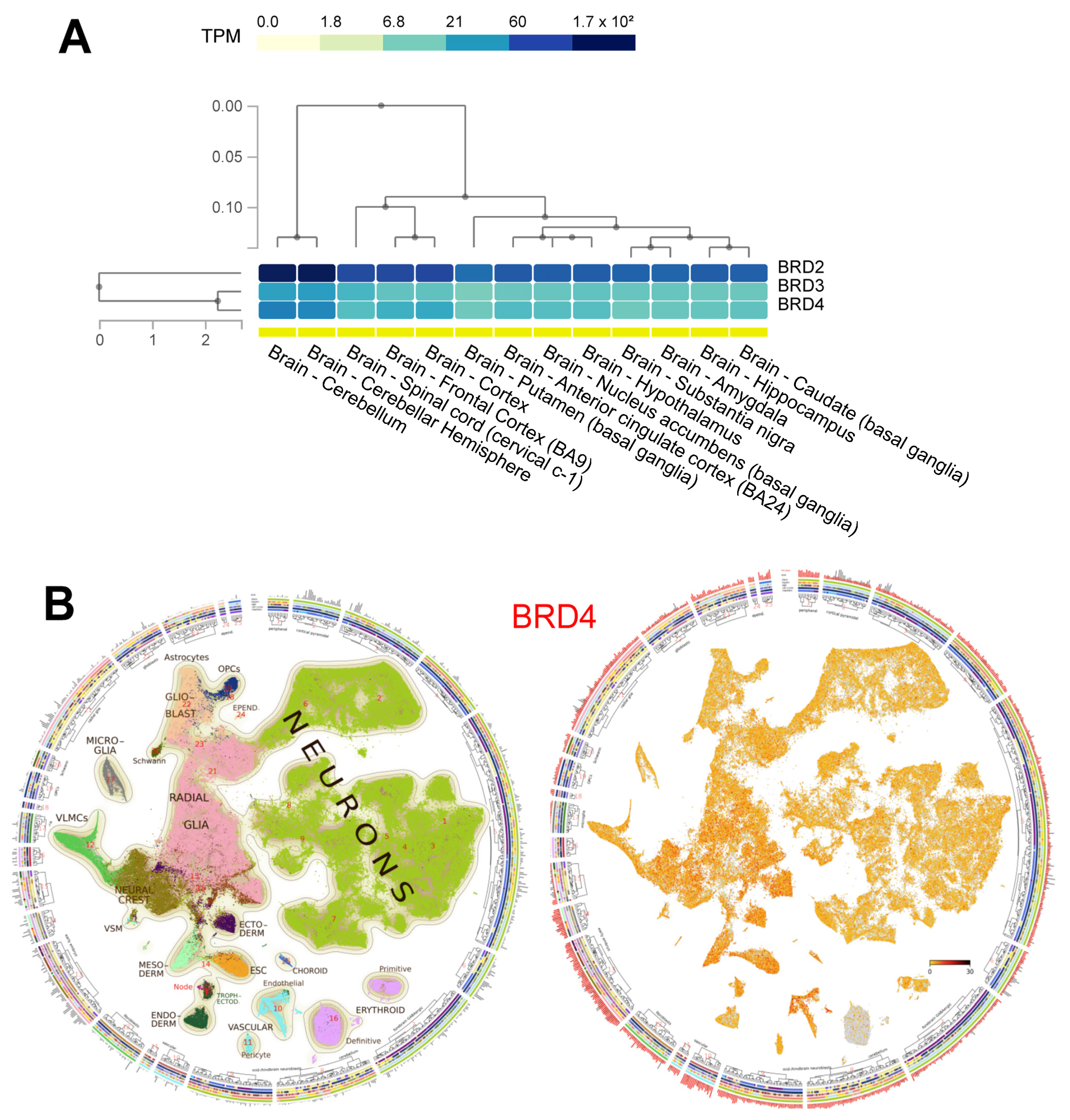{kind=link}
| Tumor | In-Vivo Cell Line/Model | BET Inhibitor | Notable Findings | Citation |
|---|---|---|---|---|
| MB | MB002, Group 3 MB | JQ1 | JQ1 was effective in broad panel of MB cell lines, induced apoptosis and G1 cell cycle arrest. RNA sequencing showed decreased MYC and MC-target expression. Orthotopic xenograft (cerebellar) showed increased survival with JQ1 treatment | Bandopadhayay, [60] |
| MB | HD-MB3, MYC amplified Group 3 MB | JQ1 | JQ1 effective in broad panel of cell lines, inducing apoptosis and G1 cell cycle arrest. Caused decreased MYC and MYC-targets’ expression, and affected components of p53 and cell cycle pathway. Flank xenograft study showed decreased tumor growth and prolonged survival | Hennsen et al. [61] |
| MB | DAOY, MYC-driven MB | JQ1 | JQ1 effective in MB cell lines, induced apoptosis and cell cycle arrest. They also showed that it induced cellular senescence, and that transcriptional programs suppressed by treatment are associated with adverse risk in MB patients. Flank xenograft study showed decreased tumor growth | Venkataraman et al. [62] |
| MB | MED1-MB, SMO-WT/SMO-D477G-MB (autochthonous derived from Ptch+/−; Tpr53−/− and Ptch+/−; lacZ mice, respectively | JQ1 | JQ1 decreased proliferation and viability of SHH-driven MB in-vitro and in-vivo (flank and cerebellar models used), even when cell lines had SMO inhibitor resistance mutations | Tang et al. [67] |
| MB | Murine Ptch+/− MB model | I-BET151 | I-BET151 decreased SHH-driven MB growth in-vivo, and decreased Gli1 expression. I-BET151 was effective in decreasing tumor growth in-vivo (subcutaneous) | Long et al. [68] |
| MB | D458 and MB002, MYC driven MB | JQ1 + LEE01 | CDK4/CDK6 inhibition delayed development of BET inhibitor resistance. Combination of JQ1 with LEE01 (a CDK4/6 inhibitor) improved survival in flank and orthotopic xenograft models of MYC-driven medulloblastoma | Bandopadhayay, [69] |
| MB | GTML2 (murine derived Group 3 MB) and MB002 | JQ1 + Milcilib | JQ1+ CDK2 inhibitor synergized to induce apoptosis and cell cycle arrest. Combination treatment in-vivo extended survival in two orthoptic (cerebellar) models of Group 3 MB | Bolin et al. [70] |
| DIPG | SF8628 | JQ1 | JQ1 impaired DIPG growth and viability in-vivo, and improved survival in an orthotopic mouse PDX model (brainstem) | Piunti et al. [76] |
