
Accurate Mass Identification of an Interfering Water Adduct and Strategies in Development and Validation of an LC-MS/MS Method for Quantification of MPI8, a Potent SARS-CoV-2 Main Protease Inhibitor, in Rat Plasma in Pharmacokinetic Studies


 ,
, 
Abstract
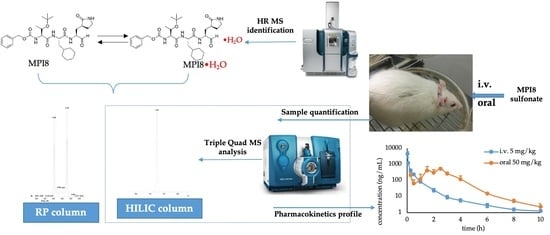
1. Introduction
2. Results and Discussion
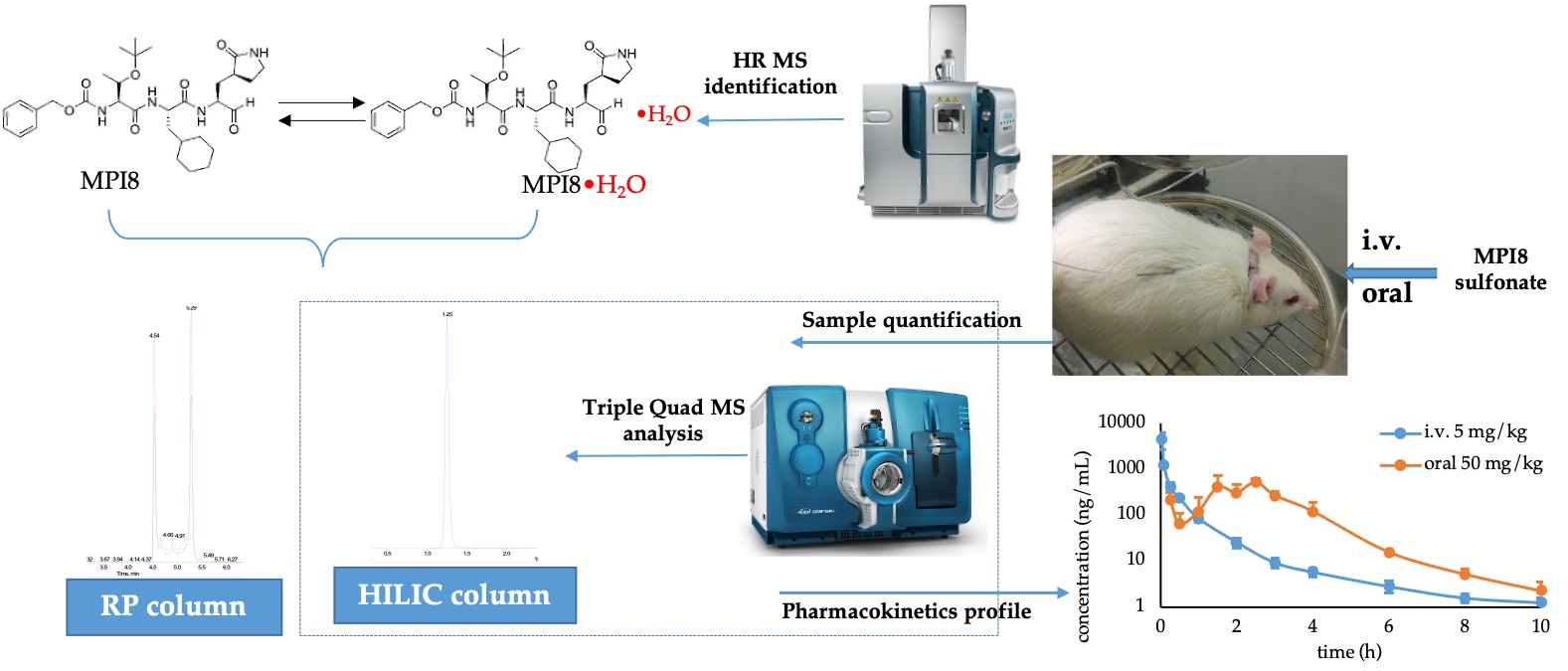
2.1. Identification of MPI8 Water Adduct (MPI8•H2O) in Rat Plasma
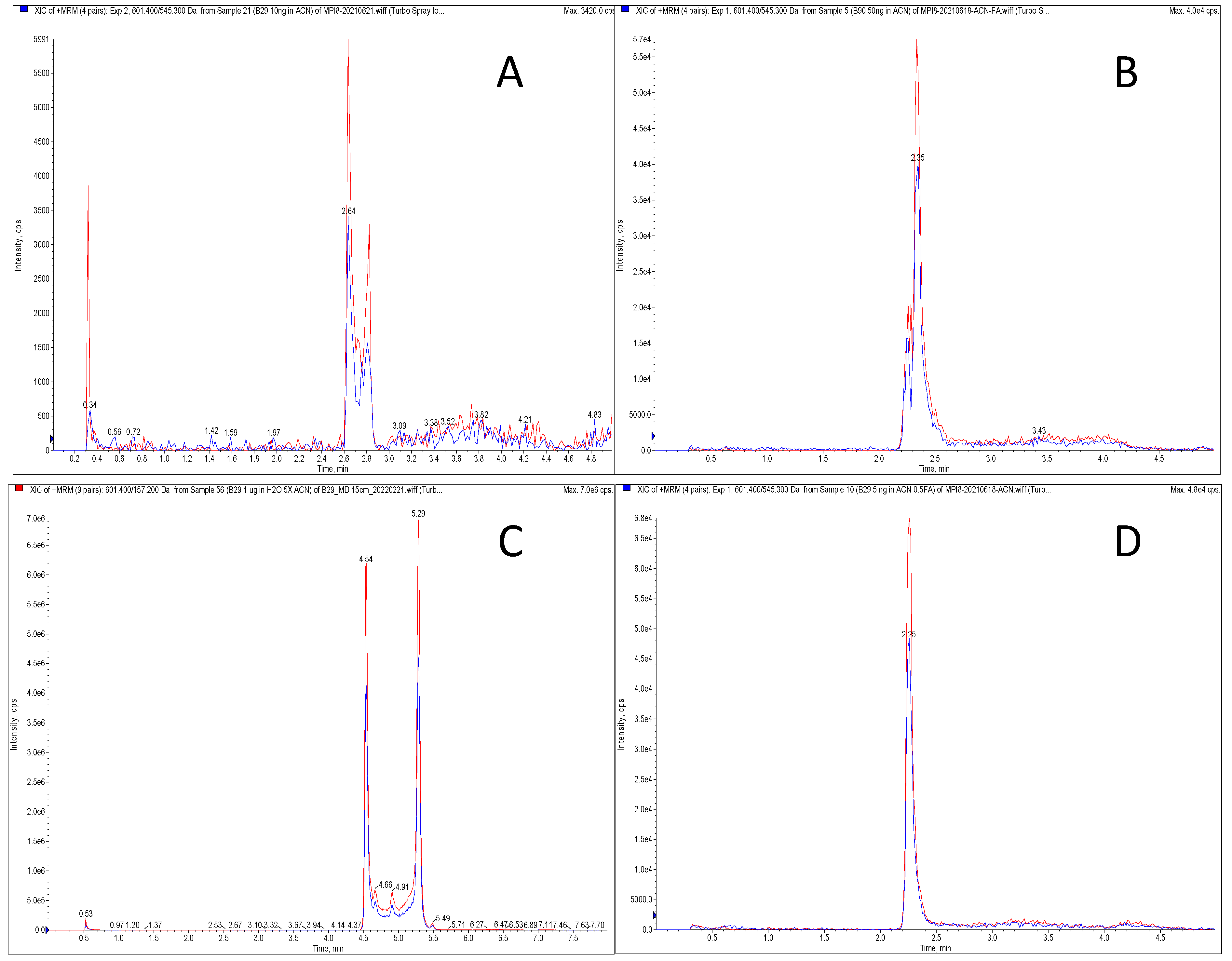
2.2. Method Development
2.3. Method Validation
2.4. Pharmacokinetic Study
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Identification of MPI8•H2O in Rat Plasma
3.3. MPI8 Quantification and Method Validation
3.3.1. Analytical Conditions
3.3.2. Preparation of Standard and QC Samples
3.3.3. Sample Processing
3.3.4. Method Validation
3.4. Animal Pharmacokinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; Nahed, A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B. 1.1. 529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.; Borella, E.W.; Smith, M.R. Monoclonal antibody therapy for COVID-19: A public health perspective from Arkansas. Open Forum Infect. Dis. 2022, 9, ofab602. [Google Scholar] [CrossRef]
- Dale, A.P.; Hudson, M.J.; Armenta, D.; Friebus, H.; Ellingson, K.D.; Davis, K.; Cullen, T.; Brady, S.; Komatsu, K.K.; Stone, N.D. Clinical outcomes of monoclonal antibody therapy during a COVID-19 outbreak in a skilled nursing facility—Arizona, 2021. J. Am. Geriatr. Soc. 2022, 70, 960–967. [Google Scholar] [CrossRef]
- Li, D.; Sempowski, G.D.; Saunders, K.O.; Acharya, P.; Haynes, B.F. SARS-CoV-2 Neutralizing Antibodies for COVID-19 Prevention and Treatment. Annu. Rev. Med. 2022, 73, 1–16. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Depcinski, S.; Sharma, M. Molnupiravir and Nirmatrelvir-Ritonavir: Oral COVID antiviral drugs. Clin. Infect. Dis. 2022, ciac180, accepted. [Google Scholar] [CrossRef]
- Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-additional-phase-23-study-results (accessed on 14 December 2021).
