
In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist

 , , , and
, , , and 
Abstract
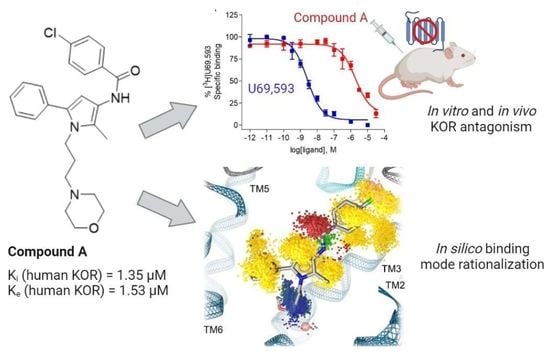
:
1. Introduction
2. Results and Discussion
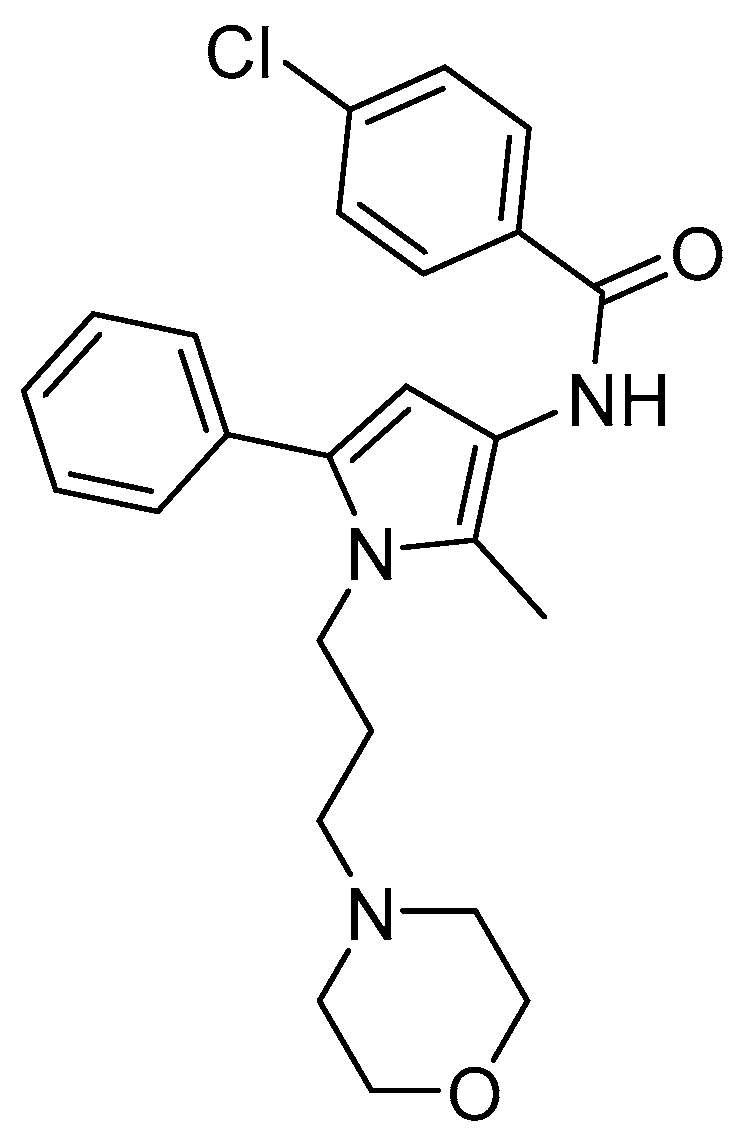
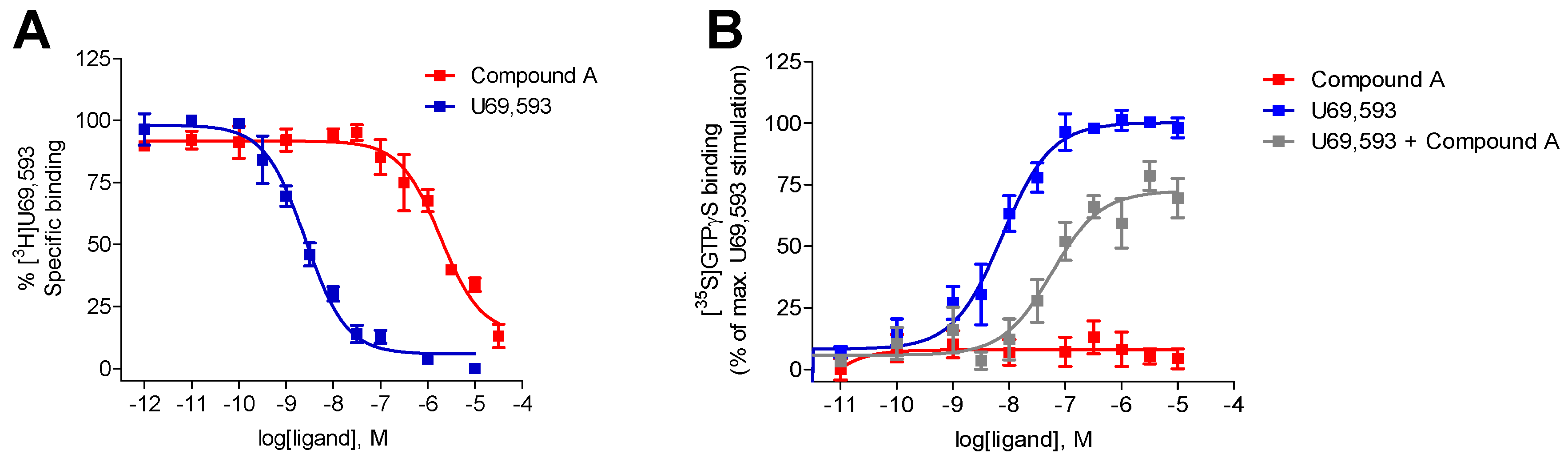
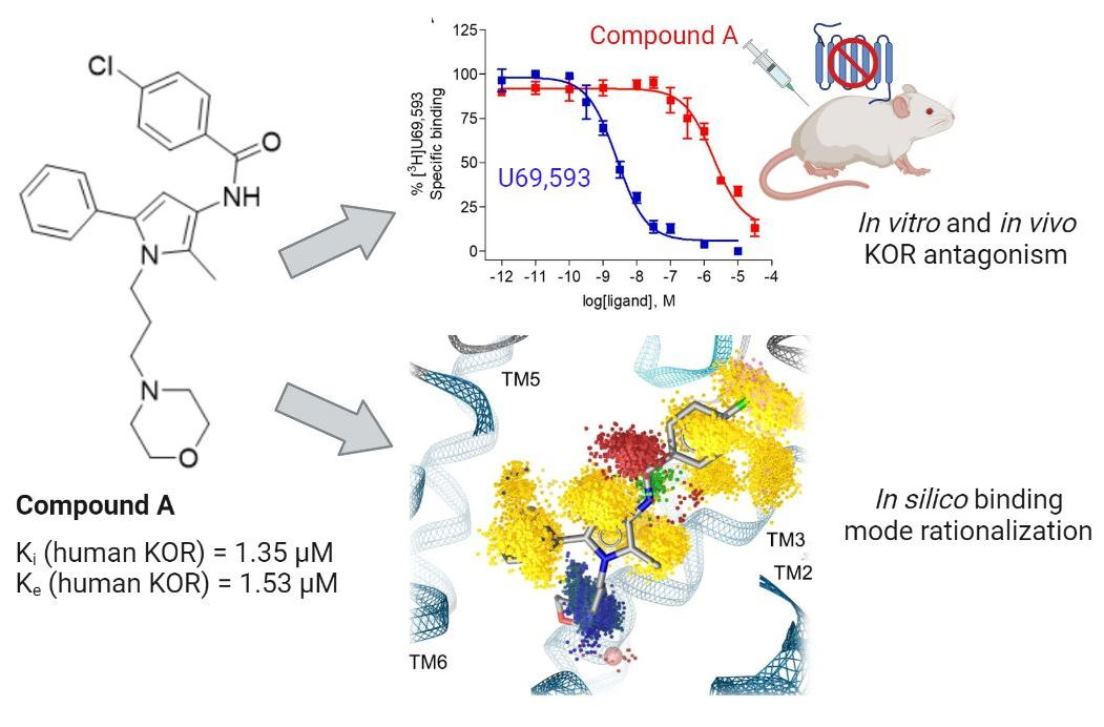
2.1. Compound A Binds at the KOR with Increased Affinity vs. MOR, Lacks Specific Binding at the DOR, and Displays KOR Antagonism In Vitro
2.2. Subcutaneous Administration of Compound A Antagonized the KOR-Mediated Antinociception Induced by U50,488 in Mice
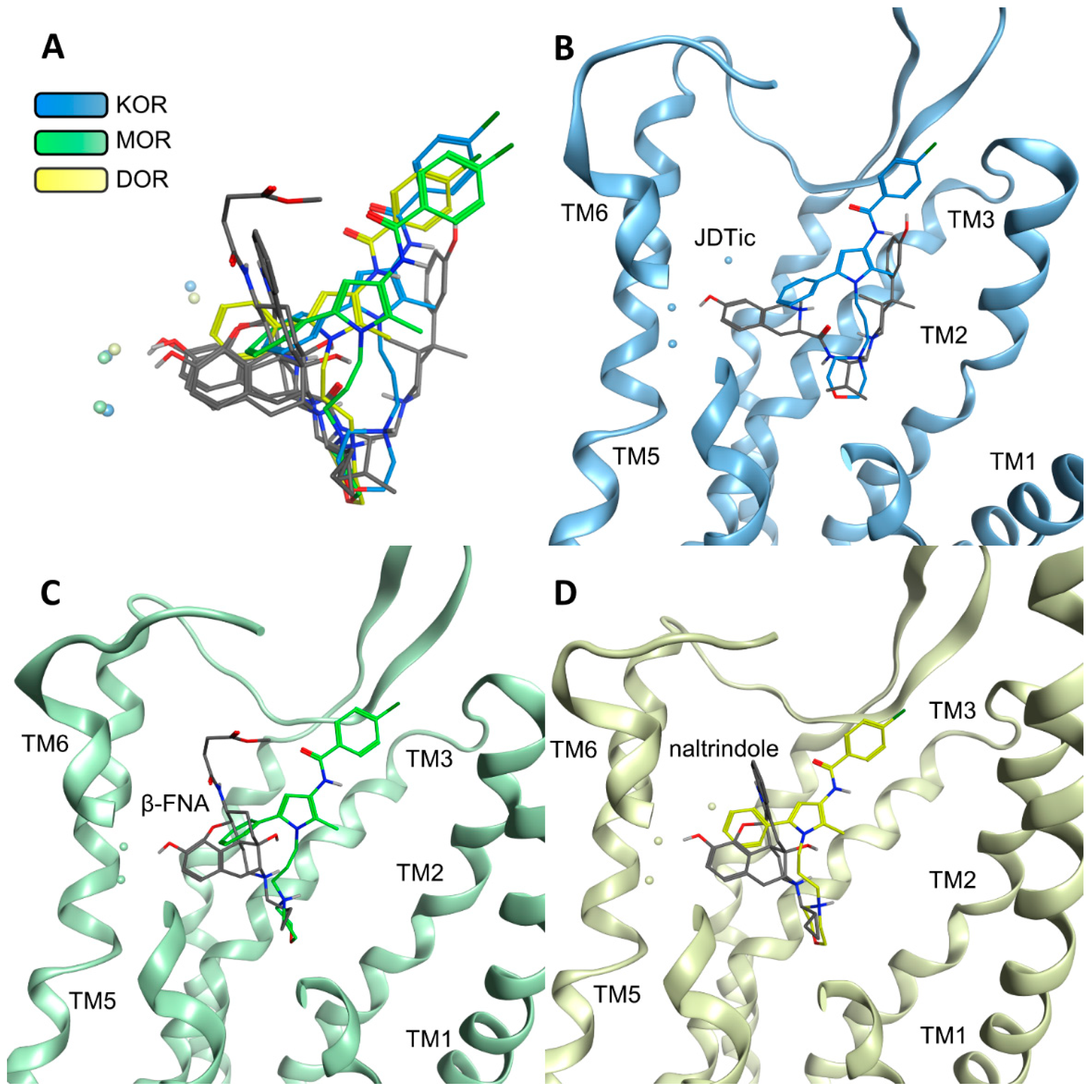
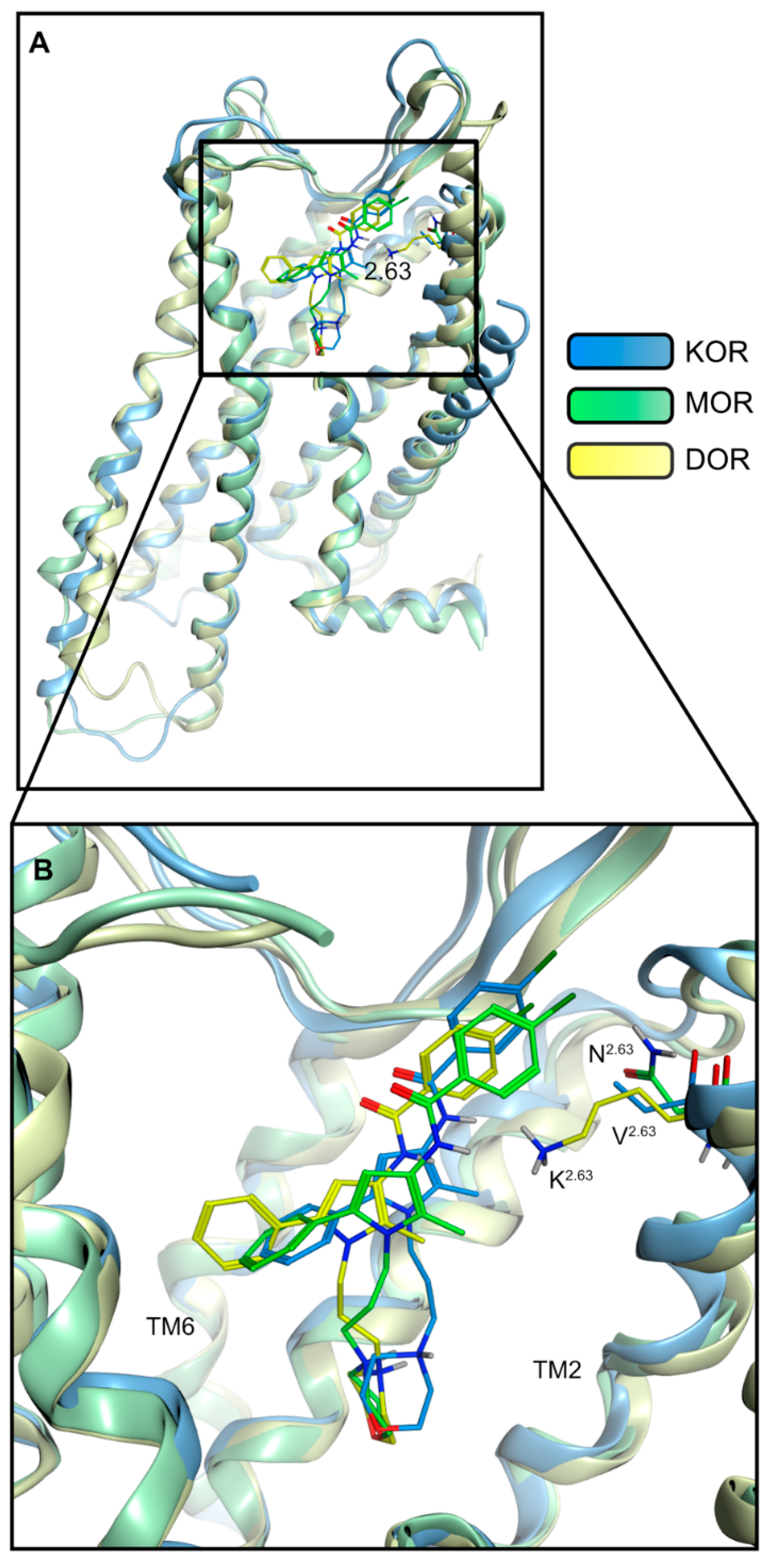
2.3. Modeling Inactive KOR Based on X-ray Crystal Structure 4DJH including Refinement of Transmembrane Helix 1
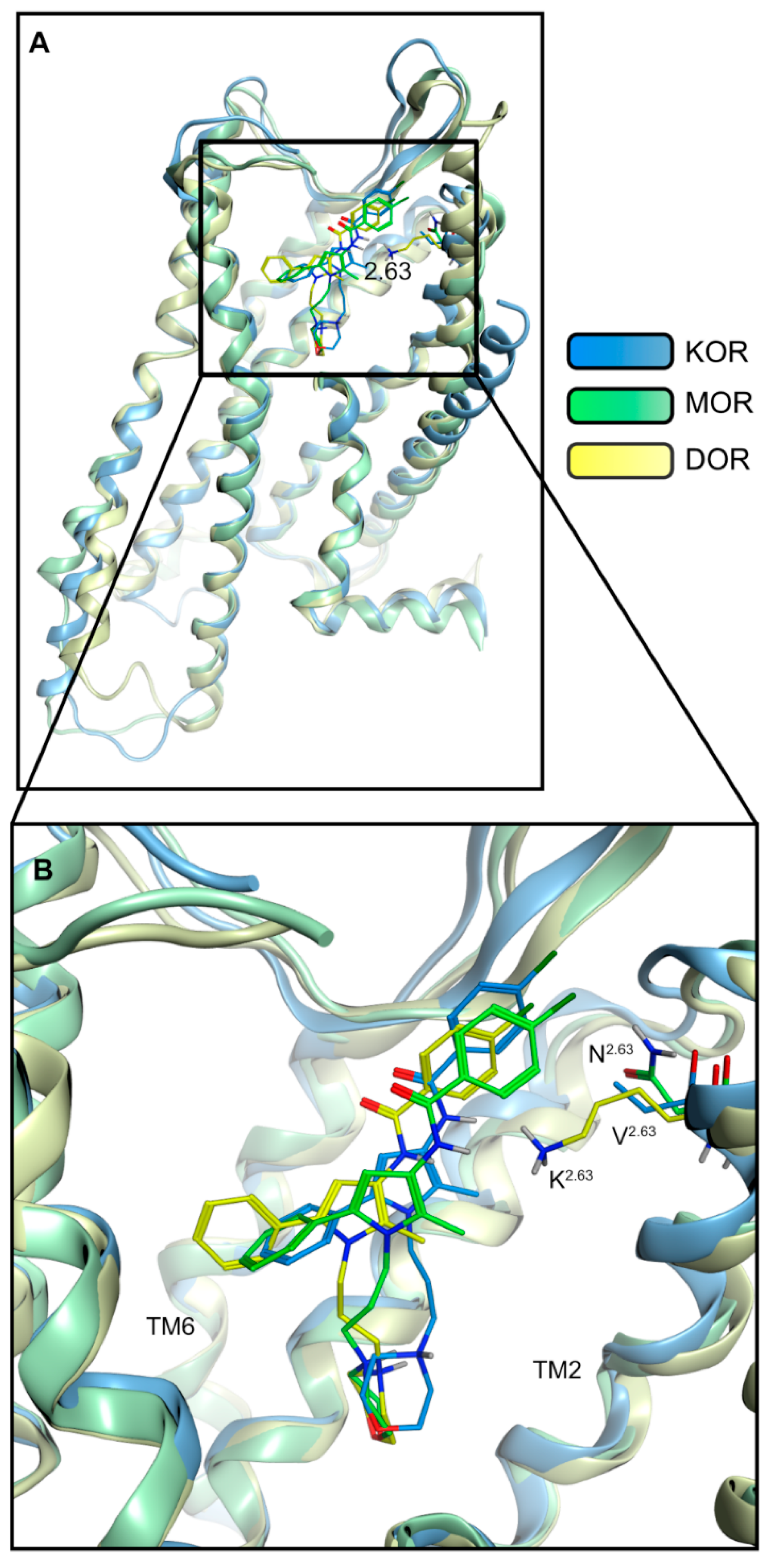
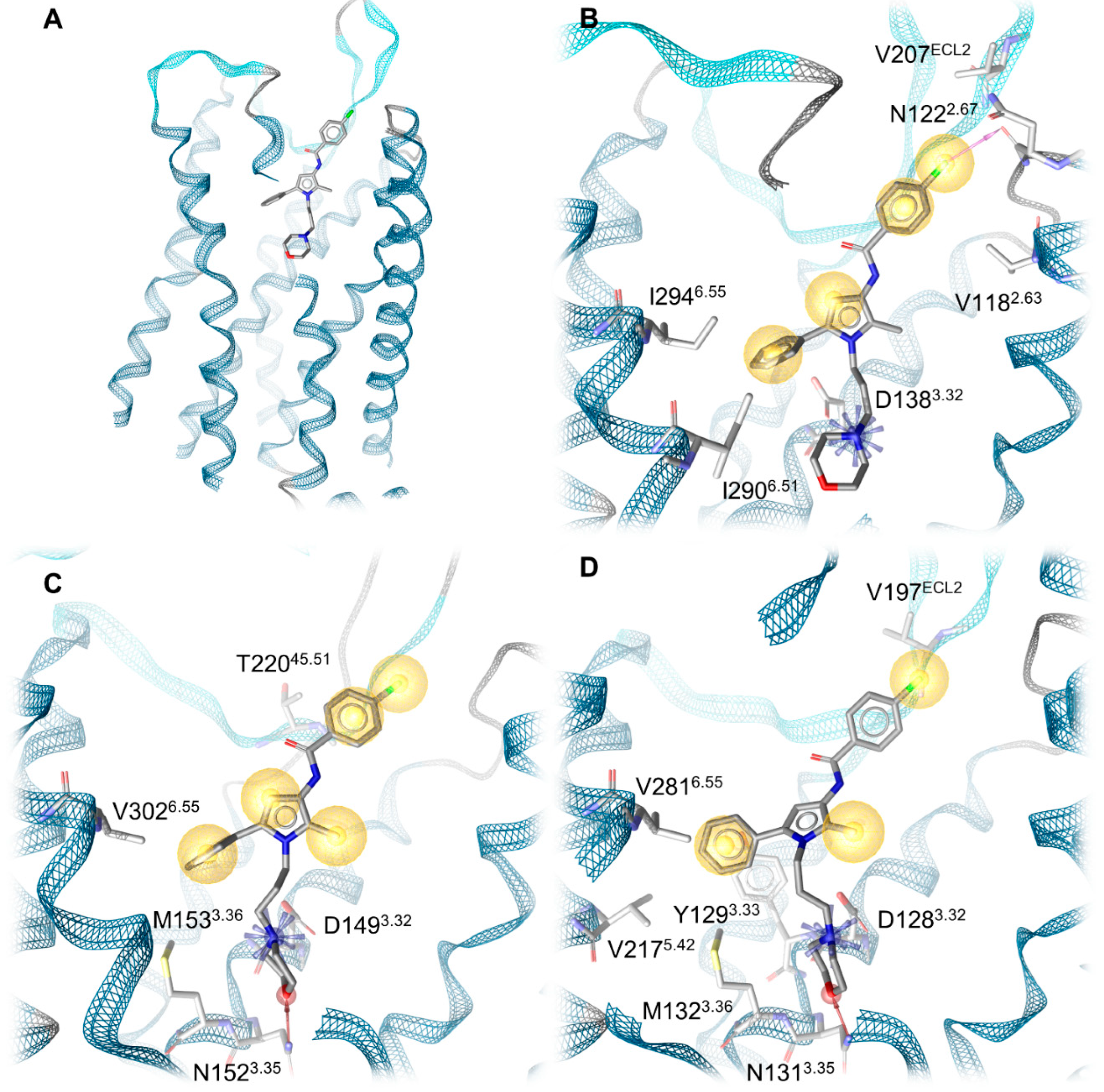
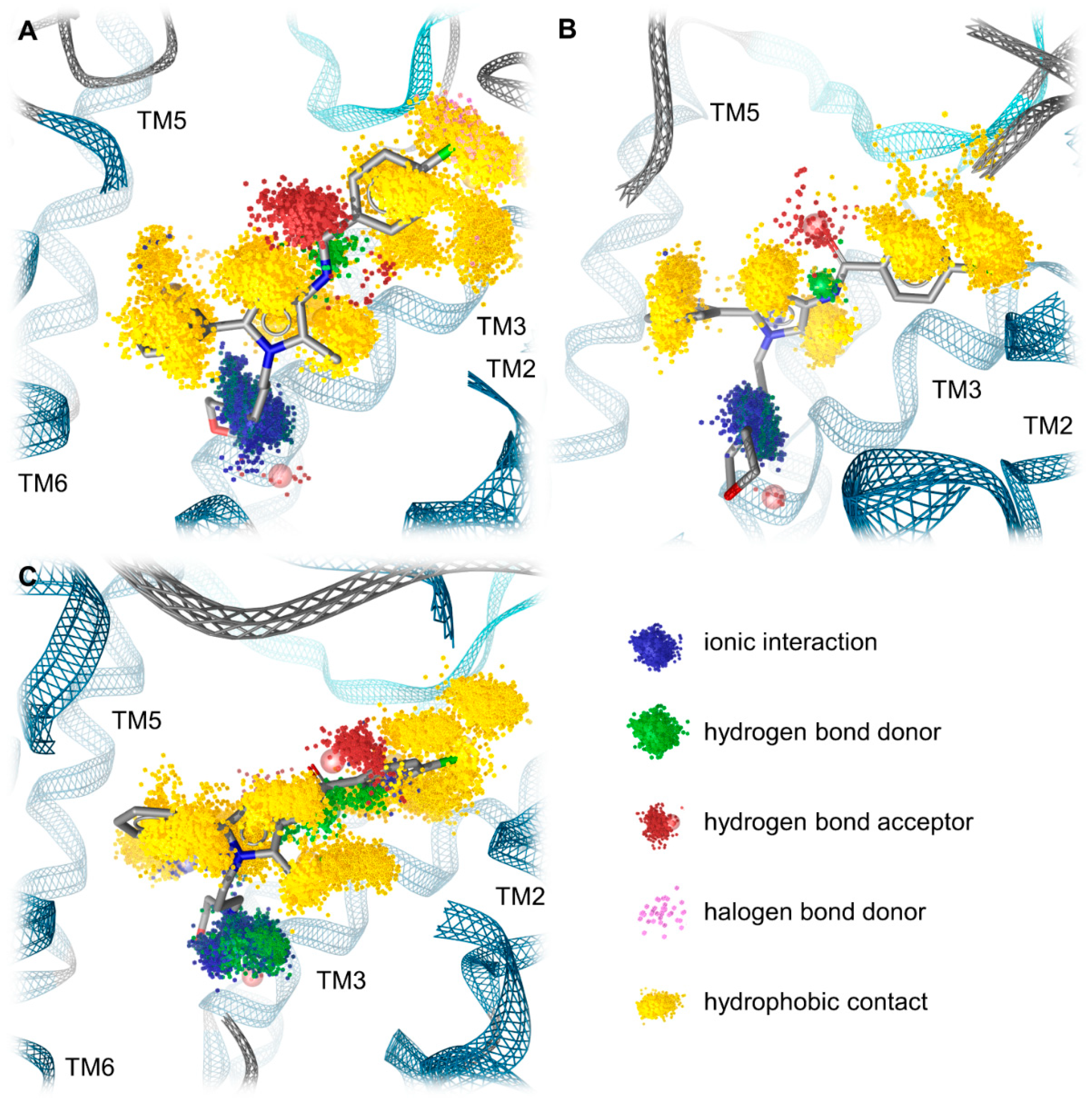
2.4. Docking Reveals Stabilizing Interactions between the Chlorophenyl Moiety of Compound A and the KOR Responsible for the Highest Subtype Affinity
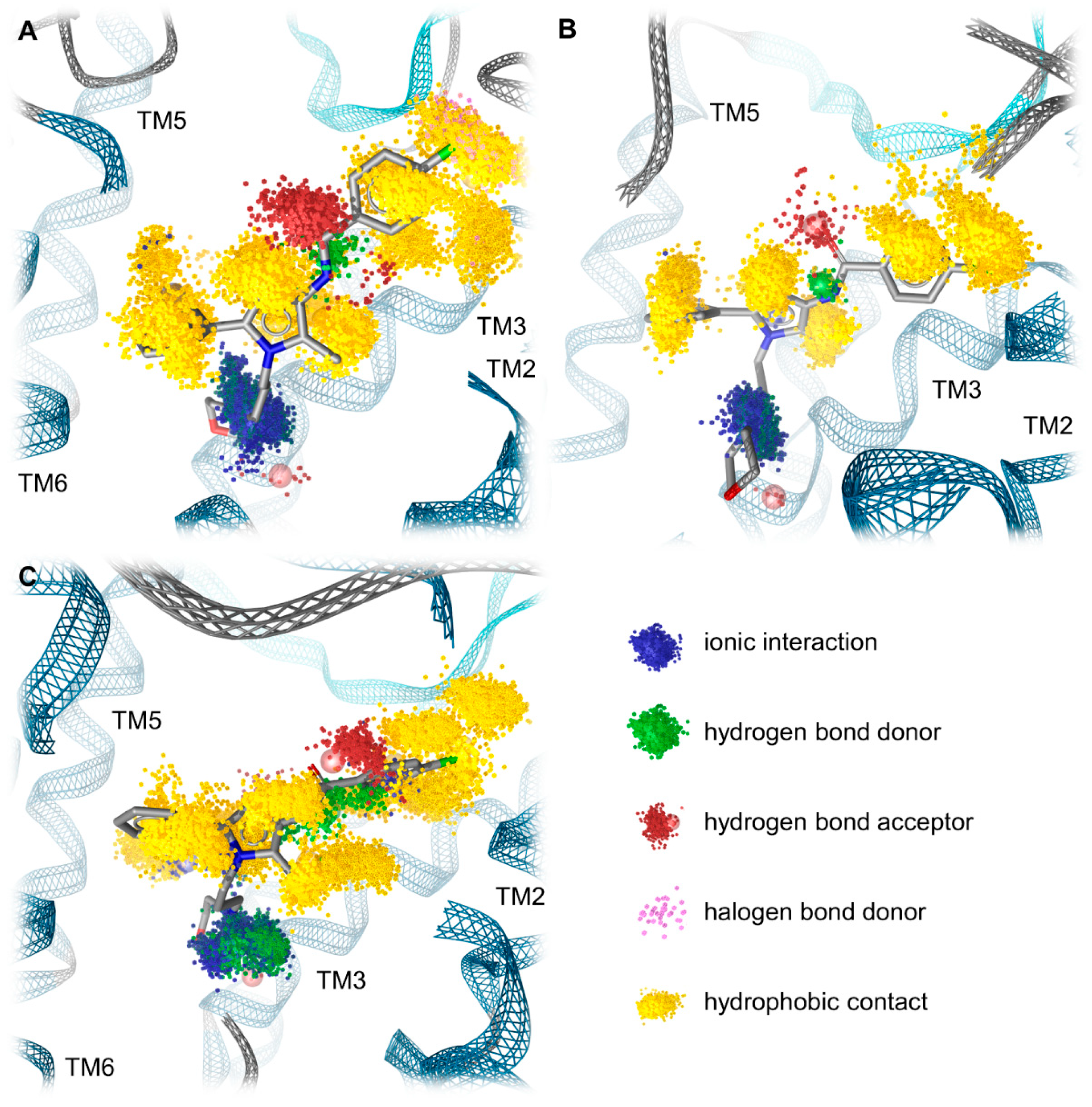
2.5. Molecular Dynamics Simulations Reveal the Most Durable and Frequent Interaction Pattern of the Chlorophenyl Moiety of Compound A in the KOR Complex
2.6. Compound A Shows Favorable Physicochemical Properties and Is a CNS Penetrant KOR Antagonist
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Cultures and Cell Membrane Preparation
3.3. Competitive Radioligand Binding Assays
3.4. [35S]GTPγS Binding Assays
3.5. Animals and Drug Administration
3.6. Acetic Acid-Induced Writhing Assay
3.7. Formalin Test
3.8. Data and Statistical Analysis
3.9. Protein Preparation
3.10. Protein-Ligand Docking Study
3.11. Molecular Dynamics Simulations and Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Bermudez, M. Allosteric coupling and biased agonism in G protein-coupled receptors. FEBS J. 2021, 288, 2513–2528. [Google Scholar] [CrossRef]