| DIPG | SU-DIPG-VI and SF7761 | BRD4 shRNA | JQ1 decreased growth of DIPG cell lines and downregulated genes associated with CNS development. Lentiviral shRNA knockdown of BRD4 extended survival of mice bearing two different orthotopic (brainstem) DIPG models | Nagaraja et al. [77] |
| DIPG | N/A (no in-vivo data) | JQ1 + ICG-001 | BET + CBP inhibition synergized to decrease growth and viability of DIPG cell lines, and preferentially downregulated super-enhancer genes | Wiese et al. [78] |
| DIPG | H3K27M/PDGFB expressing NSCs | JQ1 + Tazemtostat | BET + EZH2 inhibition was a synergistic combination in H3K27M/PDGFB transformed NSCs | Zhang et al. [80] |
| DIPG | N/A (no in-vivo data) | JQ1 + MRK003 | BET inhibition + NOTCH inhibition synergized in 2/3 DIPG models to induce apoptosis and cell death | Taylor et al. [81] |
| DIPG | N/A (no in-vivo data) | BMS986158, dBET6 | BET inhibition and degradation significantly altered the chromatin architecture of DIPG via Hi-C analysis, although the effect was more pronounced with BET degradation | Wang et al. [82] |
| Ependymoma | N/A (no in-vivo data) | JQ1 | JQ1 inhibited proliferation and viability of one supratentorial (H.EP1) and one PF-A (H.612) ependymoma cell line | Mack et al. [92] |
| Ependymoma | EPP-MI and EPV-FL-MI (PFA) | OTX015 | OTX015 induced apoptosis and cell cycle arrest in two PFA and one ST (subtype not specified) models of ependymoma. In-vivo OTX015 extended survival of the EPP-MI orthotopic intracranial PDX model, but had no improvement in the EPP-FL-MI model | Servidei et al. [93] |
| ETMR | N/A (no in-vivo data) | JQ1S (active isomer of JQ1) | JQ1S decreased growth and viability of ETMR cell lines in-vivo, and downregulated MYCN and LIN28A expression | Sin-Chan et al. [100] |
| ATRT | MAF-737 | JQ1 | JQ1 potently inhibited viability of ATRT (MYC subtype) cell lines, and decreased transcription of c-MYC targets and c-MYC itself. JQ1 prolonged survival in an orthotopic (cerebellar) ATRT model | Allimova et al. [109] |
5. BET Inhibitors in the Clinic
5.1. BET Inhibitors in CNS Malignancies