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the past: Possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Ma, X.R.; Ma, Y.; Alugubelli, Y.R.; Scott, D.A.; Vatansever, E.C.; Drelich, A.K.; Sankaran, B.; Geng, Z.Z.; Blankenship, L.R.; et al. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors. ChemMedChem 2021, 16, 942–948. [Google Scholar] [CrossRef]
- Ma, X.R.; Alugubelli, Y.R.; Ma, Y.; Vatansever, E.C.; Scott, D.A.; Qiao, Y.; Yu, G.; Xu, S.; Liu, W.R. MPI8 is potent against SARS-CoV-2 by inhibiting dually and selectively the SARS-CoV-2 main protease and the host cathepsin L. ChemMedChem 2022, 17, e202100456. [Google Scholar] [CrossRef]
- Cao, W.; Cho, C.-C.D.; Geng, Z.Z.; Shaabani, N.; Ma, X.R.; Vatansever, E.C.; Alugubelli, Y.R.; Ma, Y.; Chaki, S.P.; Ellenburg, W.H.; et al. Evaluation of SARS-CoV-2 main protease inhibitors using a novel cell-based assay. ACS Cent. Sci. 2022, 8, 192–204. [Google Scholar] [CrossRef]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef]
- Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar]
- Abranko, L.; Garcia-Reyes, J.F.; Molina-Diaz, A. In-source fragmentation and accurate mass analysis of multiclass flavonoid conjugates by electrospray ionization time-of-flight mass spectrometry. J. Mass Spectrom. 2011, 46, 478–488. [Google Scholar] [CrossRef]
- Zhai, H.; Zhang, X. A new method for differentiating adducts of common drinking water DBPs from higher molecular weight DBPs in electrospray ionization-mass spectrometry analysis. Water Res. 2009, 43, 2093–2100. [Google Scholar] [CrossRef]
- Tuytten, R.; Lemiere, F.; Esmans, E.L.; Herrebout, W.A.; van der Veken, B.J.; Dudley, E.; Newton, R.P.; Witters, E. In-source CID of guanosine: Gas phase ion-molecule reactions. J. Am. Soc. Mass Spectrom. 2006, 17, 1050–1062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Couchman, L.; Morgan, P.E. LC-MS in analytical toxicology: Some practical considerations. Biomed. Chromatogr. 2011, 25, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Dziadosz, M.; Klintschar, M.; Teske, J. Drug detection by tandem mass spectrometry on the basis of adduct formation. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 955–956, 108–109. [Google Scholar] [CrossRef]