- Qu, X.; Wang, D.; Wu, B. Progress in GPCR structure determination. In GPCRs; Jastrzebska, B., Park, P.S.H., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2020; pp. 3–22. [Google Scholar]
- Sriram, K.; Insel, P.A. G Protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid analgesia and opioid-induced adverse effects: A review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef]
- Jacobson, M.L.; Browne, C.A.; Lucki, I. Kappa opioid receptor antagonists as potential therapeutics for stress-related disorders. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 615–636. [Google Scholar] [CrossRef]
- Spetea, M.; Faheem Asim, M.; Wolber, G.; Schmidhammer, H. The μ opioid receptor and ligands acting at the μ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Pan, Y.-X. Emerging insights into mu opioid pharmacology. Subst. Use Disord. 2019, 258, 89–125. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.-X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, Ł.; Goryński, K. Pharmacological aspects of over-the-counter opioid drugs misuse. Molecules 2020, 25, 3905. [Google Scholar] [CrossRef]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2021, 26, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chen, L.-Y.; Inan, S. (Eds.) The Kappa Opioid Receptor; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar]
- Lemos, J.C.; Chavkin, C. Kappa opioid receptor function. In The Opiate Receptors; Pasternak, G., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 265–305. [Google Scholar]
- Cahill, C.; Tejeda, H.A.; Spetea, M.; Chen, C.; Liu-Chen, L.Y. Fundamentals of the dynorphins/kappa opioid receptor system: From distribution to signaling and function. Handb. Exp. Pharmacol. 