
A Comparative Study of the Pharmaceutical Properties between Amorphous Drugs Loaded-Mesoporous Silica and Pure Amorphous Drugs Prepared by Solvent Evaporation



Abstract
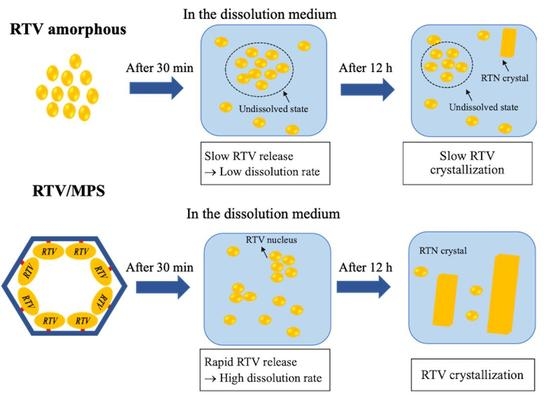
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
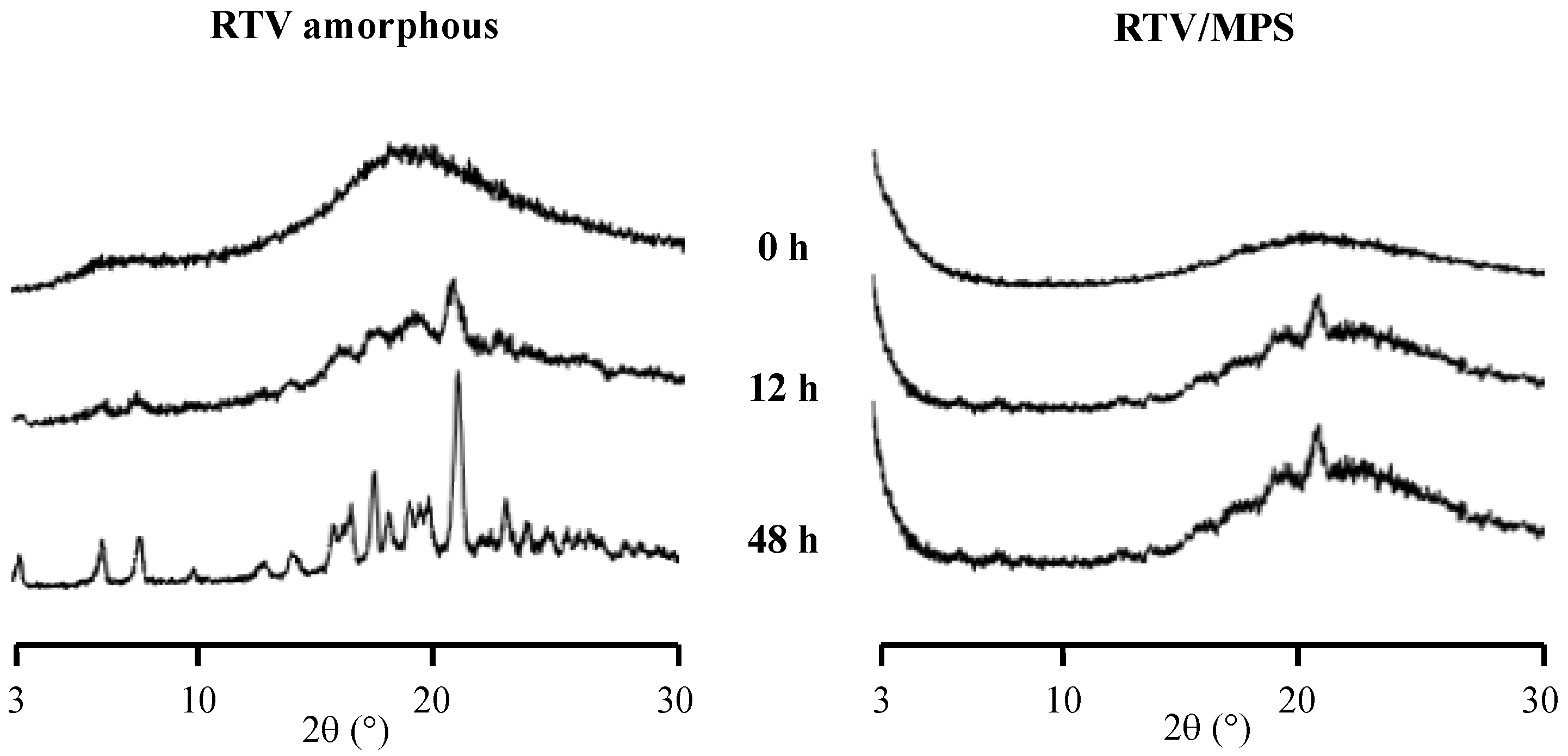{kind=link}
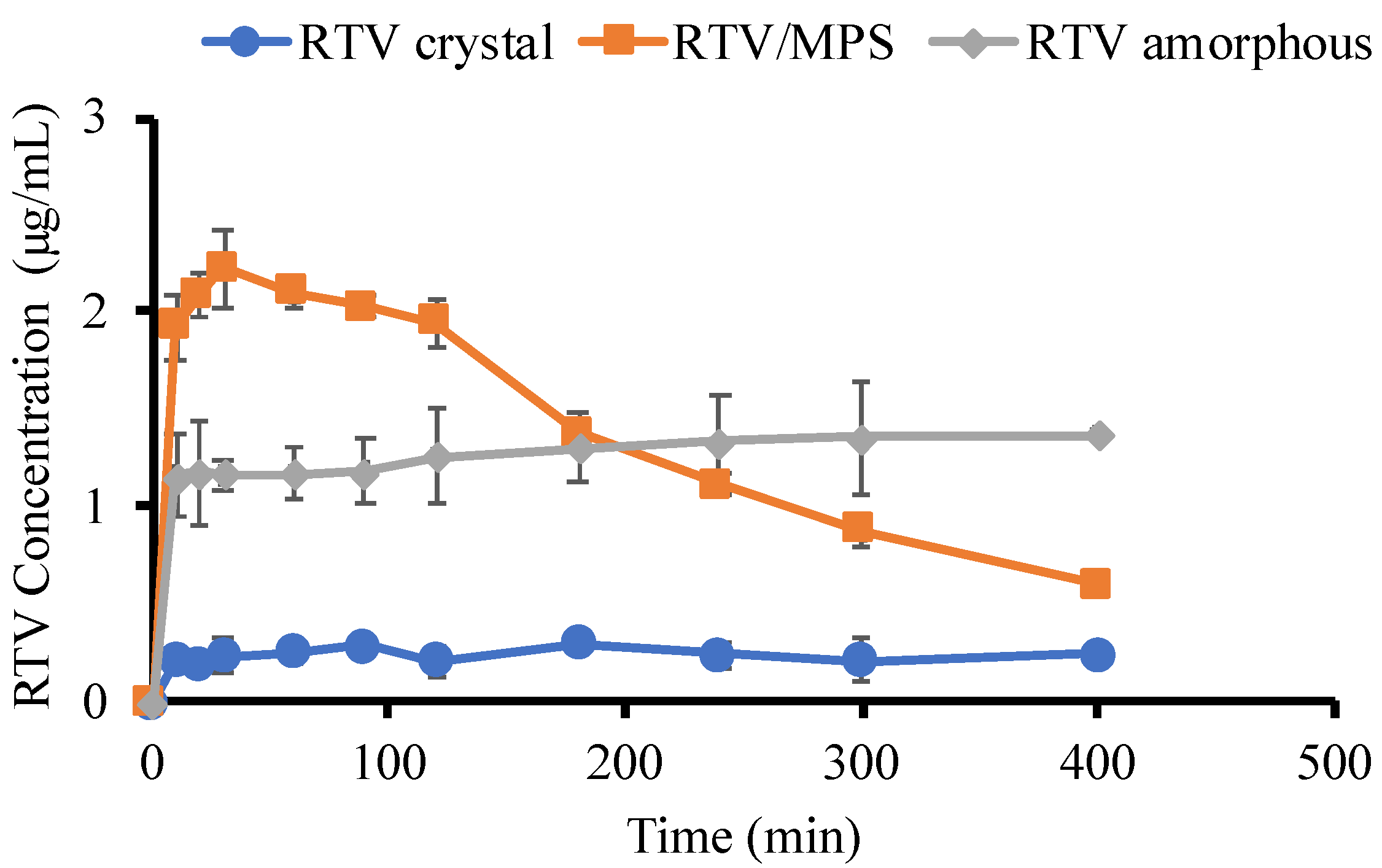{kind=link}
{kind=link}
{kind=link}
{kind=link}
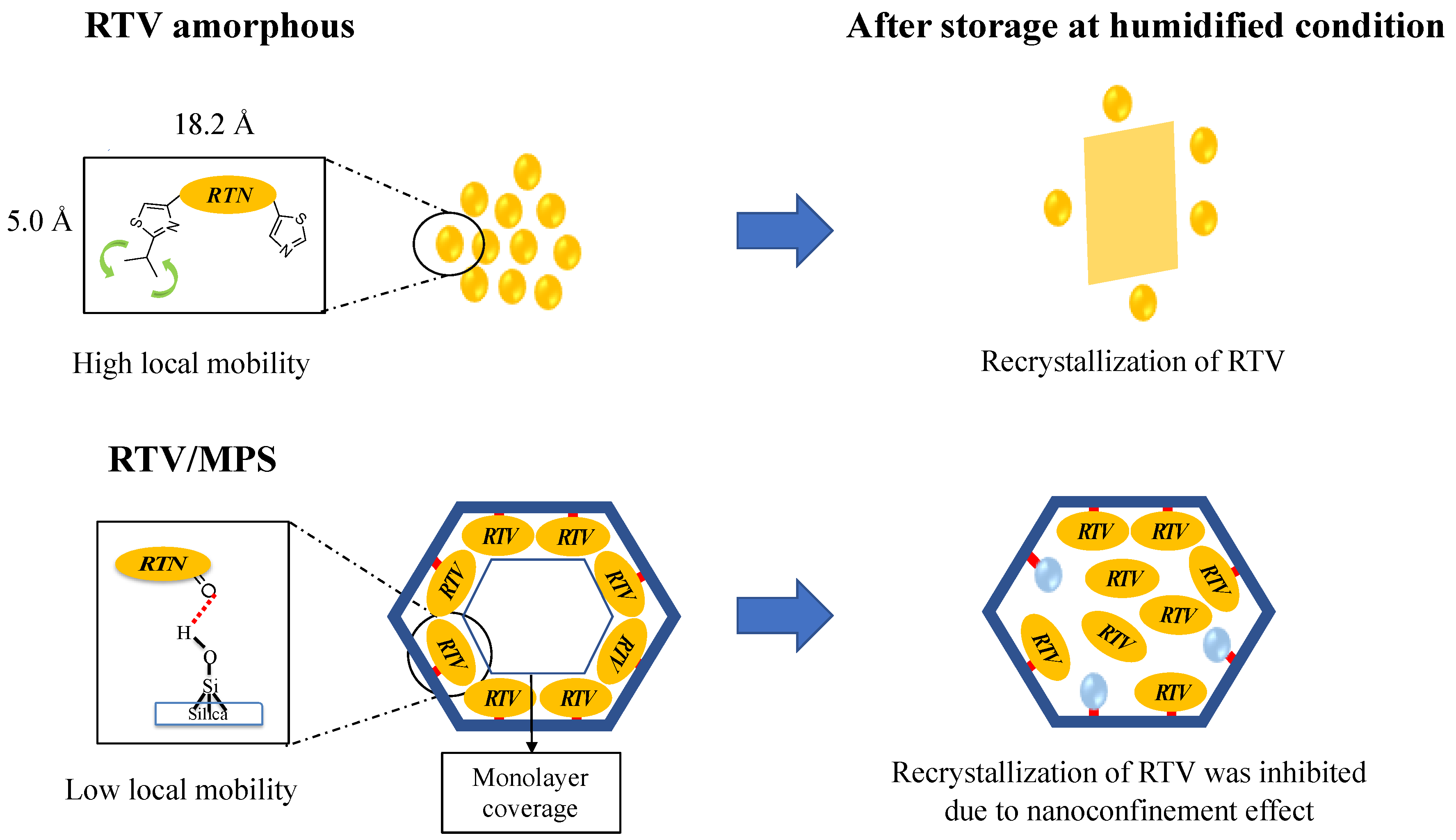{kind=link}
{kind=link}
1. Introduction
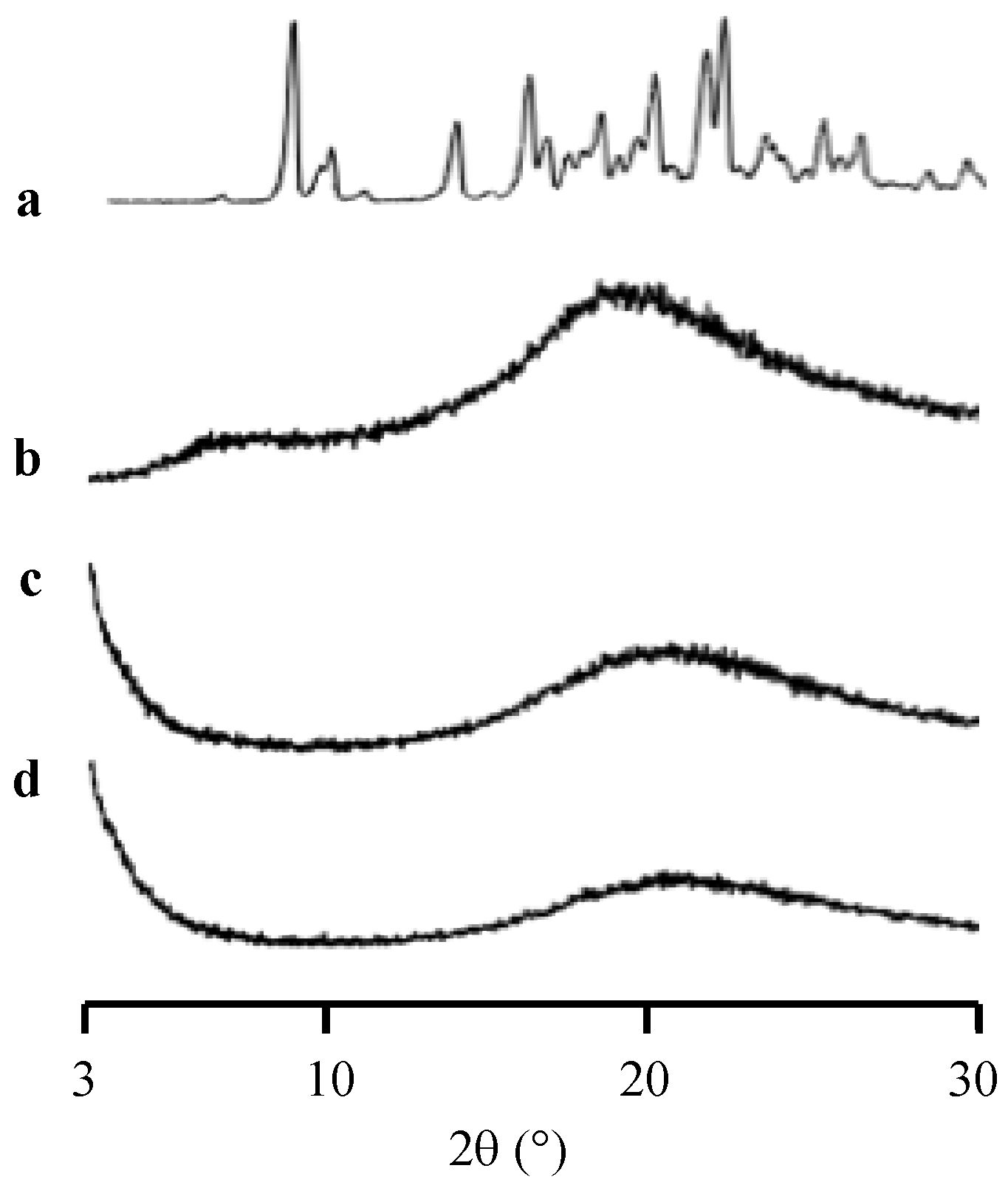
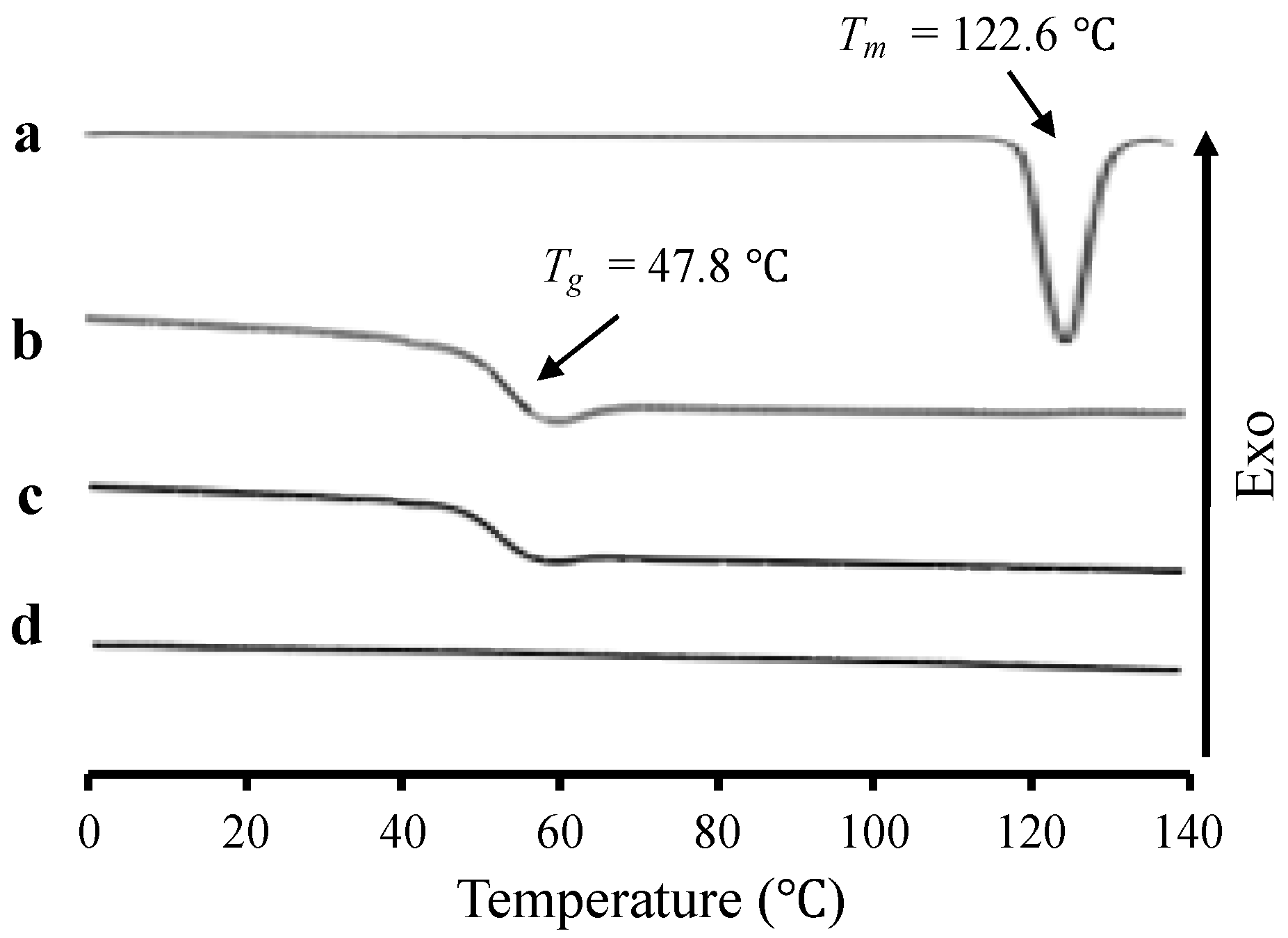
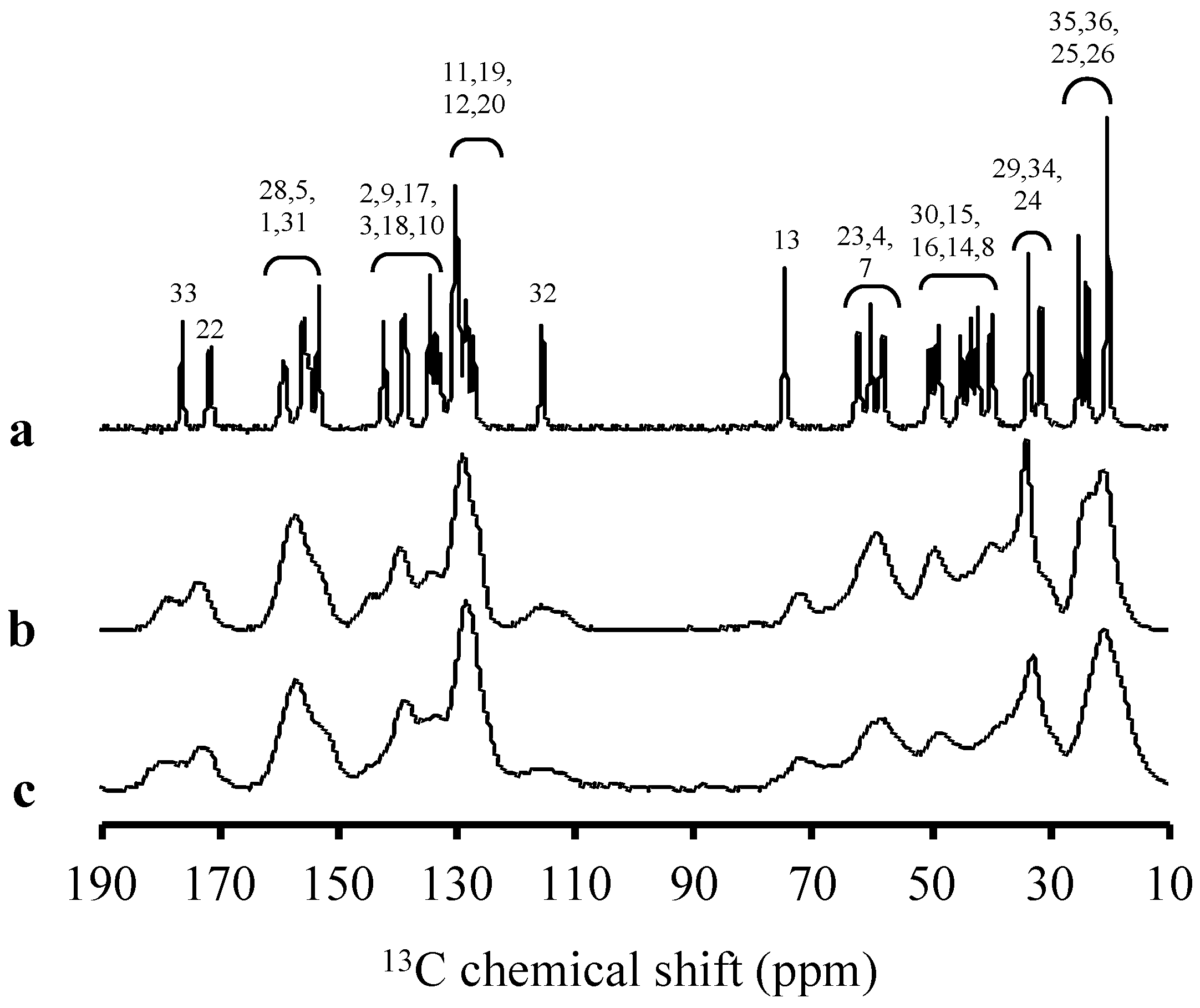
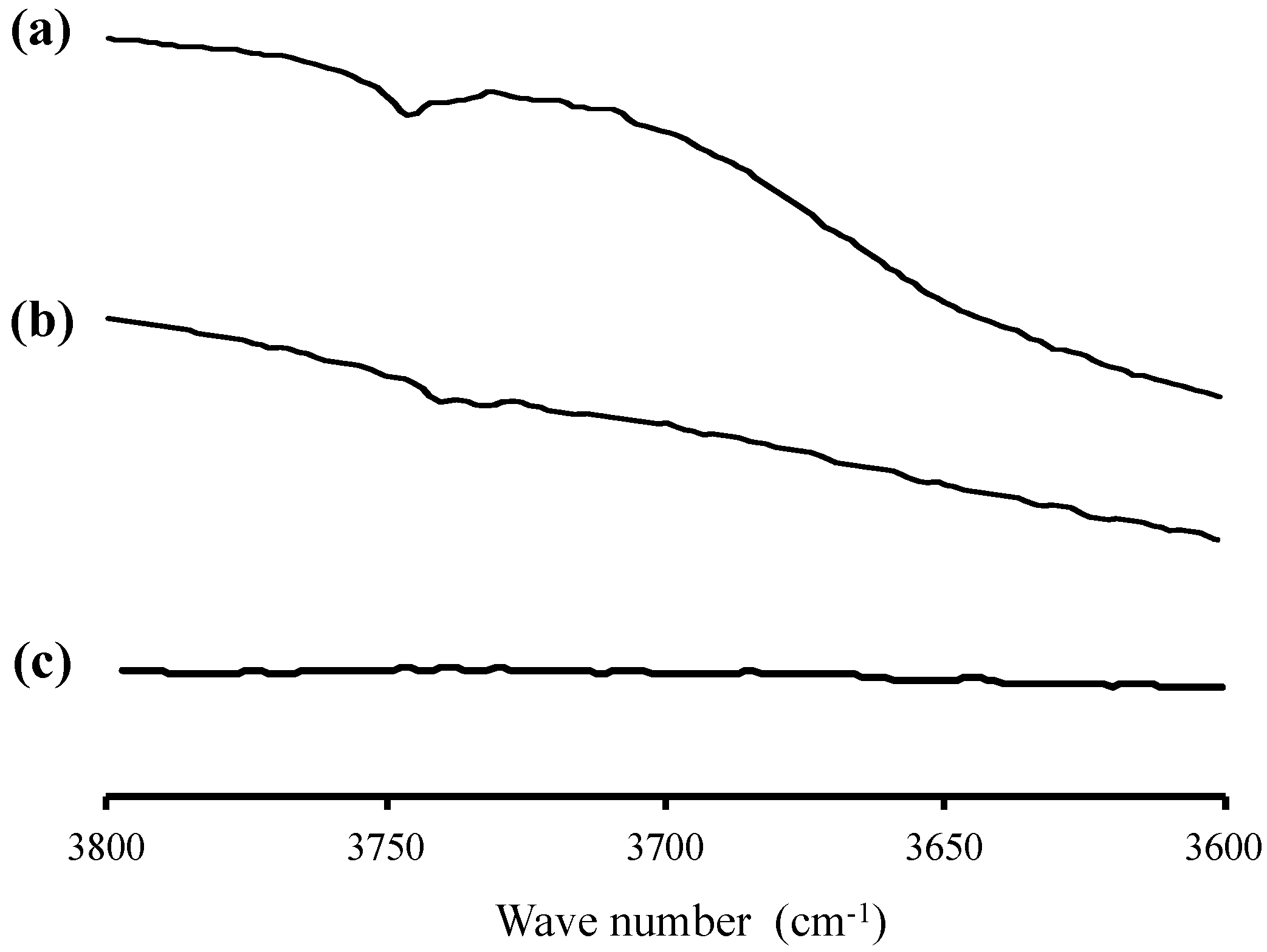
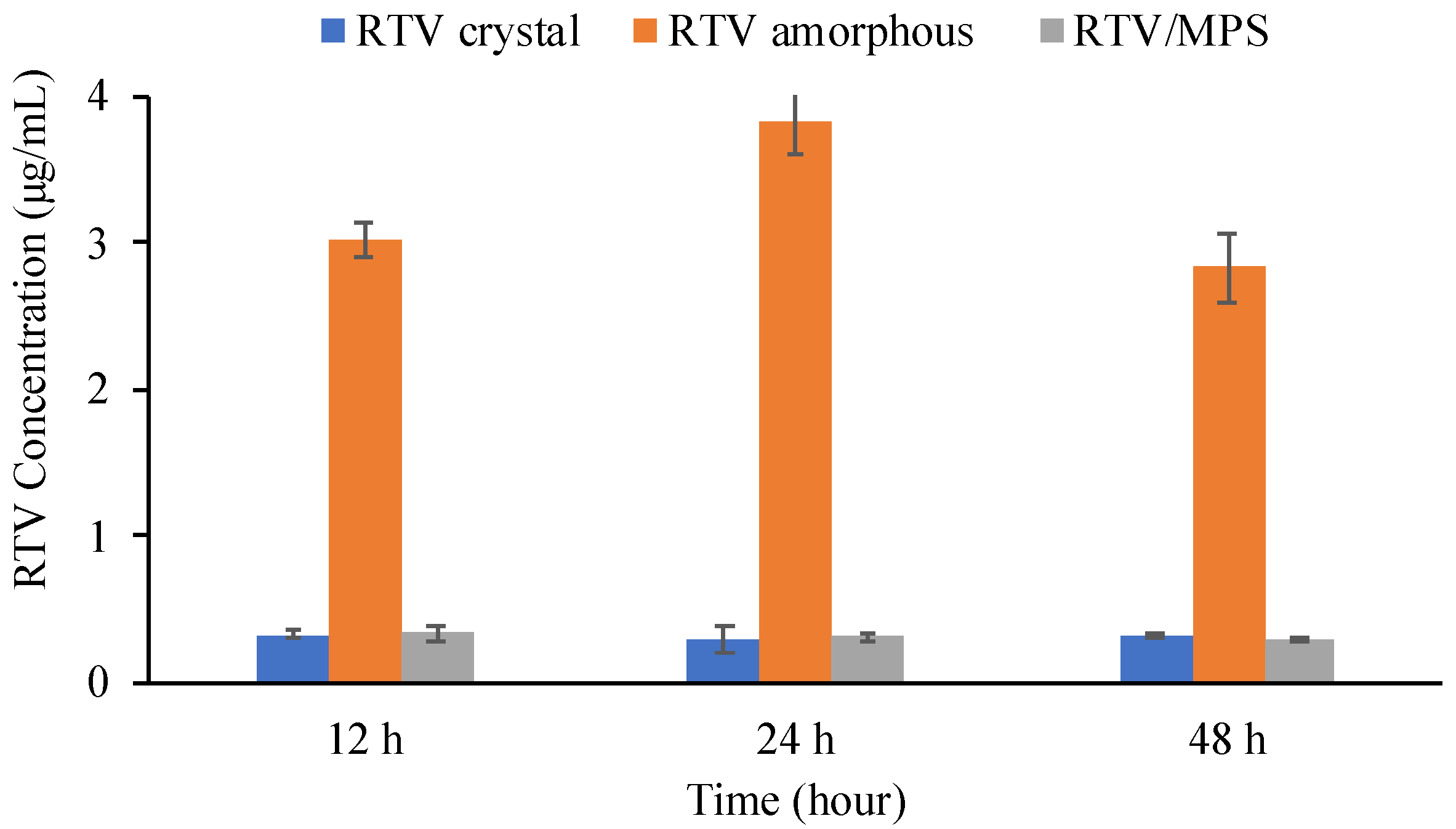
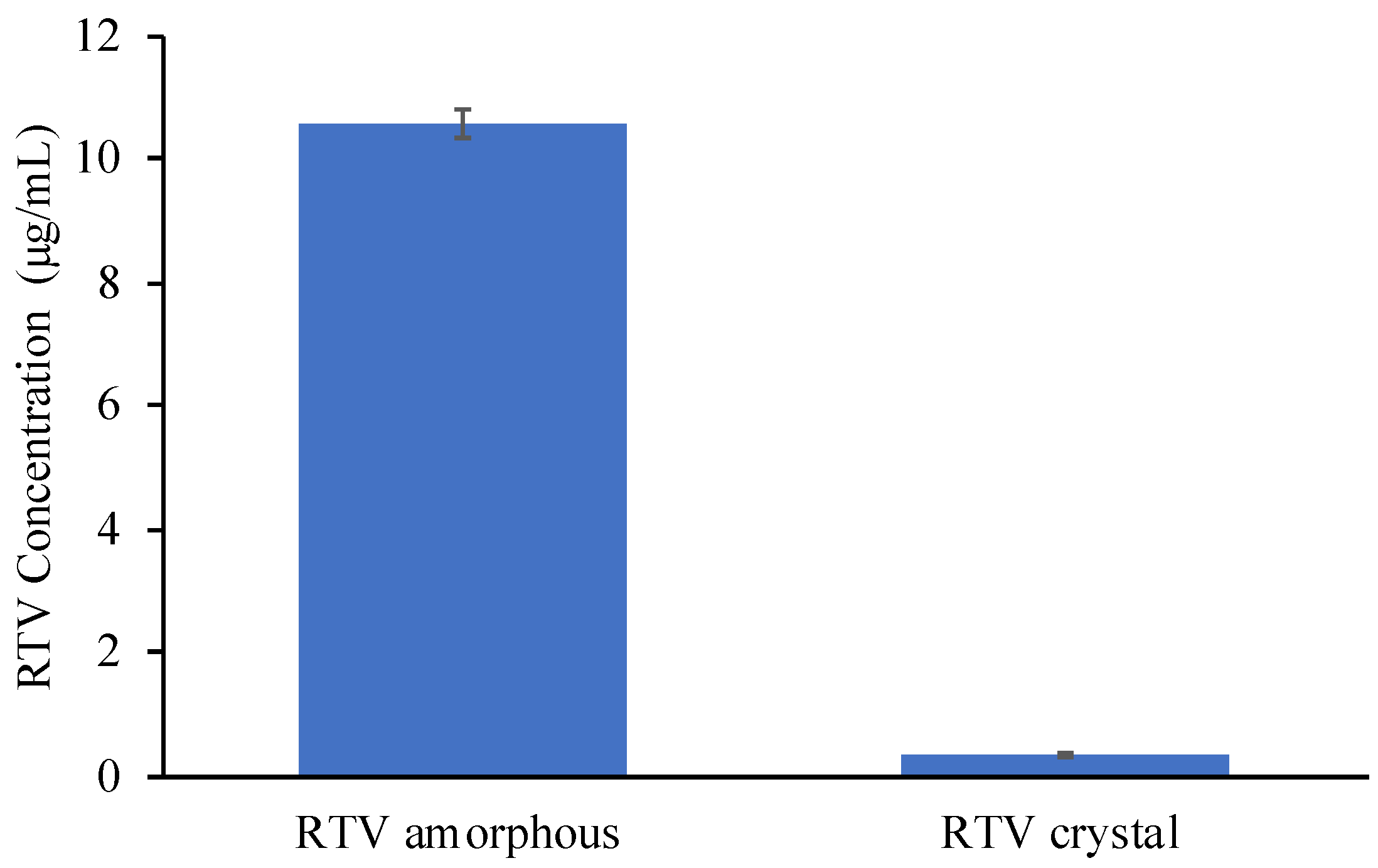
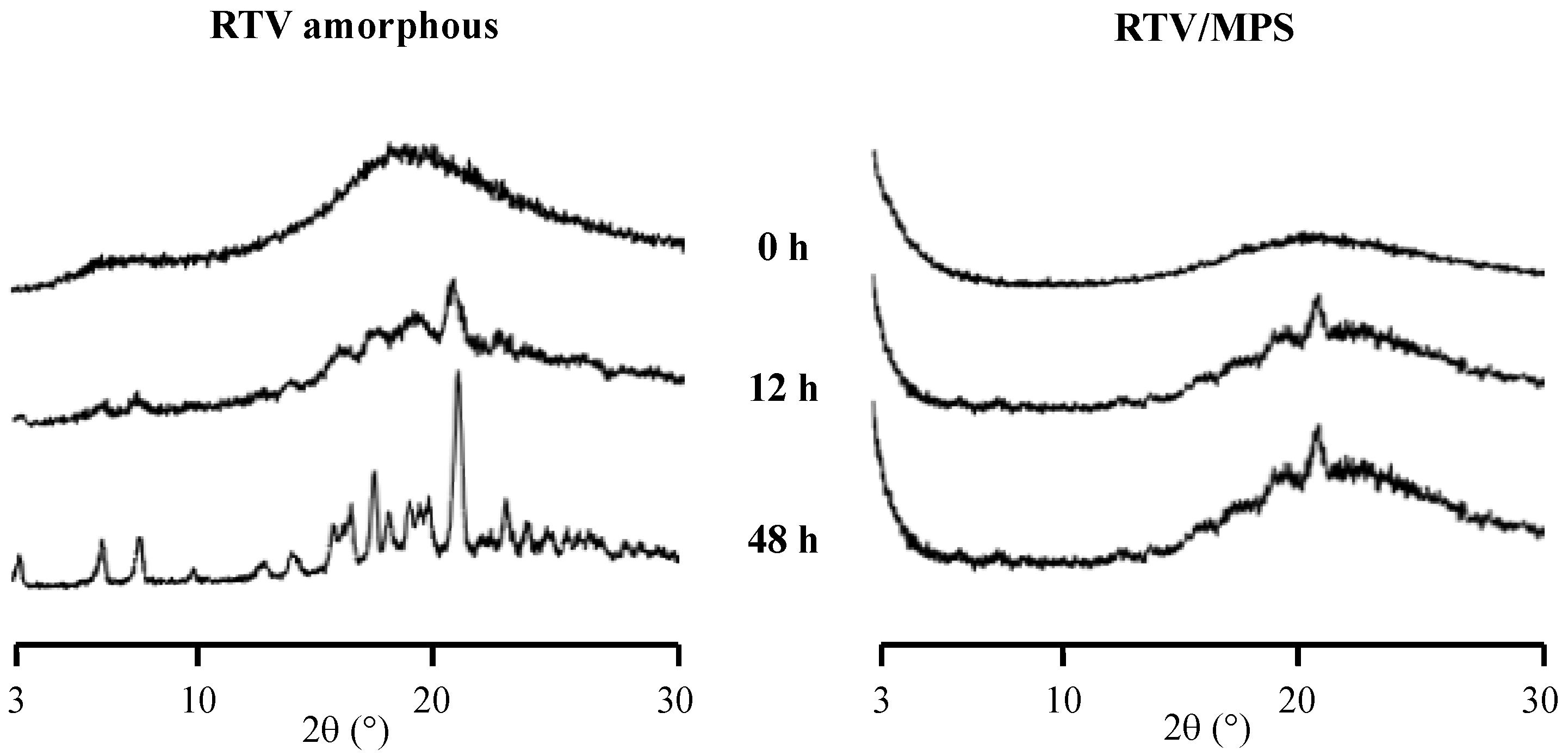
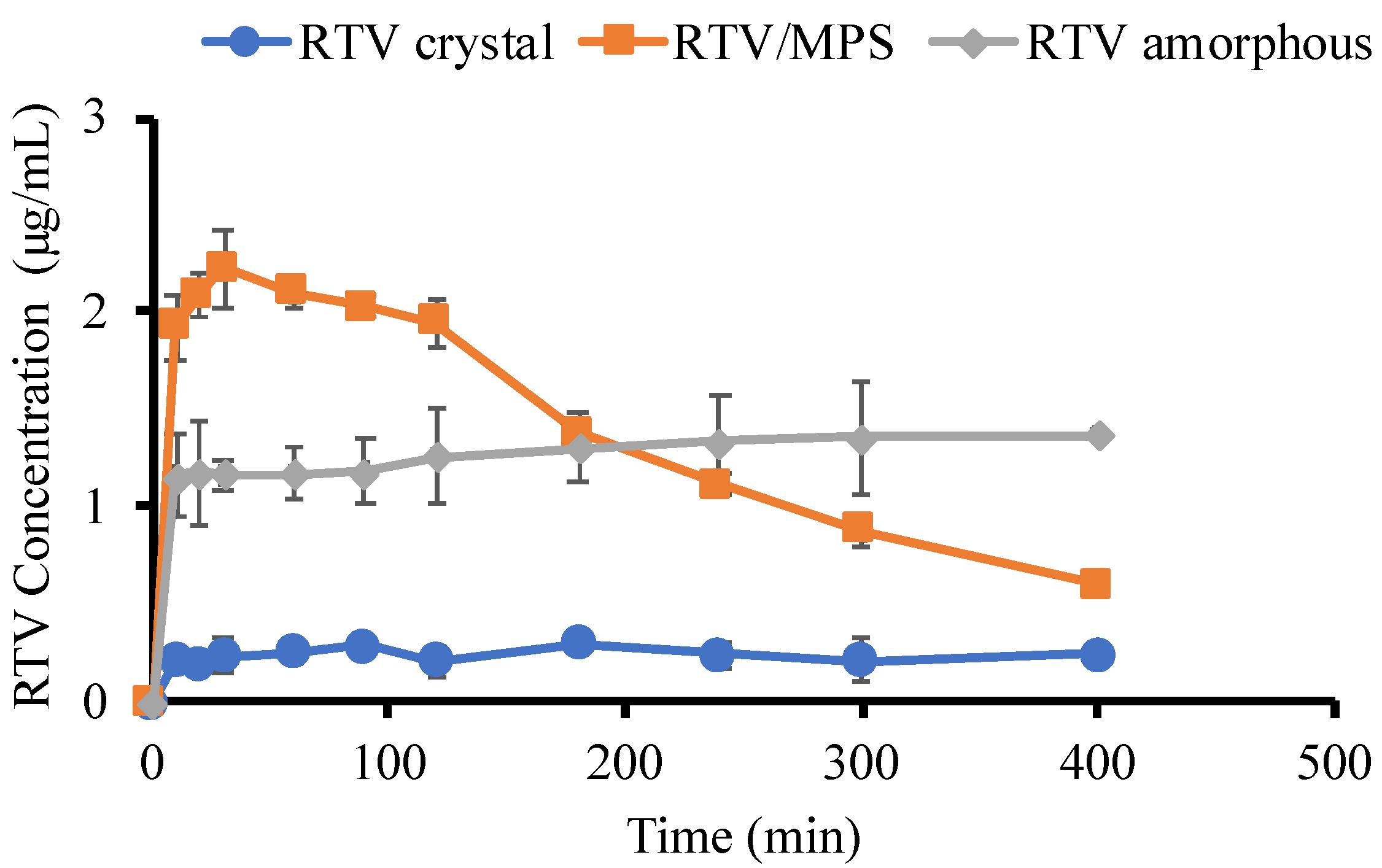
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of RTV Encapsulated into MPS
4.3. Preparation of the Pure RTV Amorphous
4.4. Powder X-ray Diffraction (PXRD)
4.5. Modulated Differential Scanning Calorimetry (MDSC)
4.6. Solid-State NMR Spectroscopy
4.7. Fourier Transform-Infrared (FT−IR) Spectroscopy
4.8. Solubility Test
4.9. Drug Loading Efficiency
4.10. Dissolution Test
4.11. High-Performance Liquid Chromatography
4.12. Physical Stability
4.13. Chemical Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skorupska, E.; Jeziorna, A.; Potrzebowski, M.J. Thermal solvent-free method of loading of pharmaceutical cocrystals into the pores of silica particles: A Case of naproxen/picolinamide cocrystal. J. Phys. Chem. C 2016, 120, 13169–13180. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Grohganz, H.; Rades, T. Aspartame as a co-former in co-amorphous systems. Int. J. Pharm. 2018, 549, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Katai, H.; Higashi, K.; Ueda, K.; Kawakami, K.; Moribe, K. Cryo-TEM and AFM Observation of the Time-Dependent Evolution of Amorphous Probucol Nanoparticles Formed by the Aqueous Dispersion of Ternary Solid Dispersions. Mol. Pharm. 2019, 16, 2184–2198. [Google Scholar] [CrossRef]
- Skorupska, E.; Paluch, P.; Jeziorna, A.; Potrzebowski, M.J. NMR study of BA/FBA cocrystal confined within mesoporous silica nanoparticles employing thermal solid phase transformation. J. Phys. Chem. C 2015, 119, 8652–8661. [Google Scholar] [CrossRef]
- Budiman, A.; Aulifa, D.L. Characterization of Drugs with Good Glass Formers in Loaded-Mesoporous Silica and Its Theoretical Value Relevance with Mesopores Surface and Pore-Filling Capacity. Pharmaceuticals 2022, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Xiao, D.; Ren, S.; Bi, S.; Wang, J.; Li, F. The binary system of ibuprofen-nicotinamide under nanoscale confinement: From cocrystal to coamorphous state. J. Pharm. Sci. 2017, 106, 3150–3155. [Google Scholar] [CrossRef]
- Mahlin, D.; Ponnambalam, S.; Heidarian Höckerfelt, M.; Bergström, C.A.S. Toward in silico prediction of glass-forming ability from molecular structure alone: A screening tool in early drug development. Mol. Pharm. 