- Li, X.-F.; Ma, M.; Scherban, K.; Tam, Y.K. Liquid chromatography-electrospray mass spectrometric studies of ginkgolides and bilobalide using simultaneous monitoring of proton, ammonium and sodium adducts. Analyst 2002, 127, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Dziadosz, M. Applicability of adduct detection in liquid chromatography–tandem mass spectrometry. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1671–1674. [Google Scholar] [CrossRef]
- Otoki, Y.; Nakagawa, K.; Kato, S.; Miyazawa, T. MS/MS and LC-MS/MS analysis of choline/ethanolamine plasmalogens via promotion of alkali metal adduct formation. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 1004, 85–92. [Google Scholar] [CrossRef]
- Dziadosz, M. Direct analysis of ethylene glycol in human serum on the basis of analyte adduct formation and liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1072, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.X.; Yang, J.Z.; Wei, W.; Liu, Y.F.; Zhang, S.S. Simultaneous determination of 13 aminoglycoside residues in foods of animal origin by liquid chromatography-electrospray ionization tandem mass spectrometry with two consecutive solid-phase extraction steps. J. Chromatogr. A 2008, 1207, 29–37. [Google Scholar] [CrossRef]
- Tuytten, R.; Lemiere, F.; Van Dongen, W.; Esmans, E.L.; Witters, E.; Herrebout, W.; Van Der Veken, B.; Dudley, E.; Newton, R.P. Intriguing mass spectrometric behavior of guanosine under low energy collision-induced dissociation: H2O adduct formation and gas-phase reactions in the collision cell. J. Am. Soc. Mass Spectrom. 2005, 16, 1291–1304. [Google Scholar] [CrossRef][Green Version]
- Millan, S.; Sampedro, M.C.; Unceta, N.; Goicolea, M.A.; Barrio, R.J. Simple and rapid determination of biogenic amines in wine by liquid chromatography-electrospray ionization ion trap mass spectrometry. Anal. Chim. Acta 2007, 584, 145–152. [Google Scholar] [CrossRef]