2022, 271, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Zangrandi, L.; Schwarzer, C. The kappa opioid receptor system in temporal lobe epilepsy. Handb. Exp. Pharmacol. 2022, 271, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Schmidhammer, H. Kappa opioid receptor ligands and pharmacology: Diphenethylamines, a class of structurally distinct, selective kappa opioid ligands. Handb. Exp. Pharmacol. 2022, 271, 163–195. [Google Scholar] [CrossRef]
- Reed, B.; Butelman, E.R.; Kreek, M.J. Kappa opioid receptor antagonists as potential therapeutics for mood and substance use disorders. Handb. Exp. Pharmacol. 2022, 271, 473–491. [Google Scholar] [CrossRef]
- Browne, C.A.; Wulf, H.; Lucki, I. Kappa Opioid receptors in the pathology and treatment of major depressive disorder. Handb. Exp. Pharmacol. 2022, 271, 493–524. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Erli, F.; Guerrieri, E.; Spetea, M. Development of diphenethylamines as selective kappa opioid receptor ligands and their pharmacological activities. Molecules 2020, 25, 5092. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Peptide Kappa opioid receptor ligands and their potential for drug development. Handb. Exp. Pharmacol. 2022, 271, 197–220. [Google Scholar] [CrossRef]
- Prisinzano, T.E. Neoclerodanes as Atypical opioid receptor ligands: 2012 David W. Robertson award for excellence in medicinal chemistry. J. Med. Chem. 2013, 56, 3435–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Hayakawa, J.; Kawamura, K.; Kawai, K.; Takezawa, Y.; Matsuura, H.; Tajima, C.; Endo, T. Discovery of a structurally novel opioid k-agonist derived from 4, 5-epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderah, T.W.; Largent-Milnes, T.; Lai, J.; Porreca, F.; Houghten, R.A.; Menzaghi, F.; Wisniewski, K.; Stalewski, J.; Sueiras-Diaz, J.; Galyean, R. Novel D-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral κ-opioid receptors. Eur. J. Pharmacol. 