
Effect of Platelet-Derived Growth Factor C on Mitochondrial Oxidative Stress Induced by High d-Glucose in Human Aortic Endothelial Cells

 , , , ,
, , , ,
Abstract
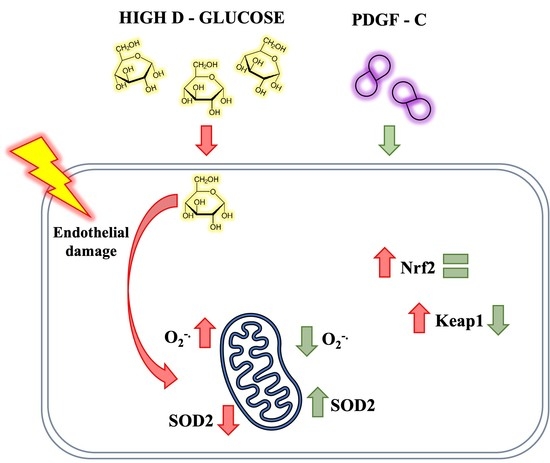
:
1. Introduction
2. Results
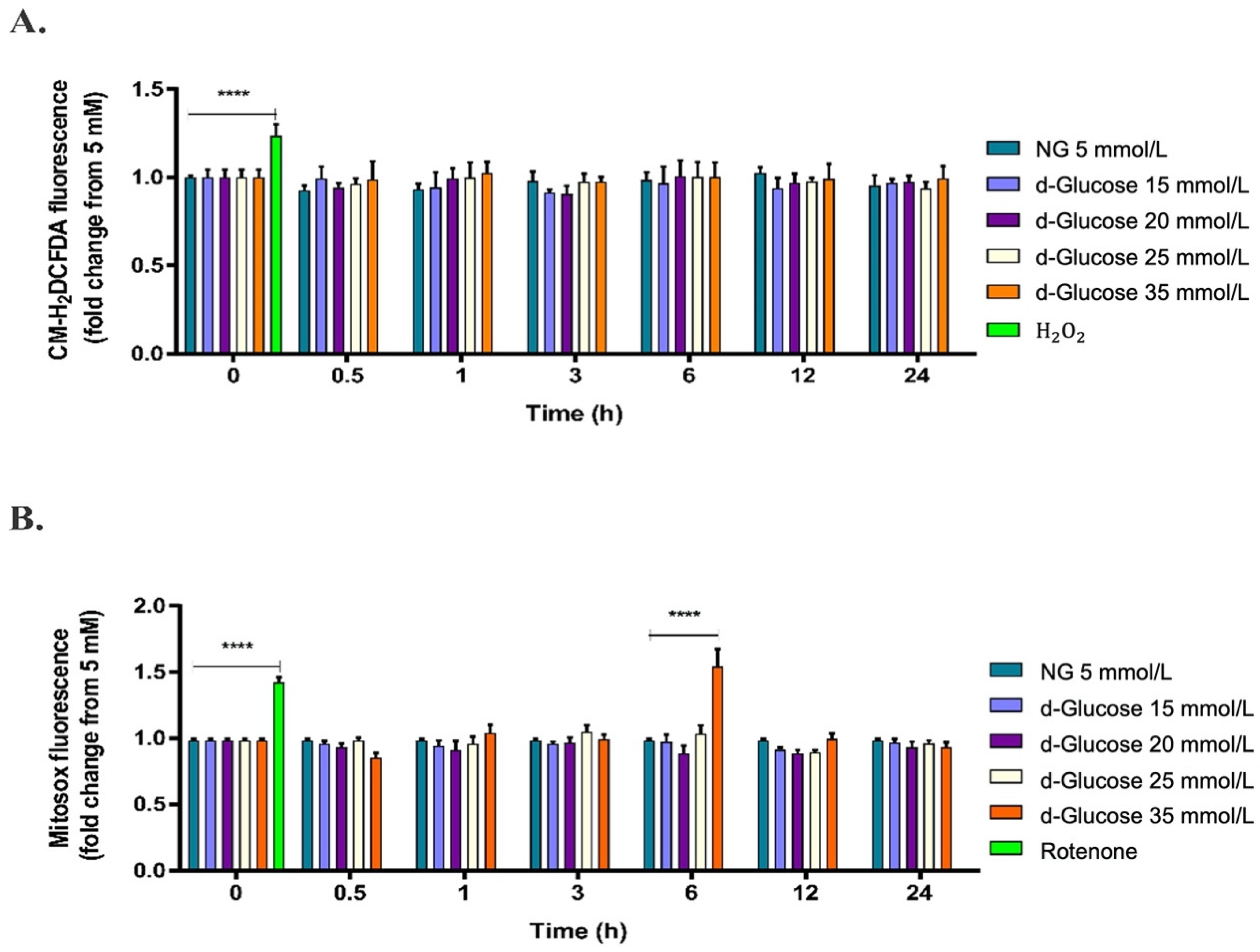