- Sultan, J. Collision induced dissociation of deprotonated guanine: Fragmentation of pyrimidine ring and water adduct formation. Int. J. Mass Spectrom. 2008, 273, 58–68. [Google Scholar] [CrossRef]
- Beuck, S.; Schwabe, T.; Grimme, S.; Schlorer, N.; Kamber, M.; Schanzer, W.; Thevis, M. Unusual mass spectrometric dissociation pathway of protonated isoquinoline-3-carboxamides due to multiple reversible water adduct formation in the gas phase. J. Am. Soc. Mass Spectrom. 2009, 20, 2034–2048. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trabelsi, H.; Bouabdallah, S.; Sabbah, S.; Raouafi, F.; Bouzouita, K. Study of the cis–trans isomerization of enalapril by reversed-phase liquid chromatography. J. Chromatogr. A 2000, 871, 189–199. [Google Scholar] [CrossRef]
- Bouabdallah, S.; Trabelsi, H.; Bouzouita, K.; Sabbah, S. Reversed-phase liquid chromatography of lisinopril conformers. J. Biochem. Biophys. Meth. 2002, 54, 391–405. [Google Scholar] [CrossRef]
- Tsakalof, A.; Bairachtari, K.; Georgarakis, M. Development of a liquid chromatography–mass spectrometry method for monitoring the angiotensin-converting enzyme inhibitor lisinopril in serum. J. Chromatogr. B 2003, 783, 425–432. [Google Scholar] [CrossRef]
- Vancea, S.; Imre, S.; Donath-Nagy, G.; Bela, T.; Nyulas, M.; Muntean, T.; Borka-Balas, R. Determination of free captopril in human plasma by liquid chromatography with mass spectrometry detection. Talanta 2009, 79, 436–441. [Google Scholar] [CrossRef]
- Kawano, S.-I.; Inohana, Y.; Hashi, Y.; Lin, J.-M. Analysis of keto-enol tautomers of curcumin by liquid chromatography/mass spectrometry. Chin. Chem. Lett. 2013, 24, 685–687. [Google Scholar] [CrossRef]
- Broglé, K.; Ornaf, R.M.; Wu, D.; Palermo, P.J. Peak fronting in reversed-phase high-performance liquid chromatography: A study of the chromatographic behavior of oxycodone hydrochloride. J. Pharm. Biomed. Anal. 1999, 19, 669–678. [Google Scholar] [CrossRef]