2008, 583, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Difelikefalin: First approval. Drugs 2021, 81, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Inan, S.; Cowan, A. Antipruritic effects of kappa opioid receptor agonists: Evidence from rodents to humans. Handb. Exp. Pharmacol. 2022, 271, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Oh, T.; Aihara, E.; Ando, A. Clinical profiles of nalfurafine hydrochloride for the treatment of pruritus patients. Handb. Exp. Pharmacol. 2022, 271, 455–472. [Google Scholar] [CrossRef]
- Albert-Vartanian, A.; Boyd, M.; Hall, A.; Morgado, S.; Nguyen, E.; Nguyen, V.; Patel, S.; Russo, L.; Shao, A.; Raffa, R. Will peripherally restricted kappa-opioid receptor agonists (pKORA s) relieve pain with less opioid adverse effects and abuse potential? J. Clin. Pharm. Ther. 2016, 41, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Lazenka, M.F. Antinociceptive Effects of kappa-opioid receptor agonists. Handb. Exp. Pharmacol. 2022, 271, 293–313. [Google Scholar] [CrossRef]
- Paton, K.F.; Atigari, D.V.; Kaska, S.; Prisinzano, T.; Kivell, B.M. Strategies for Developing κ opioid receptor agonists for the treatment of pain with fewer side effects. J. Pharmacol. Exp. Ther. 2020, 375, 332–348. [Google Scholar] [CrossRef]
- Kaski, S.W.; White, A.N.; Gross, J.D.; Siderovski, D.P. Potential for kappa-opioid receptor agonists to engineer nonaddictive analgesics: A narrative review. Anesth. Analg. 2021, 132, 406–419. [Google Scholar] [CrossRef]
- Carroll, F.I.; Carlezon, W.A., Jr. Development of κ opioid receptor antagonists. J. Med. Chem. 2013, 56, 2178–2195. [Google Scholar] [CrossRef] [PubMed]
- Portoghese, P.S.; Lipkowski, A.; Takemori, A. Binaltorphimine and nor-binaltorphimine, potent and selective k-opioid receptor antagonists. Life Sci. 1987, 40, 1287–1292. [Google Scholar] [CrossRef]