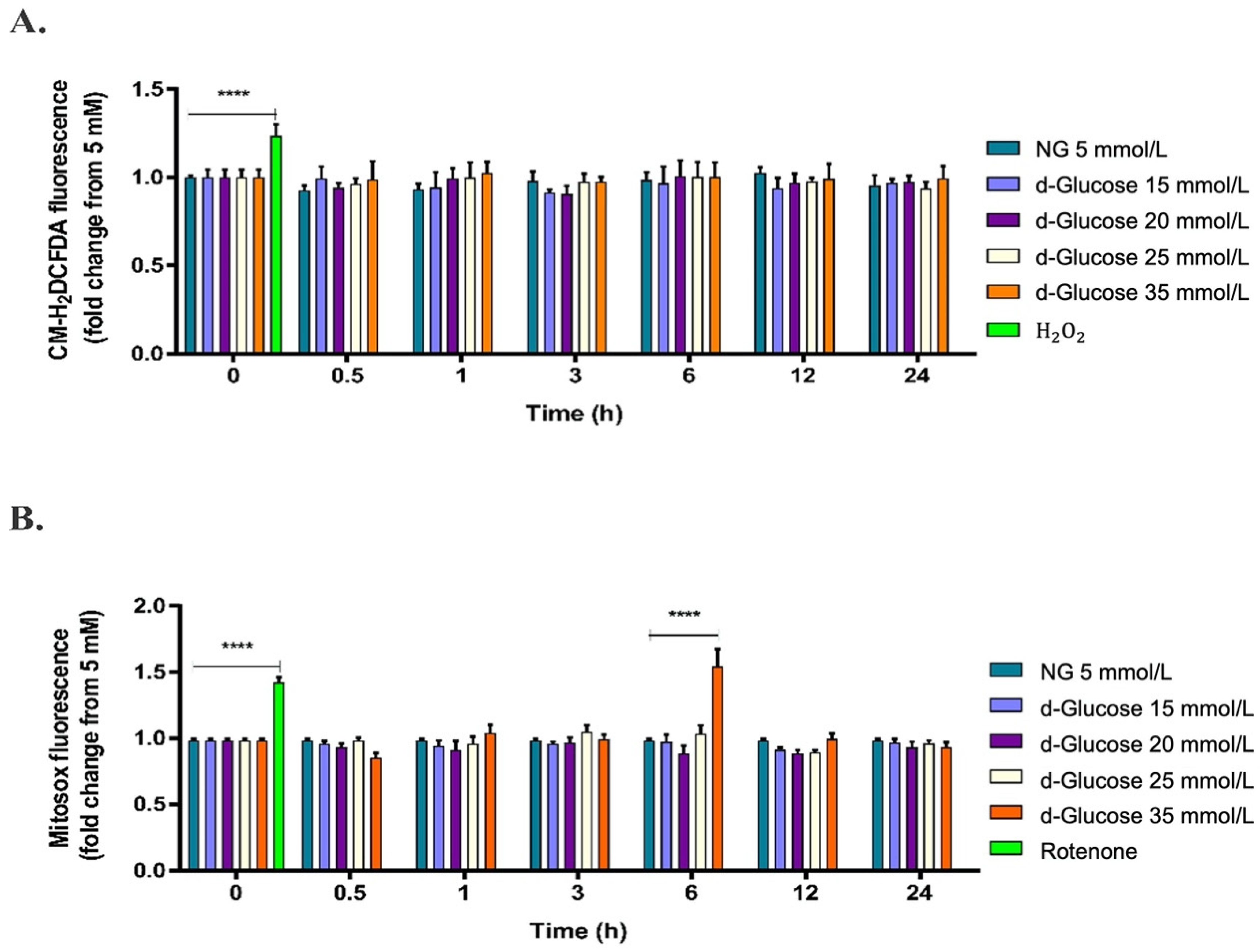
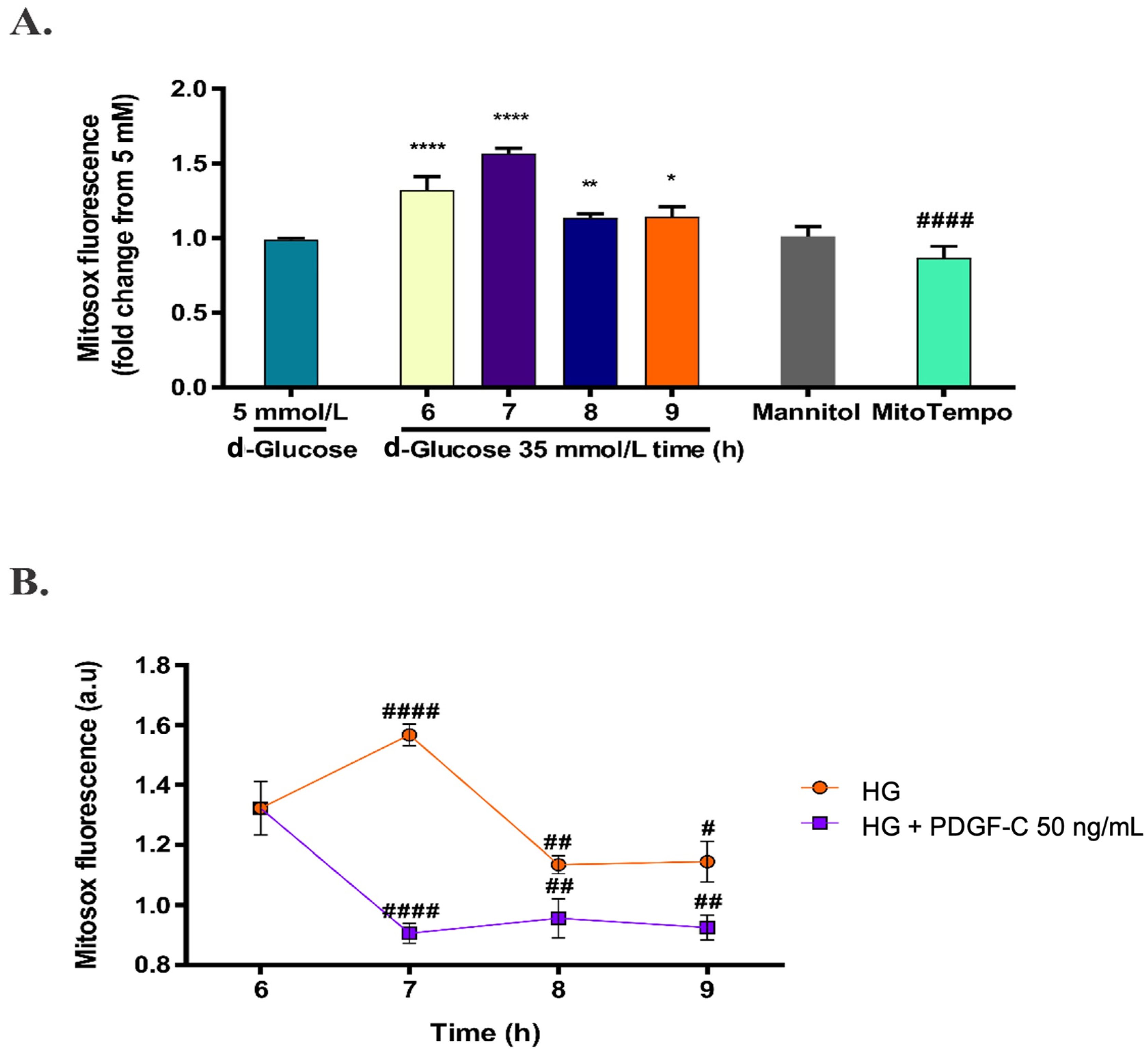
2.1. High d-Glucose Induces