5.2. BET Inhibitors in Pediatrics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro Stucklin, A.S.; Ramaswamy, V.; Daniels, C.; Taylor, M.D. Review of molecular classification and treatment implications of pediatric brain tumors. Curr. Opin. Pediatr. 2018, 30, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cruzeiro, G.A.V.; Rota, C.; Hack, O.A.; Segal, R.; Filbin, M.G. Understanding the epigenetic landscape and cellular architecture of childhood brain tumors. Neurochem. Int. 2020, 144, 104940. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Surani, M.A.; Hayashi, K.; Hajkova, P. Genetic and Epigenetic Regulators of Pluripotency. Cell 2007, 128, 747–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Khorasanizadeh, S. The Nucleosome: From Genomic Organization to Genomic Regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena Histone Acetyltransferase A: A Homolog to Yeast Gcn5p Linking Histone Acetylation to Gene Activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Kleff, S.; Andrulis, E.D.; Anderson, C.W.; Sternglanz, R. Identification of a Gene Encoding a Yeast Histone H4 Acetyltransferase. J. Biol. Chem. 1995, 270, 24674–24677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthun, M.R.; Widom, J.; Gottschling, D.E. The Major Cytoplasmic Histone Acetyltransferase in Yeast: Links to Chromatin Replication and Histone Metabolism. Cell 1996, 87, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-κB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Penas, C.; Maloof, M.E.; Stathias, V.; Long, J.; Tan, S.K.; Mier, J.; Fang, Y.; Valdes, C.; Rodriguez-Blanco, J.; Chiang, C.-M.; et al. Time series modeling of cell cycle exit identifies Brd4 dependent regulation of cerebellar neurogenesis. Nat. Commun. 2019, 10, 3028. [Google Scholar] [CrossRef]
- Velíšek, L.; Shang, E.; Velíšková, J.; Chachua, T.; Macchiarulo, S.; Maglakelidze, G.; Wolgemuth, D.J.; Greenberg, D.A. GABAergic Neuron Deficit as an Idiopathic Generalized Epilepsy Mechanism: The Role of BRD2 Haploinsufficiency in Juvenile Myoclonic Epilepsy. PLoS ONE 2011, 6, e23656. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Badimon, A.; Schaefer, U.; Ayata, P.; Gray, J.; Chung, C.-W.; Von Schimmelmann, M.; Zhang, F.; Garton, N.; Smithers, N.; et al. Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J. Exp. Med. 2015, 212, 1771–1781. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Diao, H.; Zhang, F.; Wang, Y.; Wang, K.; Wu, R. Deciphering the mechanisms of selective inhibition for the tandem BD1/BD2 in the BET-bromodomain family. Phys. Chem. Chem. Phys. 2017, 19, 23934–23941. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- GTex Portal. Available online: http://gtexportal.org/home/ (accessed on 9 March 2022).