- Jones, R.M.; Portoghese, P.S. 5′-Guanidinonaltrindole, a highly selective and potent κ-opioid receptor antagonist. Eur. J. Pharmacol. 2000, 396, 49–52. [Google Scholar] [CrossRef]
- Thomas, J.B.; Atkinson, R.N.; Rothman, R.B.; Fix, S.E.; Mascarella, S.W.; Vinson, N.A.; Xu, H.; Dersch, C.M.; Lu, Y.-F.; Cantrell, B.E. Identification of the first trans-(3 R, 4 R)-dimethyl-4-(3-hydroxyphenyl) piperidine derivative to possess highly potent and selective opioid κ receptor antagonist activity. J. Med. Chem. 2001, 44, 2687–2690. [Google Scholar] [CrossRef] [PubMed]
- Buda, J.J.; Carroll, F.I.; Kosten, T.R.; Swearingen, D.; Walters, B.B. A double-blind, placebo-controlled trial to evaluate the safety, tolerability, and pharmacokinetics of single, escalating oral doses of JDTic. Neuropsychopharmacology 2015, 40, 2059–2065. [Google Scholar] [CrossRef] [Green Version]
- Munro, T.A.; Berry, L.M.; Van’t Veer, A.; Béguin, C.; Carroll, F.; Zhao, Z.; Carlezon, W.A.; Cohen, B.M. Long-acting κ opioid antagonists nor-BNI, GNTI and JDTic: Pharmacokinetics in mice and lipophilicity. BMC Pharmacol. 2012, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Black, S.L.; Chauvignac, C.; Grundt, P.; Miller, C.N.; Wood, S.; Traynor, J.R.; Lewis, J.W.; Husbands, S.M. Guanidino N-substituted and N,N-disubstituted derivatives of the kappa-opioid antagonist GNTI. J. Med. Chem. 2003, 46, 5505–5511. [Google Scholar] [CrossRef]
- Endoh, T.; Matsuura, H.; Tanaka, C.; Nagase, H. Nor-binaltorphimine: A potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch. Int. Pharmacodyn. Ther. 1992, 316, 30–42. [Google Scholar]
- Rorick-Kehn, L.M.; Witkin, J.M.; Statnick, M.A.; Eberle, E.L.; McKinzie, J.H.; Kahl, S.D.; Forster, B.M.; Wong, C.J.; Li, X.; Crile, R.S. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 2014, 77, 131–144. [Google Scholar] [CrossRef]