2011, 8, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Kawakami, K. Crystallization tendency of pharmaceutical glasses: Relevance to compound properties, impact of formulation process, and implications for design of amorphous solid dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.; Moreno, J.; Davé, R. Stable and fast-dissolving amorphous drug composites preparation via impregnation of Neusilin® UFL2. J. Pharm. Sci. 2018, 107, 170–182. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kojima, T.; Karashima, M.; Ikeda, Y. Physicochemical evaluation and developability assessment of co-amorphouses of low soluble drugs and comparison to the co-crystals. Chem. Pharm. Bull. 2016, 64, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; Van Den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef]
- Czarnobaj, K.; Prokopowicz, M.; Greber, K. Use of materials based on polymeric silica as bone-targeted drug delivery systems for metronidazole. Int. J. Mol. Sci. 2019, 20, 1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, S.; Heng, W.; Wei, Y.; Zhang, J.; Gao, Y. Coamorphous lurasidone hydrochloride-saccharin with charge-assisted hydrogen bonding interaction shows improved physical stability and enhanced dissolution with pH-independent solubility behavior. Cryst. Growth Des. 2015, 15, 2920–2928. [Google Scholar] [CrossRef]
- Mccarthy, C.A.; Zemlyanov, D.Y.; Crean, A.M.; Taylor, L.S. A Comparison of Drug Release and Adsorption Under Supersaturating Conditions for Ordered Mesoporous Silica with Indomethacin or Indomethacin Methyl Ester. Mol. Pharm. 2020, 17, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Vraníková, B.; Niederquell, A.; Šklubalová, Z.; Kuentz, M. Relevance of the theoretical critical pore radius in mesoporous silica for fast crystallizing drugs. Int. J. Pharm. 2020, 591, 120019. [Google Scholar] [CrossRef]