- La Manno, G.; Siletti, K.; Furlan, A.; Gyllborg, D.; Vinsland, E.; Albiach, A.M.; Langseth, C.M.; Khven, I.; Lederer, A.R.; Dratva, L.M.; et al. Molecular architecture of the developing mouse brain. Nature 2021, 596, 92–96. [Google Scholar] [CrossRef]
- Mouse Brain Atlas. Available online: http://mousebrain.org/ (accessed on 12 March 2022).
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Han, J.; Wang, Z.; Li, X.; Sun, Y.; Hu, Z. Safety and Efficacy of Bromodomain and Extra-Terminal Inhibitors for the Treatment of Hematological Malignancies and Solid Tumors: A Systematic Study of Clinical Trials. Front. Pharmacol. 2020, 11, 621093. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, G.S.; Wang, L.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Pratt, J.K.; Park, C.H.; Longenecker, K.L.; Qiu, W.; Torrent, M.; et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 2020, 63, 5585–5623. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; Van Sluis, P.; Troost, D.; Meeteren, N.S.-V.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics Identifies Medulloblastoma Subgroups That Are Enriched for Specific Genetic Alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, E.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Ellison, D.W.; Kocak, M.; Dalton, J.; Megahed, H.; Lusher, M.E.; Ryan, S.L.; Zhao, W.; Nicholson, S.L.; Taylor, R.E.; Bailey, S.; et al. Definition of Disease-Risk Stratification Groups in Childhood Medulloblastoma Using Combined Clinical, Pathologic, and Molecular Variables. J. Clin. Oncol. 2011, 29, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma. Clin. Cancer Res. 2013, 20, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Slpeleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Alimova, I.; Balakrishnan, I.; Harris, P.; Birks, D.K.; Griesinger, A.; Amani, V.; Cristiano, B.; Remke, M.; Taylor, M.; et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014, 5, 2355–2371. [Google Scholar] [CrossRef] [Green Version]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened Gene Encodes a Seven-Pass Membrane Protein, a Putative Receptor for the Hedgehog Signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Oliver, T.; Grasfeder, L.L.; Carroll, A.L.; Kaiser, C.; Gillingham, C.L.; Lin, S.M.; Wickramasinghe, R.; Scott, M.P.; Wechsler-Reya, R.J. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 7331–7336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.-D.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Li, B.; Blanco, J.R.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [Green Version]
- Bandopadhayay, P.; Piccioni, F.; O’Rourke, R.; Ho, P.; Gonzalez, E.M.; Buchan, G.; Qian, K.; Gionet, G.; Girard, E.; Coxon, M.; et al. Neuronal differentiation and cell-cycle programs mediate response to BET-bromodomain inhibition in MYC-driven medulloblastoma. Nat. Commun. 2019, 10, 2400. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.M.; Van Zanten, S.E.V.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; Hamdan, F.H.; Kubiak, K.; Diederichs, C.; Gielen, G.H.; Nussbaumer, G.; Carcaboso, A.M.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020, 11, 673. [Google Scholar] [CrossRef]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, W.; Zhu, J.; Wang, L.; Wu, X.; Shan, H. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. 2017, 7, 56. [Google Scholar] [CrossRef]