- Guerrero, M.; Urbano, M.; Kim, E.-K.; Gamo, A.M.; Riley, S.; Abgaryan, L.; Leaf, N.; Van Orden, L.J.; Brown, S.J.; Xie, J.Y. Design and synthesis of a novel and selective kappa opioid receptor (KOR) antagonist (BTRX-335140). J. Med. Chem. 2019, 62, 1761–1780. [Google Scholar] [CrossRef] [Green Version]
- Patkar, K.A.; Yan, X.; Murray, T.F.; Aldrich, J.V. [Nα-BenzylTyr 1,cyclo(d-Asp5,Dap8)]-dynorphin A-(1−11)NH2 cyclized in the “Address” Domain is a novel κ-Opioid receptor antagonist. J. Med. Chem. 2005, 48, 4500–4503. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.A.; Murray, T.F.; Aldrich, J.V. Structure-Activity relationships of the peptide kappa opioid receptor antagonist zyklophin. J. Med. Chem. 2015, 58, 8783–8795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Zaidi, S.A.; Katritch, V. Structural Characterization of KOR Inactive and active states for 3D pharmacology and drug discovery. Handb. Exp. Pharmacol. 2022, 271, 41–64. [Google Scholar] [CrossRef]
- Kaserer, T.; Lantero, A.; Schmidhammer, H.; Spetea, M.; Schuster, D. μ Opioid receptor: Novel antagonists and structural modeling. Sci. Rep. 2016, 6, 21548. [Google Scholar] [CrossRef] [Green Version]
- Erli, F.; Guerrieri, E.; Ben Haddou, T.; Lantero, A.; Mairegger, M.; Schmidhammer, H.; Spetea, M. Highly Potent and selective new diphenethylamines interacting with the κ-opioid receptor: Synthesis, pharmacology, and structure-activity relationships. J. Med. Chem. 2017, 60, 7579–7590. [Google Scholar] [CrossRef]
- Dumitrascuta, M.; Bermudez, M.; Trovato, O.; De Neve, J.; Ballet, S.; Wolber, G.; Spetea, M. Antinociceptive efficacy of the µ-opioid/nociceptin peptide-based hybrid KGNOP1 in inflammatory pain without rewarding effects in mice: An experimental assessment and molecular docking. Molecules 2021, 26, 3267. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: San Diego, CA, USA; London, UK, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Fenalti, G.; Zatsepin, N.A.; Betti, C.; Giguere, P.; Han, G.W.; Ishchenko, A.; Liu, W.; Guillemyn, K.; Zhang, H.; James, D.; et al. Structural basis for bifunctional peptide recognition at human δ-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, Q.N.; Mahinthichaichan, P.; Shen, J.; Ellis, C.R. How μ-opioid receptor recognizes fentanyl. Nat. Commun. 2021, 12, 984. [Google Scholar] [CrossRef] [PubMed]
- Sydow, D. Dynophores: Novel Dynamic Pharmacophores Implementation of Pharmacophore Generation Based on Molecular Dynamics Trajectories and Their Graphical Representation; Freie Universität Berlin: Berlin, Germany, 2015. [Google Scholar]
- Avdeef, A.; Testa, B. Physicochemical profiling in drug research: A brief survey of the state-of-the-art of experimental techniques. Cell. Mol. Life Sci. 2002, 59, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Faller, B. Physicochemical profiling in early drug discovery: New challenges at the age of high-throughput screen and combinatorial chemistry. In Chemistry and Molecular Aspects of Drug Design and Action; Rekka, E.A., Kourounakis, P.N., Eds.; CRC Press: Boca Raton, FL, USA, 2008; pp. 303–312. [Google Scholar]
- Habgood, M.; Begley, D.; Abbott, N. Determinants of passive drug entry into the central nervous system. Cell. Mol. Neurobiol. 2000, 20, 231–253. [Google Scholar] [CrossRef]
- ACD/Percepta, Version 2021; Advanced Chemistry Development (ACD/Labs), Inc.: Toronto, ON, Canada, 2021. Available online: https://www.acdlabs.com/products/percepta/(accessed on 3 May 2022).
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), C.C.G.U., Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021. Available online: https://www.chemcomp.com/Products.htm (accessed on 24 January 2022).