- Andersson, J.; Rosenholm, J.; Areva, S.; Lindén, M. Influences of material characteristics on ibuprofen drug loading and release profiles from ordered micro- and mesoporous silica matrices. Chem. Mater. 2004, 16, 4160–4167. [Google Scholar] [CrossRef]
- Antonino, R.S.C.M.Q.; Ruggiero, M.; Song, Z.; Nascimento, T.L.; Lima, E.M.; Bohr, A.; Knopp, M.M.; Löbmann, K. Impact of drug loading in mesoporous silica-amorphous formulations on the physical stability of drugs with high recrystallization tendency. Int. J. Pharm. X 2019, 1, 100026. [Google Scholar] [CrossRef]
- Bavnhøj, C.G.; Knopp, M.M.; Madsen, C.M.; Löbmann, K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int. J. Pharm. X 2019, 1, 100008. [Google Scholar] [CrossRef]
- Gonzalez, G.; Sagarzazu, A.; Cordova, A.; Gomes, M.E.; Salas, J.; Contreras, L.; Noris-Suarez, K.; Lascano, L. Comparative study of two silica mesoporous materials (SBA-16 and SBA-15) modified with a hydroxyapatite layer for clindamycin controlled delivery. Microporous Mesoporous Mater. 2018, 256, 251–265. [Google Scholar] [CrossRef]
- Alazzawi, H.F.; Salih, I.K.; Albayati, T.M. Drug delivery of amoxicillin molecule as a suggested treatment for covid-19 implementing functionalized mesoporous SBA-15 with aminopropyl groups. Drug Deliv. 2021, 28, 856–864. [Google Scholar] [CrossRef]