- Taylor, I.C.; Hütt-Cabezas, M.; Brandt, W.D.; Kambhampati, M.; Nazarian, J.; Chang, H.T.; Warren, K.E.; Eberhart, C.G.; Raabe, E.H. Disrupting NOTCH Slows Diffuse Intrinsic Pontine Glioma Growth, Enhances Radiation Sensitivity, and Shows Combinatorial Efficacy With Bromodomain Inhibition. J. Neuropathol. Exp. Neurol. 2015, 74, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, T.Y.-T.; Hou, Y.; Bartom, E.; Lu, X.; Shilatifard, A.; Yue, F.; Saratsis, A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci. Adv. 2021, 7, eabg4126. [Google Scholar] [CrossRef]
- Elsamadicy, A.A.; Koo, A.B.; David, W.B.; Lee, V.; Zogg, C.K.; Kundishora, A.J.; Hong, C.S.; DeSpenza, T.; Reeves, B.; Kahle, K.T.; et al. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neuro Oncol. Adv. 2020, 2, vdaa019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tihan, T.; Zhou, T.; Holmes, E.; Burger, P.C.; Ozuysal, S.; Rushing, E.J. The prognostic value of histological grading of posterior fossa ependymomas in children: A Children’s Oncology Group study and a review of prognostic factors. Mod. Pathol. 2007, 21, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitanza, N.A.; Partap, S. Pediatric Ependymoma. J. Child Neurol. 2016, 31, 1354–1366. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). Maintenance Chemotherapy or Observation Following Induction Chemotherapy and Radiation Therapy in Treating Patients with Newly Diagnosed Ependymoma. March 2010. Identifier NCT01096368. Available online: https://clinicaltrials.gov/ct2/show/NCT01096368 (accessed on 10 March 2022).
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Arabzade, A.; Zhao, Y.; Varadharajan, S.; Chen, H.-C.; Jessa, S.; Rivas, B.; Stuckert, A.J.; Solis, M.; Kardian, A.; Tlais, D.; et al. ZFTA–RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021, 11, 2200–2215. [Google Scholar] [CrossRef] [PubMed]
- Kupp, R.; Ruff, L.; Terranova, S.; Nathan, E.; Ballereau, S.; Stark, R.; Chilamakuri, C.S.R.; Hoffmann, N.; Wickham-Rahrmann, K.; Widdess, M.; et al. ZFTA Translocations Constitute Ependymoma Chromatin Remodeling and Transcription Factors. Cancer Discov. 2021, 11, 2216–2229. [Google Scholar] [CrossRef]
- Zheng, T.; Ghasemi, D.R.; Okonechnikov, K.; Korshunov, A.; Sill, M.; Maass, K.K.; da Silva, P.B.G.; Ryzhova, M.; Gojo, J.; Stichel, D.; et al. Cross-Species Genomics Reveals Oncogenic Dependencies in ZFTA/C11orf95 Fusion–Positive Supratentorial Ependymomas. Cancer Discov. 2021, 11, 2230–2247. [Google Scholar] [CrossRef]
- Bockmayr, M.; Harnisch, K.; Pohl, L.C.; Schweizer, L.; Mohme, T.; Körner, M.; Alawi, M.; Suwala, A.K.; Dorostkar, M.M.; Monoranu, C.M.; et al. Comprehensive profiling of myxopapillary ependymomas identifies a distinct molecular subtype with relapsing disease. Neuro Oncol. 2022. Available online: https://pubmed.ncbi.nlm.nih.gov/35380708/ (accessed on 10 March 2022). [CrossRef]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2017, 553, 101–105. [Google Scholar] [CrossRef]
- Servidei, T.; Meco, D.; Martini, M.; Battaglia, A.; Granitto, A.; Buzzonetti, A.; Babini, G.; Massimi, L.; Tamburrini, G.; Scambia, G.; et al. The BET inhibitor OTX015 exhibits in vitro and in vivo antitumor activity in pediatric ependymoma stem cell models. Int. J. Mol. Sci. 2021, 22, 1877. [Google Scholar] [CrossRef]