- LoBrutto, R.; Bereznitski, Y.; Novak, T.J.; DiMichele, L.; Pan, L.; Journet, M.; Kowal, J.; Grinberg, N. Kinetic analysis and subambient temperature on-line on-column derivatization of an active aldehyde. J. Chromatogr. A 2003, 995, 67–78. [Google Scholar] [CrossRef]
- Inaba, S. Theoretical study of decomposition of methanediol in aqueous solution. J. Phys. Chem. A 2015, 119, 5816–5825. [Google Scholar] [CrossRef]
- Matubayasi, N.; Morooka, S.; Nakahara, M.; Takahashi, H. Chemical equilibrium of formaldehyde and methanediol in hot water: Free-energy analysis of the solvent effect. J. Mol. Liq. 2007, 134, 58–63. [Google Scholar] [CrossRef]
- De Bruyn, M.; Budarin, V.L.; Misefari, A.; Shimizu, S.; Fish, H.; Cockett, M.; Hunt, A.J.; Hofstetter, H.; Weckhuysen, B.M.; Clark, J.H.; et al. Geminal Diol of Dihydrolevoglucosenone as a Switchable Hydrotrope: A Continuum of Green Nanostructured Solvents. ACS Sustain. Chem. Eng. 2019, 7, 7878–7883. [Google Scholar] [CrossRef] [PubMed]
- Raschke, T.M. Water structure and interactions with protein surfaces. Curr. Opin. Struct. Boil. 2006, 16, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, K.; Qiu, X.; Wang, C. Visualizing cyclic peptide hydration at the single-molecule level. Sci. Rep. 2013, 3, 2461. [Google Scholar] [CrossRef]
- Gregurick, S.K.; Fredj, E.; Elber, R.; Gerber, R.B. Vibrational spectroscopy of peptides and peptide−water complexes: Anharmonic coupled-mode calculations. J. Phys. Chem. B 1997, 101, 8595–8606. [Google Scholar] [CrossRef]
- Nemukhin, A.V.; Grigorenko, B.L.; Bochenkova, A.V.; Kovba, V.M.; Epifanovsky, E.M. Structures of the peptide–water complexes studied by the hybrid quantum mechanical—molecular mechanical (QM/MM) technique. Struct. Chem. 2004, 15, 3–9. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry; US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Washington, DC, USA, 2018; pp. 1–41. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
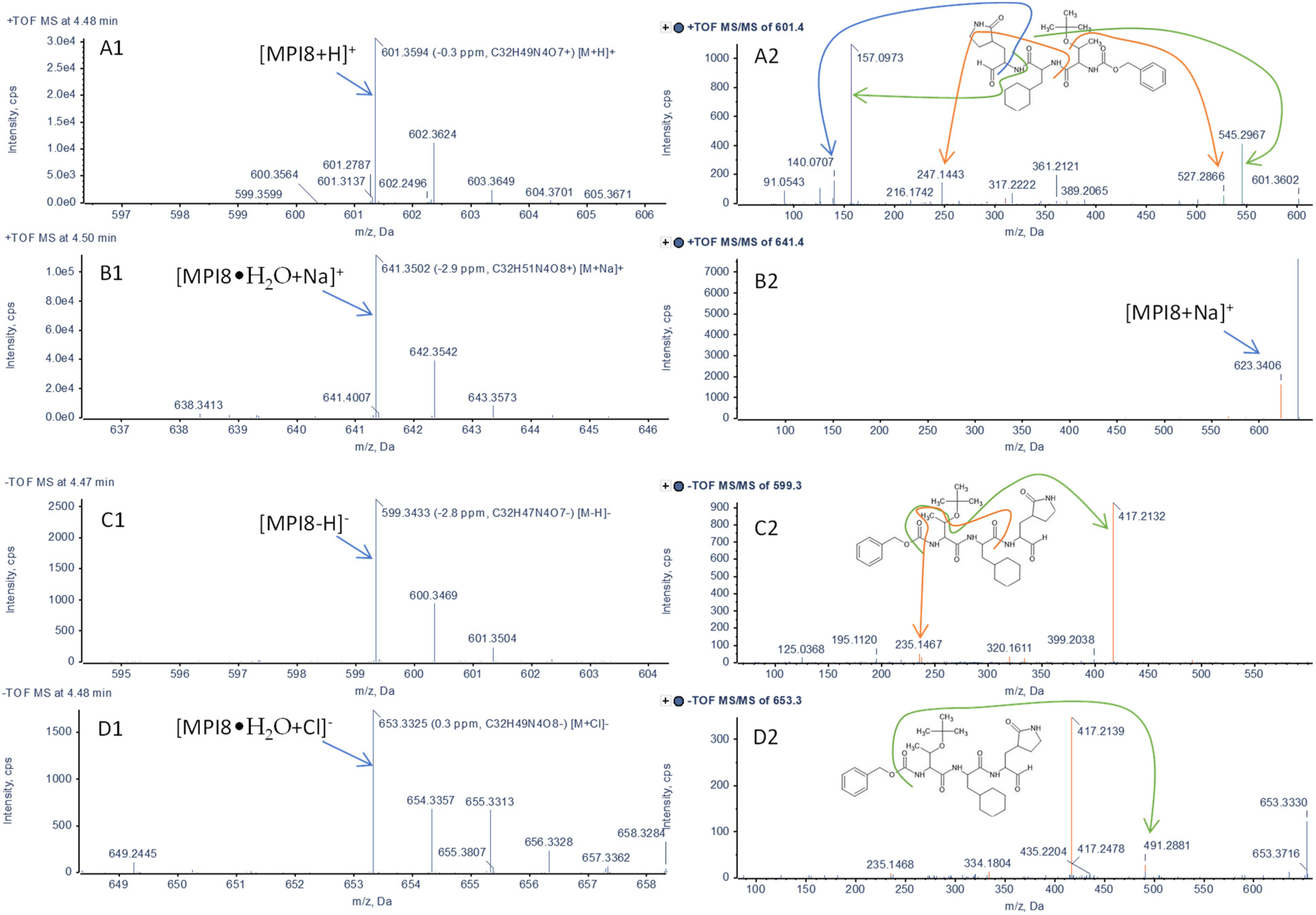{kind=link}
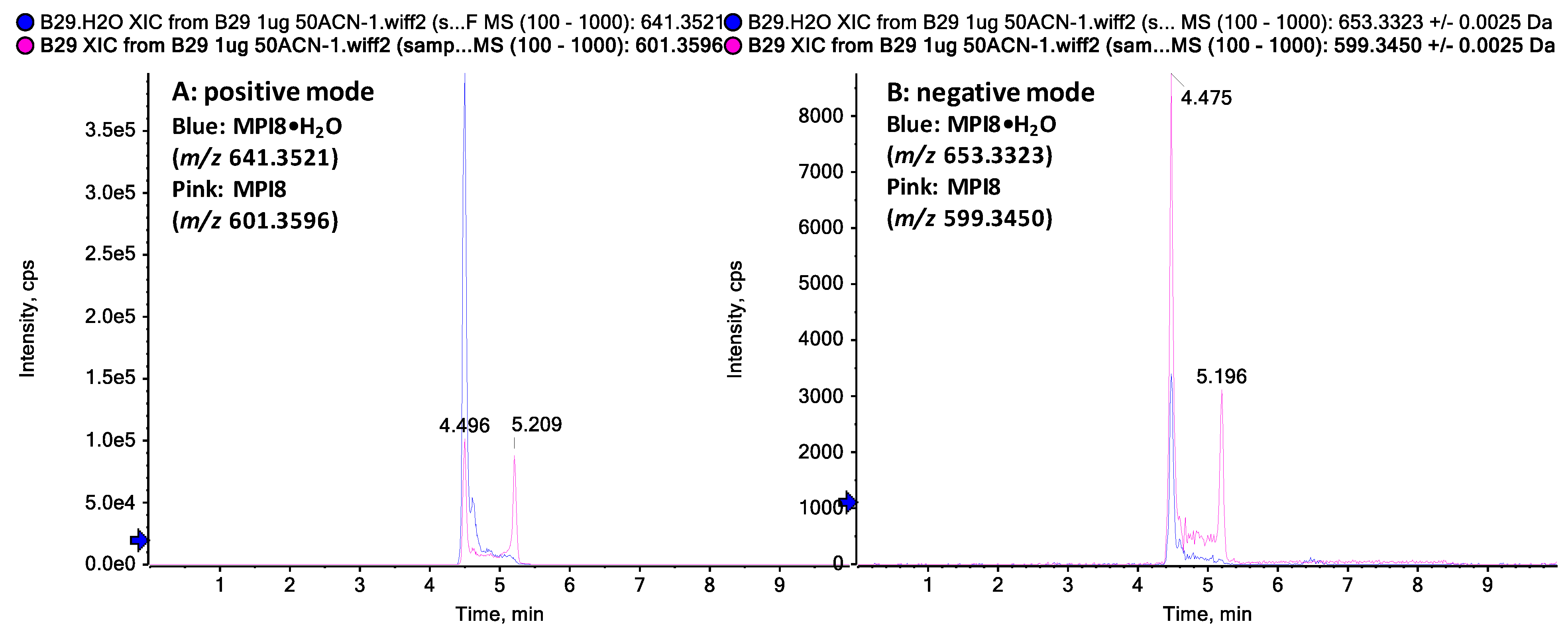{kind=link}