Mitochondrial ROS Production in HAECs
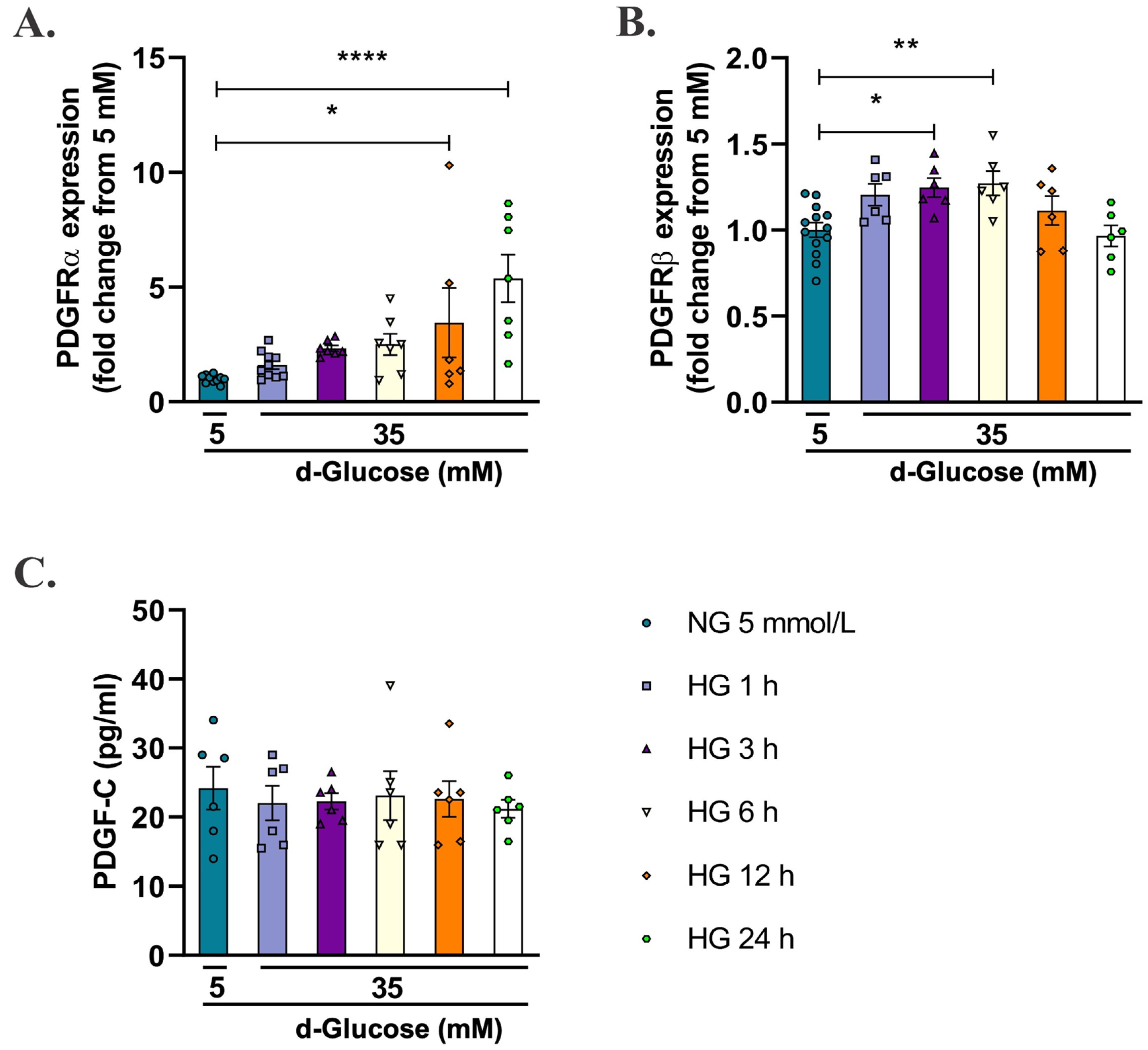
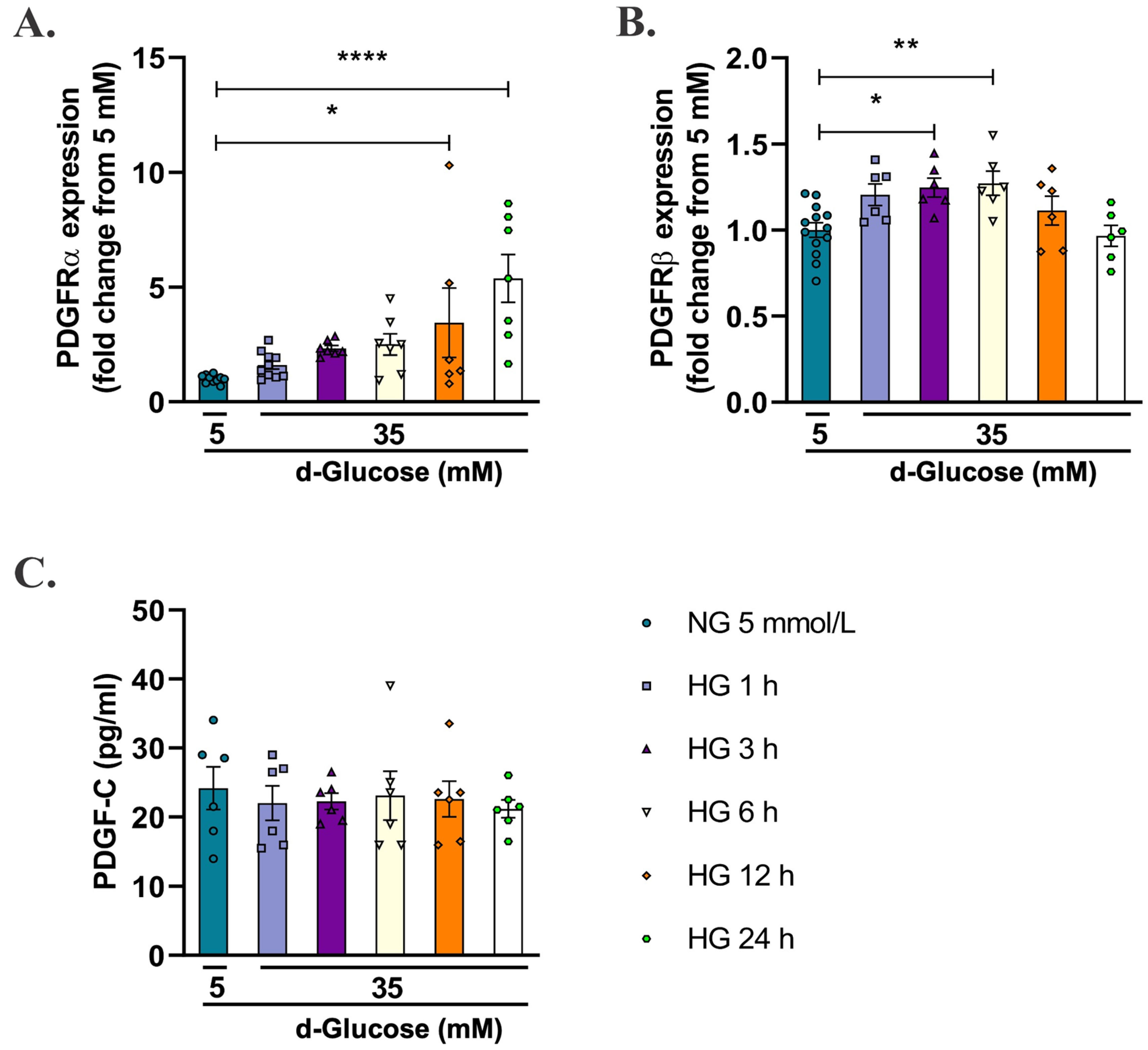