- Eberhart, C.G.; Brat, D.J.; Cohen, K.J.; Burger, P.C. Pediatric Neuroblastic Brain Tumors Containing Abundant Neuropil and True Rosettes. Pediatr. Dev. Pathol. 2000, 3, 346–352. [Google Scholar] [CrossRef]
- Li, M.; Lee, K.F.; Lu, Y.; Clarke, I.; Shih, D.J.H.; Eberhart, C.; Collins, V.P.; Van Meter, T.; Picard, D.; Zhou, L.; et al. Frequent Amplification of a chr19q13.41 MicroRNA Polycistron in Aggressive Primitive Neuroectodermal Brain Tumors. Cancer Cell 2009, 16, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.; Remke, M.; Castoldi, M.; Bai, A.H.C.; Muckenthaler, M.; Kulozik, A.; von Deimling, A.; Pscherer, A.; Lichter, P.; Korshunov, A. Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol. 2008, 117, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Lambo, S.; Gröbner, S.N.; Rausch, T.; Waszak, S.; Schmidt, C.; Gorthi, A.; Romero, J.C.; Mauermann, M.; Brabetz, S.; Krausert, S.; et al. The molecular landscape of ETMR at diagnosis and relapse. Nature 2019, 576, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, M.; Dufour, C.; Leblond, P.; Bourdeaut, F.; Faure-Conter, C.; Bertozzi, A.-I.; Delisle, M.B.; Palenzuela, G.; Jouvet, A.; Scavarda, D.; et al. Embryonal tumors with multilayered rosettes in children: The SFCE experience. Child’s Nerv. Syst. 2015, 32, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Hanson, D.; Hoffman, L.M.; Nagabushan, S.; Goumnerova, L.C.; Rathmann, A.; Vogel, T.; Ziegler, D.S.; Chi, S. A modified IRS-III chemotherapy regimen leads to prolonged survival in children with embryonal tumor with multilayer rosettes. Neuro Oncol. Adv. 2020, 2, vdaa120. [Google Scholar] [CrossRef]
- Sin-Chan, P.; Mumal, I.; Suwal, T.; Ho, B.; Infante, E.R.; Singh, I.; Du, Y.; Lu, M.; Patel, N.; Torchia, J.; et al. A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor. Cancer Cell 2019, 36, 51–67.e7. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Ginn, K.F.M.; Gajjar, A.M. Atypical Teratoid Rhabdoid Tumor: Current Therapy and Future Directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [CrossRef] [Green Version]
- Biegel, J.A.; Tan, L.; Zhang, F.; Wainwright, L.; Russo, P.; Rorke, L.B. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002, 8, 3461–3467. [Google Scholar]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.-F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R.; et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.; Sturm, D.; Hermann, C.; Wang, M.S.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchia, J.; Picard, D.; Lafay-Cousin, L.; Hawkins, C.E.; Kim, S.-K.; Letourneau, L.; Ra, Y.-S.; Ho, K.C.; Chan, T.S.Y.; Sin-Chan, P.; et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: An integrated genomic and clinicopathological analysis. Lancet Oncol. 2015, 16, 569–582. [Google Scholar] [CrossRef]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.; Pierce, A.; Danis, E.; Donson, A.; Birks, D.K.; Griesinger, A.; Foreman, N.K.; Santi, M.; Soucek, L.; Venkataraman, S.; et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int. J. Cancer 2019, 144, 1983–1995. [Google Scholar] [CrossRef]
- Berenguer-Daizé, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Sanson, M.; Moyal, E.; Delord, J.-P.; De Micheli, R.; Rezai, K.; Leung, A.C.; Perez, S.; Bekradda, M.; Lachaux, N.; et al. Dose optimization of MK-8628 (OTX015), a small molecule inhibitor of bromodomain and extra-terminal (BET) proteins, in patients (pts) with recurrent glioblastoma (GB). J. Clin. Oncol. 2016, 34, e14123. [Google Scholar] [CrossRef]