- Santos, L.F.S.; de Jesus, R.A.; Costa, J.A.S.; Gouveia, L.G.T.; de Mesquita, M.E.; Navickiene, S. Evaluation of MCM-41 and MCM-48 mesoporous materials as sorbents in matrix solid phase dispersion method for the determination of pesticides in soursop fruit (Annona muricata). Inorg. Chem. Commun. 2019, 101, 45–51. [Google Scholar] [CrossRef]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Tho, I.; Liepold, B.; Rosenberg, J.; Maegerlein, M.; Brandl, M.; Fricker, G. Formation of nano/micro-dispersions with improved dissolution properties upon dispersion of ritonavir melt extrudate in aqueous media. Eur. J. Pharm. Sci. 2010, 40, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Effect of binary additive combinations on solution crystal growth of the poorly water-soluble drug, ritonavir. Cryst. Growth Des. 2012, 12, 6050–6060. [Google Scholar] [CrossRef]
- Dengale, S.J.; Ranjan, O.P.; Hussen, S.S.; Krishna, B.S.M.; Musmade, P.B.; Gautham Shenoy, G.; Bhat, K. Preparation and characterization of co-amorphous ritonavir-indomethacin systems by solvent evaporation technique: Improved dissolution behavior and physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Sci. 2014, 62, 57–64. [Google Scholar] [CrossRef]
- Morissette, S.L.; Soukasene, S.; Levinson, D.; Cima, M.J.; Almarsson, Ö. Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellaerts, R.; Jammaer, J.A.G.; Van Speybroeck, M.; Chen, H.; Van Humbeeck, J.; Augustijns, P.; Van Den Mooter, G.; Martens, J.A. Physical state of poorly water soluble therapeutic molecules loaded into SBA-15 ordered mesoporous silica carriers: A case study with itraconazole and ibuprofen. Langmuir 2008, 24, 8651–8659. [Google Scholar] [CrossRef] [PubMed]