2.2. High d-Glucose Increases PDGFRα and PDGFRβ Receptors, but Does Not Induces Changes in PDGF-C in HAECs
2.3. PDGF-C Reduces the Increase of Mitochondrial ROS Production Induced by High d-Glucose
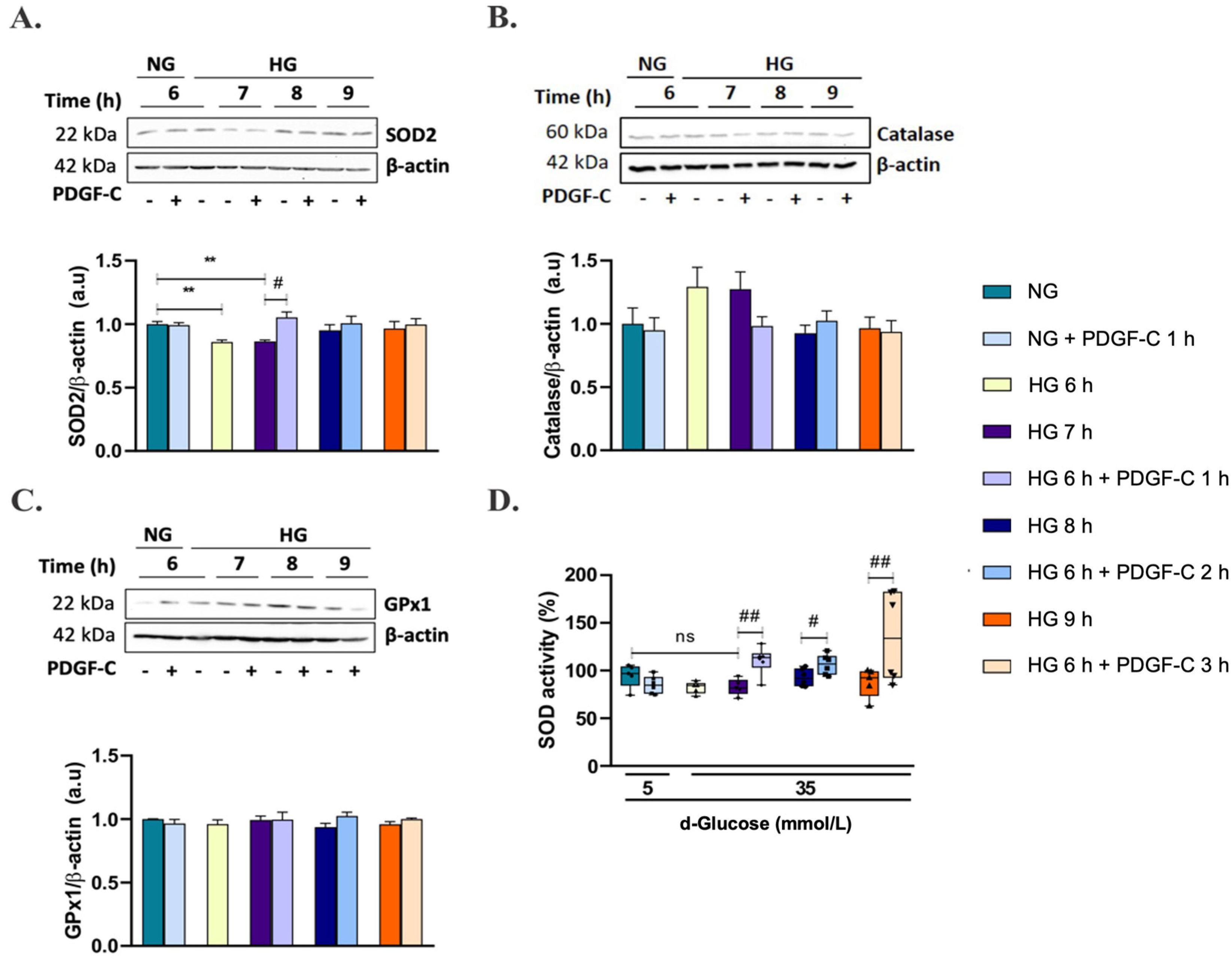
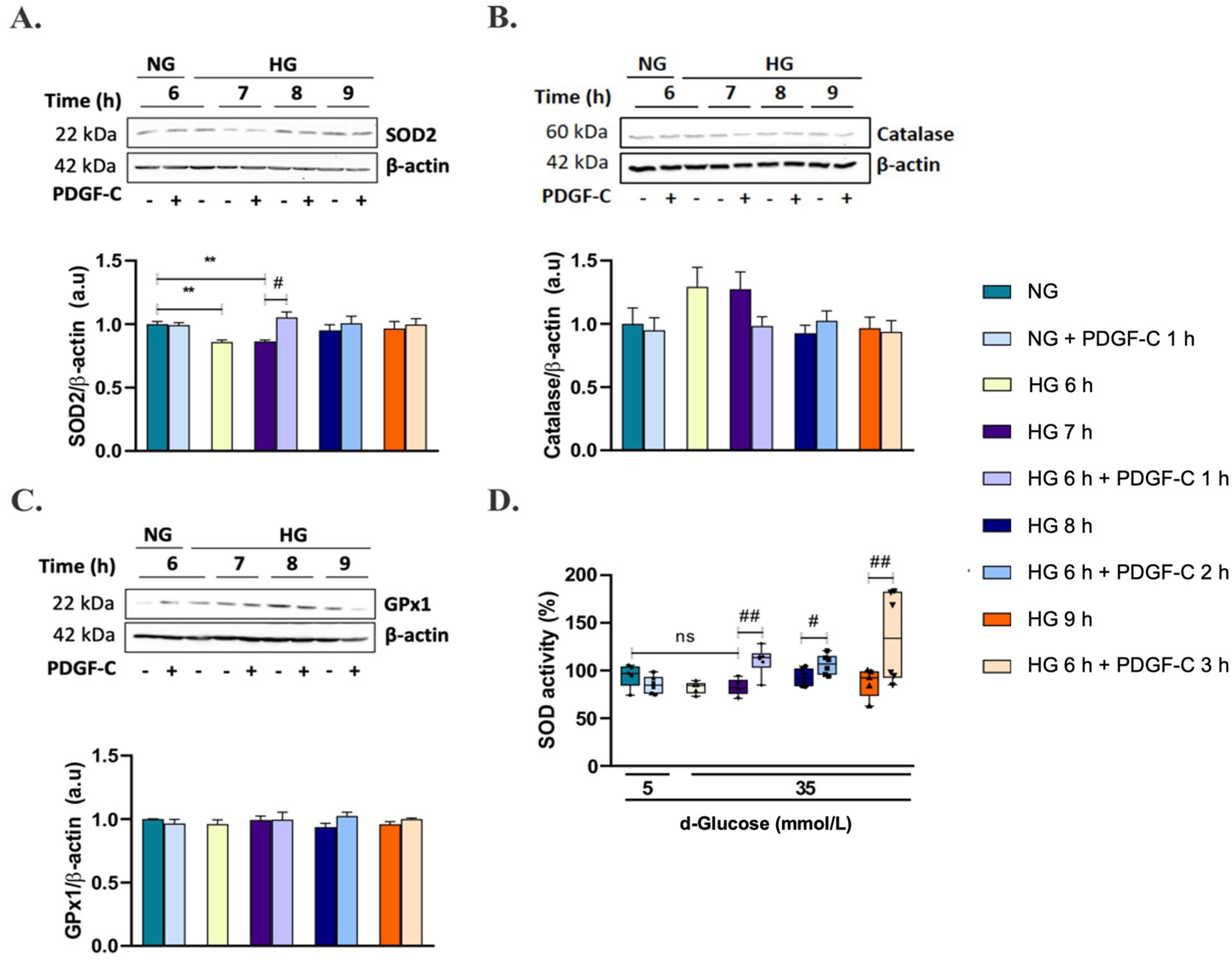