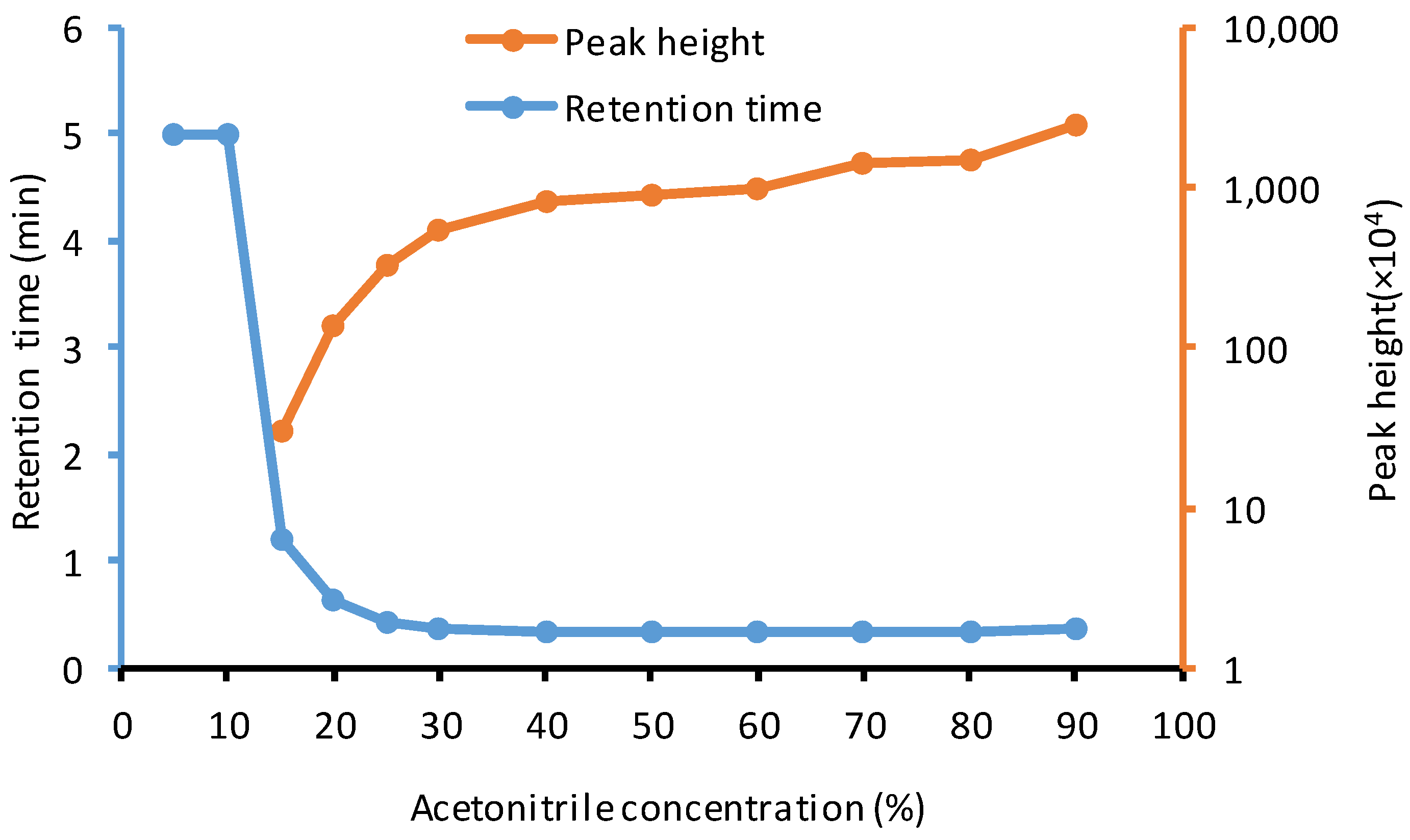{kind=link}
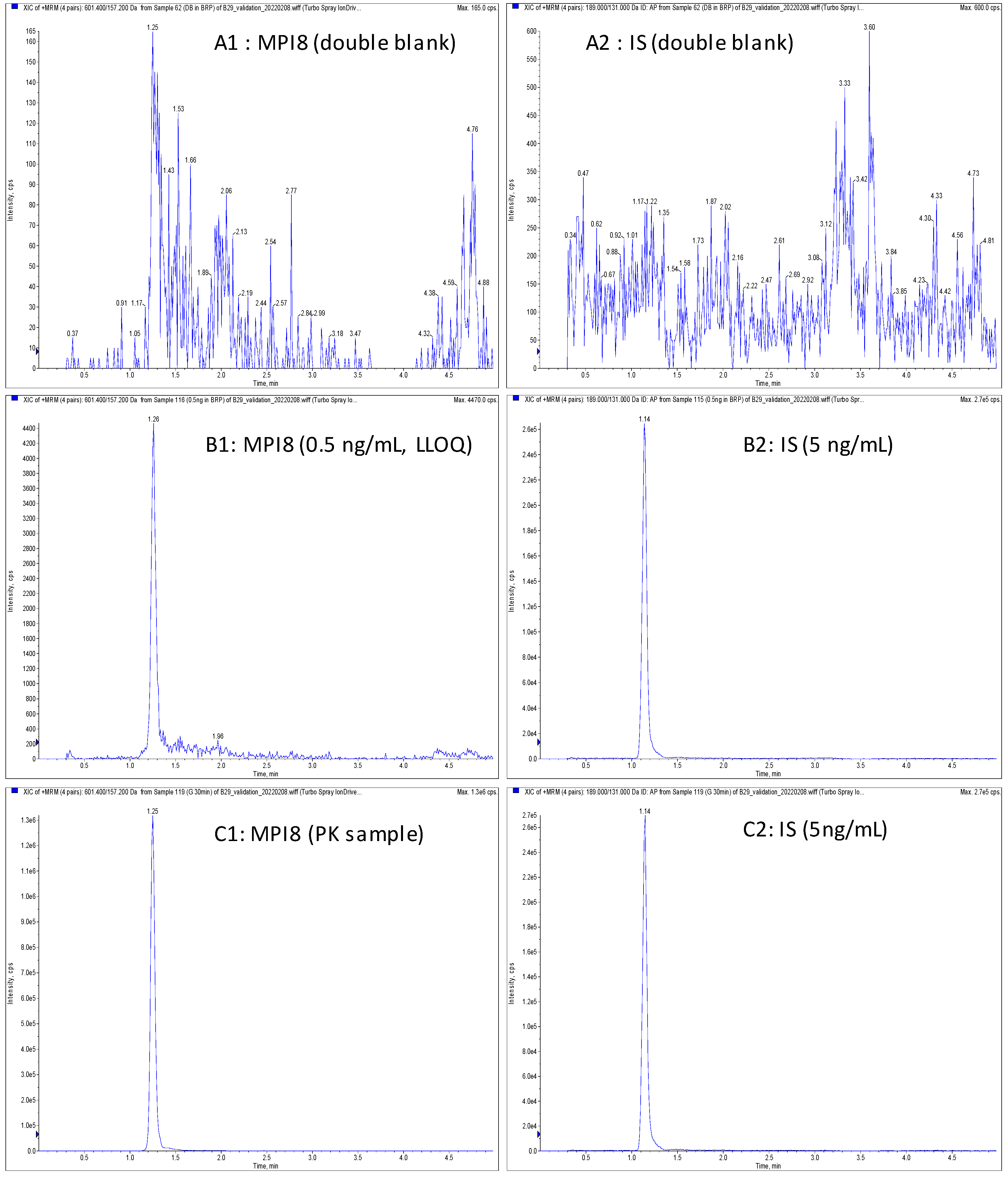{kind=link}
{kind=link}
| Q1 (m/z) (Da) | Q3 (m/z) (Da) | DP (v) | EP (v) | CE (v) | CXP (v) | |
|---|---|---|---|---|---|---|
| MPI8 | 601.4 | 157.2 * | 60 | 5 | 30 | 5 |
| 545.3 | 60 | 5 | 20 | 5 | ||
| AP (IS) | 189 | 131 | 70 | 5 | 28 | 5 |
| Nominal Concentration (ng/mL) | Intra-Day (n = 6) | Inter-Day (n = 18) | ||||
|---|---|---|---|---|---|---|
| Observed Concentration (Mean ± SD) | Accuracy (RE%) | Precision (CV%) | Observed Concentration (Mean ± SD) | Accuracy (RE%) | Precision (CV%) | |
| 0.5 (LLOQ) | 0.44 ± 0.04 | −12.03 | 10.10 | 0.46 ± 0.06 | −8.44 | 12.03 |
| 1.5 (LQC) | 1.48 ± 0.15 | −1.44 | 9.92 | 1.51 ± 0.16 | 0.93 | 10.53 |
| 25 (MQC) | 26.88 ± 2.07 | 7.53 | 7.69 | 25.54 ± 3.07 | 2.16 | 12.01 |
| 400 (HQC) | 378.17 ± 28.05 | −5.46 | 7.42 | 393.61 ± 52.43 | −1.60 | 13.32 |
| Nominal Concentration (ng/mL) | Matrix Effect (%) (n = 4) | Recovery (%) (n = 4) | |
|---|---|---|---|
| MPI8 | 1.5 | 135.92 ± 8.87 | 79.88 ± 5.66 |
| 25 | 138.43 ± 8.45 | 70.92 ± 3.76 | |
| 400 | 142.78 ± 3.48 | 68.22 ± 2.22 | |
| AP (IS) | 5 | 94.54 ± 6.55 | 97.62 ± 6.12 |
| Stability Test | Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) (n = 6) | ||
|---|---|---|---|---|
| Mean ± SD | (RE%) | (CV%) | ||
| Auto-sampler (10 °C 24 h) | 1.5 | 1.44 ± 0.11 | −4.33 | 7.81 |
| 25 | 26.8 ± 1.91 | 7.20 | 7.14 | |
| 400 | 458.5 ± 47.18 | 14.63 | 10.29 | |
| Bench top (4 h, 4 °C) | 1.5 | 1.35 ± 0.11 | −9.87 | 8.18 |
| 25 | 21.3 ± 0.12 | −14.80 | 0.57 | |
| 400 | 340.25 ± 14.03 | −14.94 | 4.12 | |
| Freeze and thaw (−80 °C to 4 °C, 3 cycles) | 1.5 | 1.33 ± 0.08 | −11.17 | 6.05 |
| 25 | 23.5 ± 1.41 | −6.00 | 6.02 | |
| 400 | 362.2 ± 11.56 | −9.45 | 3.19 | |
| Long-term (−80 °C, 20 days) | 1.5 | 1.44 ± 0.19 | −4.22 | 12.96 |
| 25 | 25.18 ± 1.37 | 5.43 | 0.73 | |
| 400 | 399 ± 17.61 | −0.25 | 4.41 | |
| Parameters | Mean ± SD | |
|---|---|---|
| i.v. (5 mg/kg) (n = 3) | Oral (50 mg/kg) (n = 3) | |
| Cmax | - | 600.67 ± 75.04 |