- Mellaerts, R.; Aerts, C.A.; Van Humbeeck, J.; Augustijns, P.; Van Den Mooter, G.; Martens, J.A. Enhanced release of itraconazole from ordered mesoporous SBA-15 silica materials. Chem. Commun. 2007, 13, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Acquasaliente, M.; Houllemare, D.; Zhang, G.; Singam, P.; Morris, J.; Marsh, K.; Babcock, M.; Pavlina, J.; Shi, Y.; Gong, Y. Ritonavir Bis-Hydrochloride. US Patent 8598216 B1, 3 December 2013. [Google Scholar]
- Liu, N.; Higashi, K.; Kikuchi, J.; Ando, S.; Kameta, N.; Ding, W.; Masuda, M.; Shimizu, T.; Ueda, K.; Yamamoto, K.; et al. Molecular-Level Understanding of the Encapsulation and Dissolution of Poorly Water-Soluble Ibuprofen by Functionalized Organic Nanotubes Using Solid-State NMR Spectroscopy. J. Phys. Chem. B 2016, 120, 4496–4507. [Google Scholar] [CrossRef]
- Budiman, A.; Aulifa, D.L. Encapsulation of drug into mesoporous silica by solvent evaporation: A comparative study of drug characterization in mesoporous silica with various molecular weights. Heliyon 2021, 7, e08627. [Google Scholar] [CrossRef]
- Dening, T.J.; Zemlyanov, D.; Taylor, L.S. Application of an adsorption isotherm to explain incomplete drug release from ordered mesoporous silica materials under supersaturating conditions. J. Control Release 2019, 307, 186–199. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Taylor, L.S. Liquid-liquid phase separation in highly supersaturated aqueous solutions of poorly water-soluble drugs: Implications for solubility enhancing formulations. Cryst. Growth Des. 2013, 13, 1497–1509. [Google Scholar] [CrossRef]