- Moreno, V.; Sepulveda, J.; Vieito, M.; Hernández-Guerrero, T.; Doger, B.; Saavedra, O.; Ferrero, O.; Sarmiento, R.; Arias, M.; De Alvaro, J.; et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. 2020, 31, 780–788. [Google Scholar] [CrossRef]
- Pearson, A.D.; DuBois, S.G.; Buenger, V.; Kieran, M.; Stegmaier, K.; Bandopadhayay, P.; Bennett, K.; Bourdeaut, F.; Brown, P.A.; Chesler, L.; et al. Bromodomain and extra-terminal inhibitors—A consensus prioritisation after the Paediatric Strategy Forum for medicinal product development of epigenetic modifiers in children—ACCELERATE. Eur. J. Cancer 2021, 146, 115–124. [Google Scholar] [CrossRef]
| Agent | Selectivity | Phase | Disease Focus; Comments | Structure |
|---|---|---|---|---|
| ABBV-075 | Pan-BET | I | Solid tumors, AML, multiple myeloma, myelofibrosis | 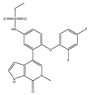 |
| ABBV-744 | BDII selective | I | AML, myelofibrosis | 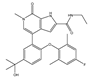 |
| AZDZ5153 | Pan-BET | I/II | AML; Unique bivalent binding mode | 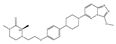 |
| BI-894999 | Pan-BET | I | Advanced solid tumors, DLBCL, or NMC | 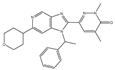 |
| BMS-986158 | Pan-BET | I/II | Advanced solid tumors and hematologic malignancies; Ongoing pediatric study | 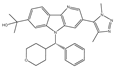 |
| BMS-986378 (CC-90010) | Pan-BET | I | Advanced solid tumors, NHL; Ongoing pediatric study, good CNS penetration | 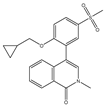 |
| CPI-0610 | Pan-BET | I/II | Lymphoma, Multiple myeloma, AML, MPNST | 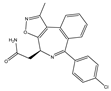 |
| I-BET762 (GSK525762) | Pan-BET | I/II | Hematologic malignancies, solid tumors | 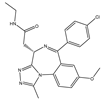 |
| INCB57643 | Pan-BET | I/II | Hematologic malignancies, solid tumors | 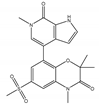 |
| NEO2734 | Pan-BET and P300 | I/II | Hematologic malignancies, solid tumors | 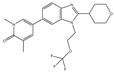 |
| OTX015 | Pan-BET | I/II | Hematologic malignancies, solid tumors, GBM | 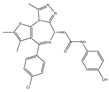 |
| PLX51107 | Pan-BET | I | Hematologic malignancies, solid tumors | 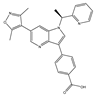 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groves, A.; Clymer, J.; Filbin, M.G. Bromodomain and Extra-Terminal Protein Inhibitors: Biologic Insights and Therapeutic Potential in Pediatric Brain Tumors. Pharmaceuticals 2022, 15, 665. https://doi.org/10.3390/ph15060665
Groves A, Clymer J, Filbin MG. Bromodomain and Extra-Terminal Protein Inhibitors: Biologic Insights and Therapeutic Potential in Pediatric Brain Tumors. Pharmaceuticals. 2022; 15(6):665. https://doi.org/10.3390/ph15060665
Chicago/Turabian StyleGroves, Andrew, Jessica Clymer, and Mariella G. Filbin. 2022. "Bromodomain and Extra-Terminal Protein Inhibitors: Biologic Insights and Therapeutic Potential in Pediatric Brain Tumors" Pharmaceuticals 15, no. 6: 665. https://doi.org/10.3390/ph15060665
APA StyleGroves, A., Clymer, J., & Filbin, M. G. (2022). Bromodomain and Extra-Terminal Protein Inhibitors: Biologic Insights and Therapeutic Potential in Pediatric Brain Tumors. Pharmaceuticals, 15(6), 665. https://doi.org/10.3390/ph15060665