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Zhu, S. Validation of the Generalized Force Fields GAFF, CGenFF, OPLS-AA, and PRODRGFF by Testing Against Experimental Osmotic Coefficient Data for Small Drug-Like Molecules. J. Chem. Inf. Model. 2019, 59, 4239–4247. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Vardy, E.; Mosier, P.D.; Frankowski, K.J.; Wu, H.; Katritch, V.; Westkaemper, R.B.; Aubé, J.; Stevens, R.C.; Roth, B.L. Chemotype-selective modes of action of κ-opioid receptor agonists. J. Biol. Chem. 2013, 288, 34470–34483. [Google Scholar] [CrossRef] [Green Version]
- Chavkin, C.; McLaughlin, J.P.; Celver, J.P. Regulation of opioid receptor function by chronic agonist exposure: Constitutive activity and desensitization. Mol. Pharmacol. 2001, 60, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [Green Version]
- Befort, K.; Zilliox, C.; Filliol, D.; Yue, S.; Kieffer, B.L. Constitutive activation of the delta opioid receptor by mutations in transmembrane domains III and VII. J. Biol. Chem. 1999, 274, 18574–18581. [Google Scholar] [CrossRef] [Green Version]
- Décaillot, F.M.; Befort, K.; Filliol, D.; Yue, S.; Walker, P.; Kieffer, B.L. Opioid receptor random mutagenesis reveals a mechanism for G protein-coupled receptor activation. Nat. Struct. Biol. 2003, 10, 629–636. [Google Scholar] [CrossRef]
- 3D Structure Generator CORINA Classic. Molecular Networks GmbH, Nuremberg, Germany. Available online: https://mn-am.com/products/corina/ (accessed on 3 February 2022).
- Gasteiger, J.; Rudolph, C.; Sadowski, J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comput. Methodol. 1990, 3, 537–547. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, A.; Hessler, G.; Matter, H.; Klabunde, T. Virtual screening of biogenic amine-binding G-protein coupled receptors: Comparative evaluation of protein- and ligand-based virtual screening protocols. J. Med. Chem. 2005, 48, 5448–5465. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94. J. Comput. Chem. 1996, 17, 553–586. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release-4: Maestro, Version 2020-4; Schrödinger LLC.: New York, NY, USA, 2020.
- Jorgensen, W.L.; Schyman, P. Treatment of Halogen Bonding in the OPLS-AA Force Field: Application to Potent Anti-HIV Agents. J. Chem. Theory Comput. 2012, 8, 3895–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponder, J.W.; Case, D.A. Force Fields for protein simulations. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 2003; Volume 66, pp. 27–85. [Google Scholar]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; Amici, M.d.; Lohse, M.J.; et al. Ligand binding ensembles determine graded agonist efficacies at a G protein-coupled receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Opioid Receptor Binding (Ki, µM) a | [35S]GTPγS Binding, KOR b | |||||
|---|---|---|---|---|---|---|
| KOR | MOR | DOR | EC50 (µM) | % stim. | Ke (µM) | |
| Compound A | 1.35 ± 0.32 | 10.7 ± 4.7 c | - d | - e | - e | 1.53 ± 0.38 |
| U69,593 | 0.0019 ± 0.0004 | n.d. | n.d. | 0.011 ± 0.004 | 100 | n.a. |
| Receptor | Interaction | Functional Group | Involved Residues |
|---|---|---|---|
| KOR | Ionic interaction | Morpholine nitrogen | D1383.32 |
| Halogen bond | Chlorine | N1222.67 | |
| Hydrophobic contacts | Chlorophenyl | V207ECL2 V1182.63 | |
| 2-Methylpyrrole | Y3127.35 | ||
| Phenyl moiety | I2906.51 I2946.55 I3167.39 | ||
| MOR | Ionic interaction | Morpholine nitrogen | D1493.32 |
| Hydrogen bond | Morpholine oxygen | N1523.35 | |
| Hydrophobic contacts | Chlorophenyl | T22045.51/ECL2 | |
| 2-Methylpyrrole | I3247.39 | ||
| Phenyl moiety | M1533.36 V3026.55 I3247.39 | ||
| DOR | Ionic interaction + hydrogen bond | Morpholine nitrogen | D1283.32 |
| Hydrogen bond | Morpholine oxygen | N1313.35 | |
| Hydrophobic contacts | Chlorophenyl | V197ECL2 | |
| 2-Methylpyrrol | V3047.39 | ||
| Phenyl moiety | Y1293.33 M1323.36 V2175.42 V2816.55 |
| Receptor | Interaction | Functional Group | Mean Frequency (n = 3) |
|---|---|---|---|
| KOR | Hydrophobic contacts | Chloride | 99.1% |
| Halogen bond | Chloride | 13.3% | |
| Hydrophobic contacts | Chlorophenyl | 98.4% | |
| MOR | Hydrophobic contacts | Chloride | 99.8% |
| Hydrophobic contacts | Chlorophenyl | 99.2% | |
| DOR | Hydrophobic contacts | Chloride | 100% |
| Hydrophobic contacts | Chlorophenyl | 83.2% |
| Ligand | clogP a | clogD7.4a |
|---|---|---|
| Compound A | 4.2 | 4.09 |
| nor-BNI | 3.13 | 1.55 |
| 5′-GNTI | 1.72 | −0.55 |
| JDTic | 3.43 | 1.78 |
| JNJ-67953964 | 4.97 | 3.24 |
| CYM-53003/BTRX-335140 | 3.82 | 2.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puls, K.; Olivé-Marti, A.-L.; Pach, S.; Pinter, B.; Erli, F.; Wolber, G.; Spetea, M. In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist. Pharmaceuticals 2022, 15, 680. https://doi.org/10.3390/ph15060680
Puls K, Olivé-Marti A-L, Pach S, Pinter B, Erli F, Wolber G, Spetea M. In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist. Pharmaceuticals. 2022; 15(6):680. https://doi.org/10.3390/ph15060680
Chicago/Turabian StylePuls, Kristina, Aina-Leonor Olivé-Marti, Szymon Pach, Birgit Pinter, Filippo Erli, Gerhard Wolber, and Mariana Spetea. 2022. "In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist" Pharmaceuticals 15, no. 6: 680. https://doi.org/10.3390/ph15060680
APA StylePuls, K., Olivé-Marti, A.-L., Pach, S., Pinter, B., Erli, F., Wolber, G., & Spetea, M. (2022). In Vitro, In Vivo and In Silico Characterization of a Novel Kappa-Opioid Receptor Antagonist. Pharmaceuticals, 15(6), 680. https://doi.org/10.3390/ph15060680