- Sliwinska-bartkowiak, M.; Dudziak, G.; Gras, R. Freezing behavior in porous glasses and MCM-41. Colloids Surfaces A Physicochem. Eng. Asp. 2001, 188, 523–529. [Google Scholar] [CrossRef]
- Ambrogi, V.; Perioli, L.; Marmottini, F.; Giovagnoli, S.; Esposito, M.; Rossi, C. Improvement of dissolution rate of piroxicam by inclusion into MCM-41 mesoporous silicate. Eur. J. Pharm. Sci. 2007, 32, 216–222. [Google Scholar] [CrossRef]
- Cheng, S.; McKenna, G.B. Nanoconfinement effects on the glass transition and crystallization behaviors of nifedipine. Mol. Pharm. 2019, 16, 856–866. [Google Scholar] [CrossRef]
- Kissi, E.O.; Ruggiero, M.T.; Hempel, N.J.; Song, Z.; Grohganz, H.; Rades, T.; Löbmann, K. Characterising glass transition temperatures and glass dynamics in mesoporous silica-based amorphous drugs. Phys. Chem. Chem. Phys. 2019, 21, 19686–19694. [Google Scholar] [CrossRef]
- Li, Q.; Simon, S.L. Curing of bisphenol M dicyanate ester under nanoscale constraint. Macromolecules 2008, 41, 1310–1317. [Google Scholar] [CrossRef]
- Li, W.; Quan, P.; Zhang, Y.; Cheng, J.; Liu, J.; Cun, D.; Xiang, R.; Fang, L. Influence of drug physicochemical properties on absorption of water insoluble drug nanosuspensions. Int. J. Pharm. 2014, 460, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Dening, T.J.; Taylor, L.S. Supersaturation potential of ordered mesoporous silica delivery systems. Part 1: Dissolution performance and drug membrane transport Rates. Mol. Pharm. 