| Tmax | - | 2.17 ± 0.58 |
| AUC0−10 h (h∙ng/mL) | 613.06 ± 15.40 | 1177.26 ± 290.10 |
| AUC0−inf (h∙ng/mL) | 617.84 ± 15.68 | 1181.62 ± 287.53 |
| T1/2 (h) | 2.75 ± 0.53 | 1.22 ± 0.46 |
| CL/F (mL/h/kg) | 6606.93 ± 213.82 | 43,984.84 ± 10,389.13 |
| Vz/F (ml/kg) | 26,324.66 ± 5882.81 | 81,570.63 ± 48,751.79 |
| MRT (h) | 0.54 ± 0.10 | 2.52 ± 0.16 |
| F (%) | - | 15.76 ± 4.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Xie, H.; Alugubelli, Y.R.; Ma, Y.; Xu, S.; Ma, J.; Liu, W.R.; Liang, D. Accurate Mass Identification of an Interfering Water Adduct and Strategies in Development and Validation of an LC-MS/MS Method for Quantification of MPI8, a Potent SARS-CoV-2 Main Protease Inhibitor, in Rat Plasma in Pharmacokinetic Studies. Pharmaceuticals 2022, 15, 676. https://doi.org/10.3390/ph15060676
Wang Y, Xie H, Alugubelli YR, Ma Y, Xu S, Ma J, Liu WR, Liang D. Accurate Mass Identification of an Interfering Water Adduct and Strategies in Development and Validation of an LC-MS/MS Method for Quantification of MPI8, a Potent SARS-CoV-2 Main Protease Inhibitor, in Rat Plasma in Pharmacokinetic Studies. Pharmaceuticals. 2022; 15(6):676. https://doi.org/10.3390/ph15060676
Chicago/Turabian StyleWang, Yang, Huan Xie, Yugendar R. Alugubelli, Yuying Ma, Shiqing Xu, Jing Ma, Wenshe R. Liu, and Dong Liang. 2022. "Accurate Mass Identification of an Interfering Water Adduct and Strategies in Development and Validation of an LC-MS/MS Method for Quantification of MPI8, a Potent SARS-CoV-2 Main Protease Inhibitor, in Rat Plasma in Pharmacokinetic Studies" Pharmaceuticals 15, no. 6: 676. https://doi.org/10.3390/ph15060676
APA StyleWang, Y., Xie, H., Alugubelli, Y. R., Ma, Y., Xu, S., Ma, J., Liu, W. R., & Liang, D. (2022). Accurate Mass Identification of an Interfering Water Adduct and Strategies in Development and Validation of an LC-MS/MS Method for Quantification of MPI8, a Potent SARS-CoV-2 Main Protease Inhibitor, in Rat Plasma in Pharmacokinetic Studies. Pharmaceuticals, 15(6), 676. https://doi.org/10.3390/ph15060676