2018, 15, 3489–3501. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Collman, B.M.; Greene, L.R.; Grant, D.J.W.; Blackburn, A.C. Identifying the stable polymorph early in the drug discovery-development process. Pharm. Dev. Technol. 2005, 10, 291–297. [Google Scholar] [CrossRef]
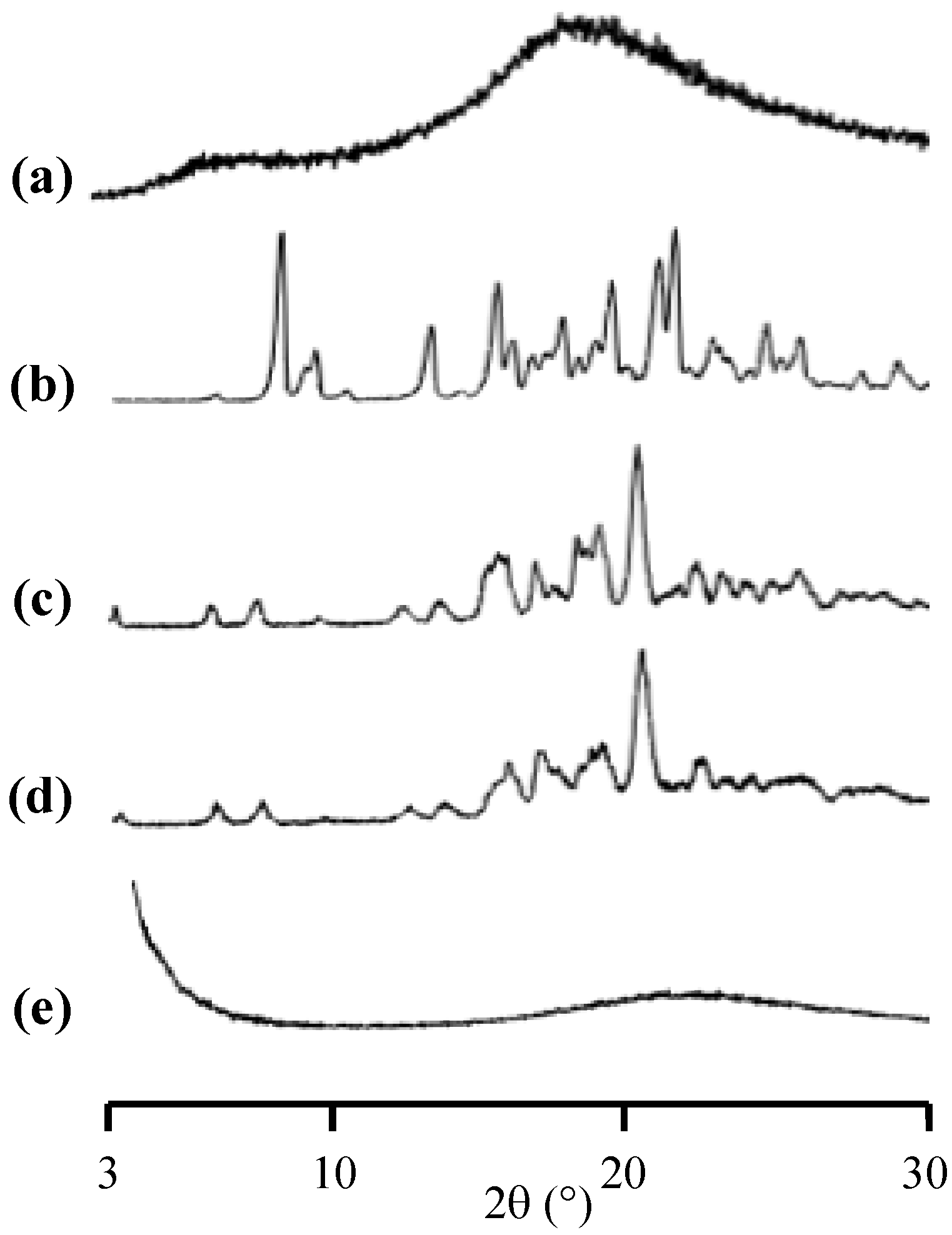
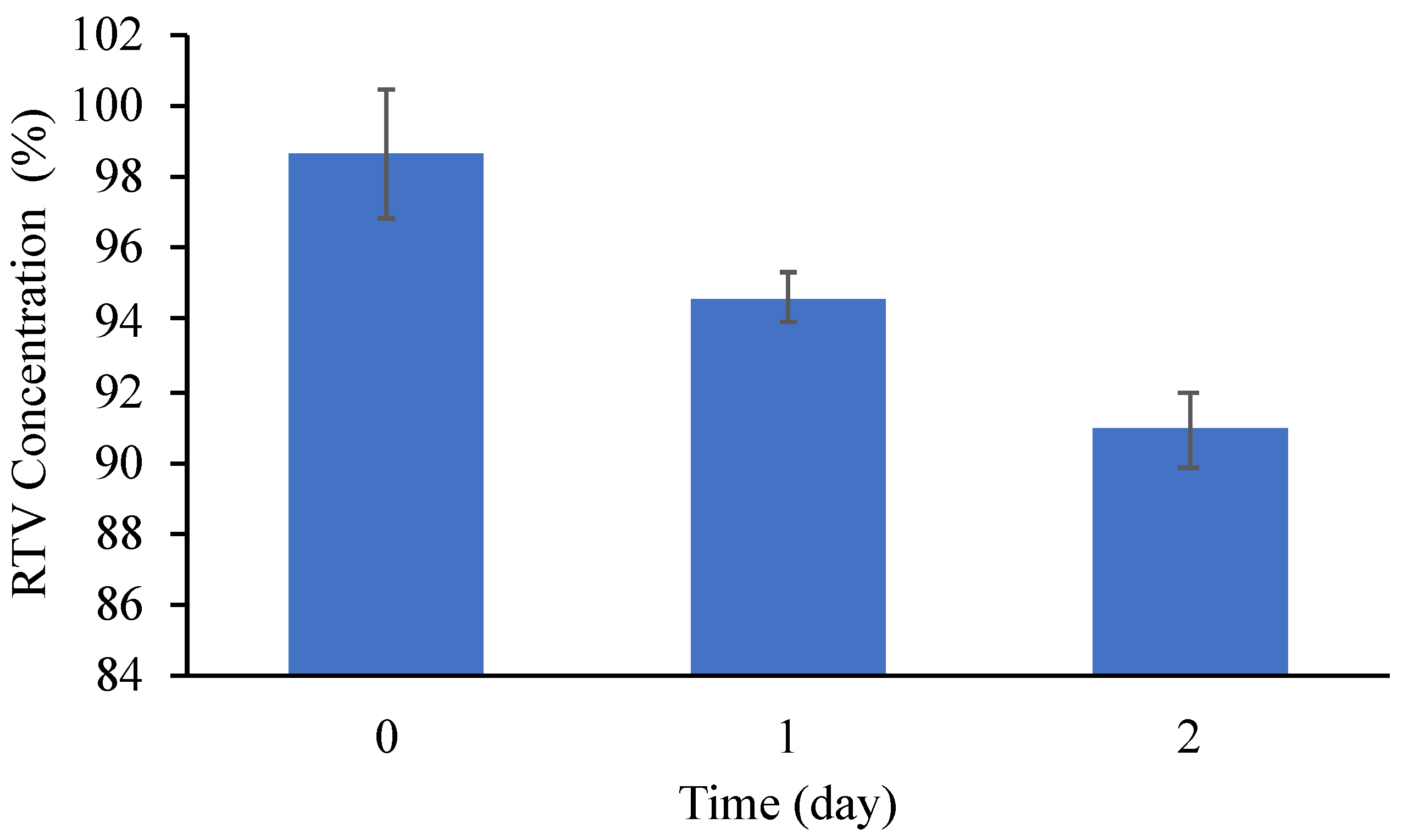
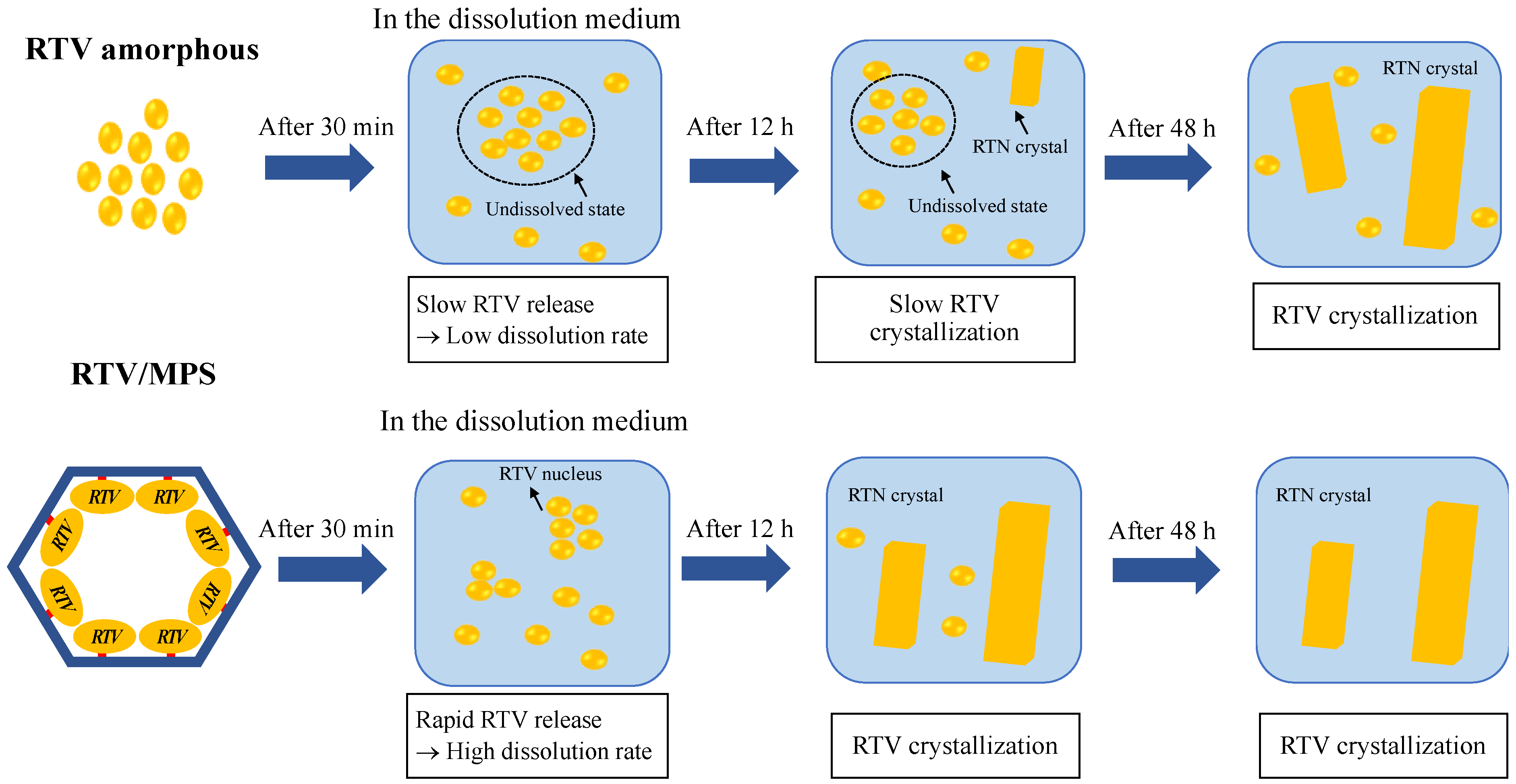
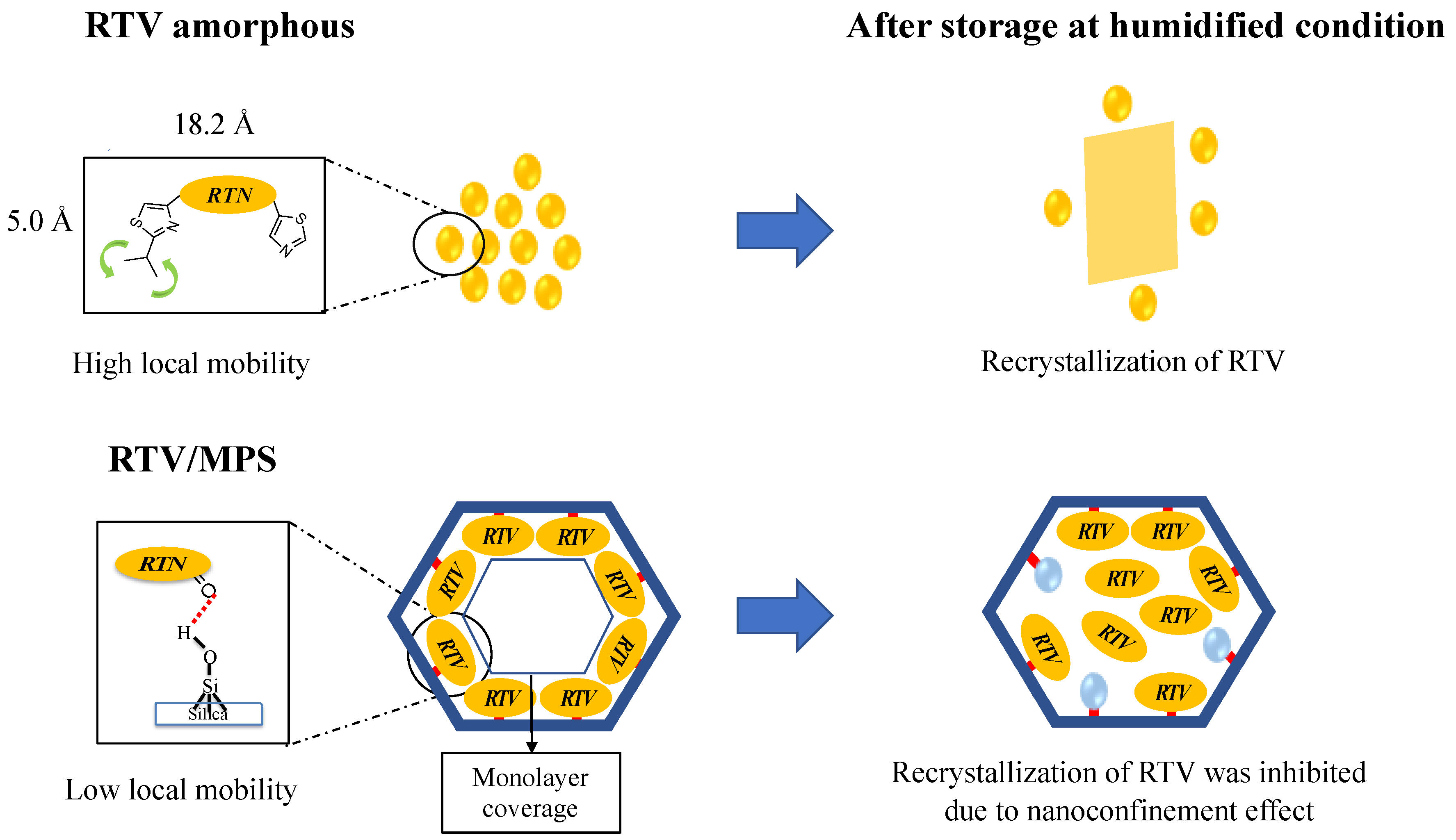
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budiman, A.; Aulifa, D.L. A Comparative Study of the Pharmaceutical Properties between Amorphous Drugs Loaded-Mesoporous Silica and Pure Amorphous Drugs Prepared by Solvent Evaporation. Pharmaceuticals 2022, 15, 730. https://doi.org/10.3390/ph15060730
Budiman A, Aulifa DL. A Comparative Study of the Pharmaceutical Properties between Amorphous Drugs Loaded-Mesoporous Silica and Pure Amorphous Drugs Prepared by Solvent Evaporation. Pharmaceuticals. 2022; 15(6):730. https://doi.org/10.3390/ph15060730
Chicago/Turabian StyleBudiman, Arif, and Diah Lia Aulifa. 2022. "A Comparative Study of the Pharmaceutical Properties between Amorphous Drugs Loaded-Mesoporous Silica and Pure Amorphous Drugs Prepared by Solvent Evaporation" Pharmaceuticals 15, no. 6: 730. https://doi.org/10.3390/ph15060730
APA StyleBudiman, A., & Aulifa, D. L. (2022). A Comparative Study of the Pharmaceutical Properties between Amorphous Drugs Loaded-Mesoporous Silica and Pure Amorphous Drugs Prepared by Solvent Evaporation. Pharmaceuticals, 15(6), 730. https://doi.org/10.3390/ph15060730