2.4. PDGF-C Up-Regulates SOD2 Antioxidant Enzyme Expression and SOD Activity in HAECs Going on High d-Glucose
2.5. PDGF-C Up-Regulates SOD2 Antioxidant Enzyme Expression and SOD Activity in HAECs Going on High d-Glucose
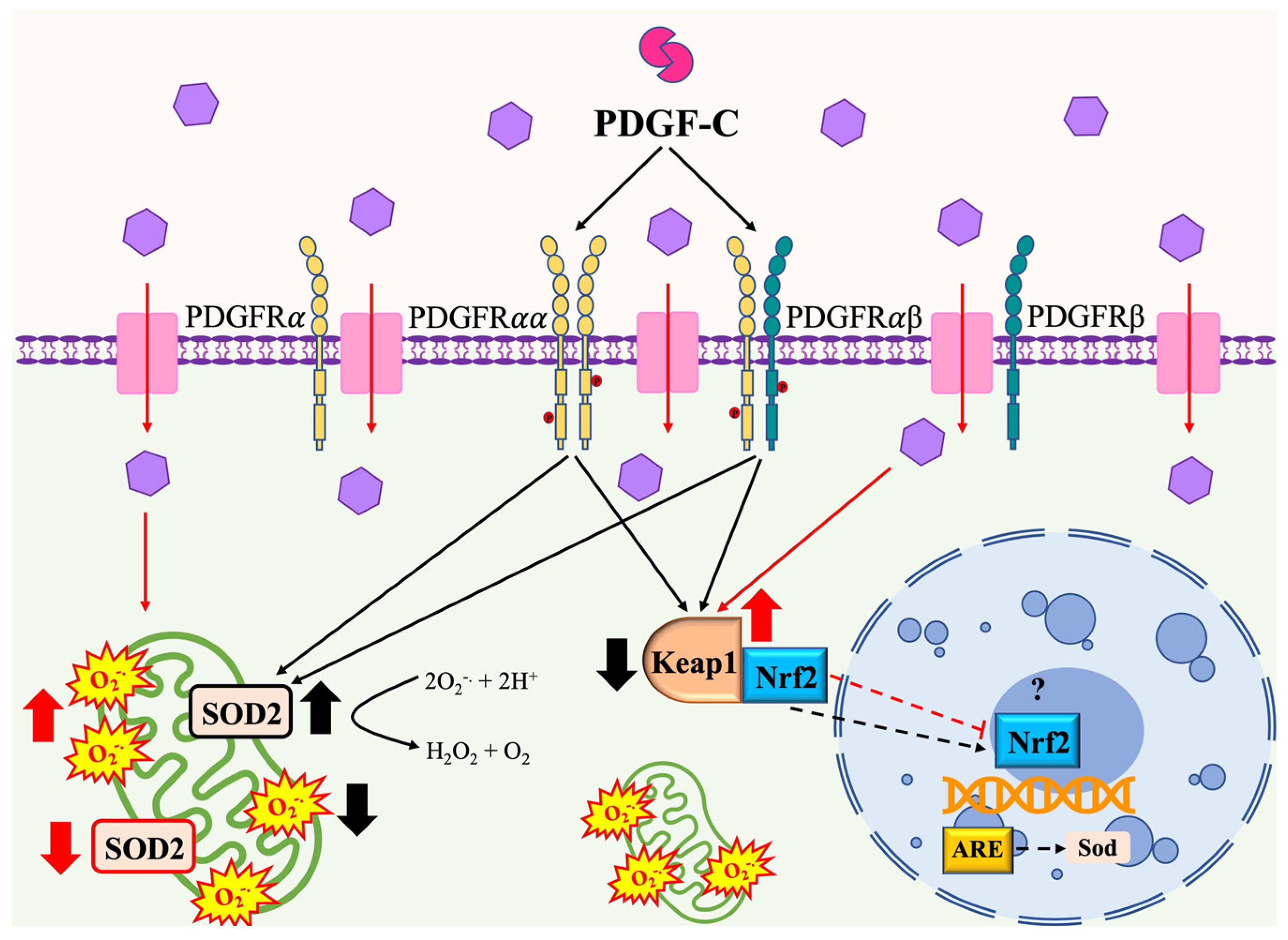
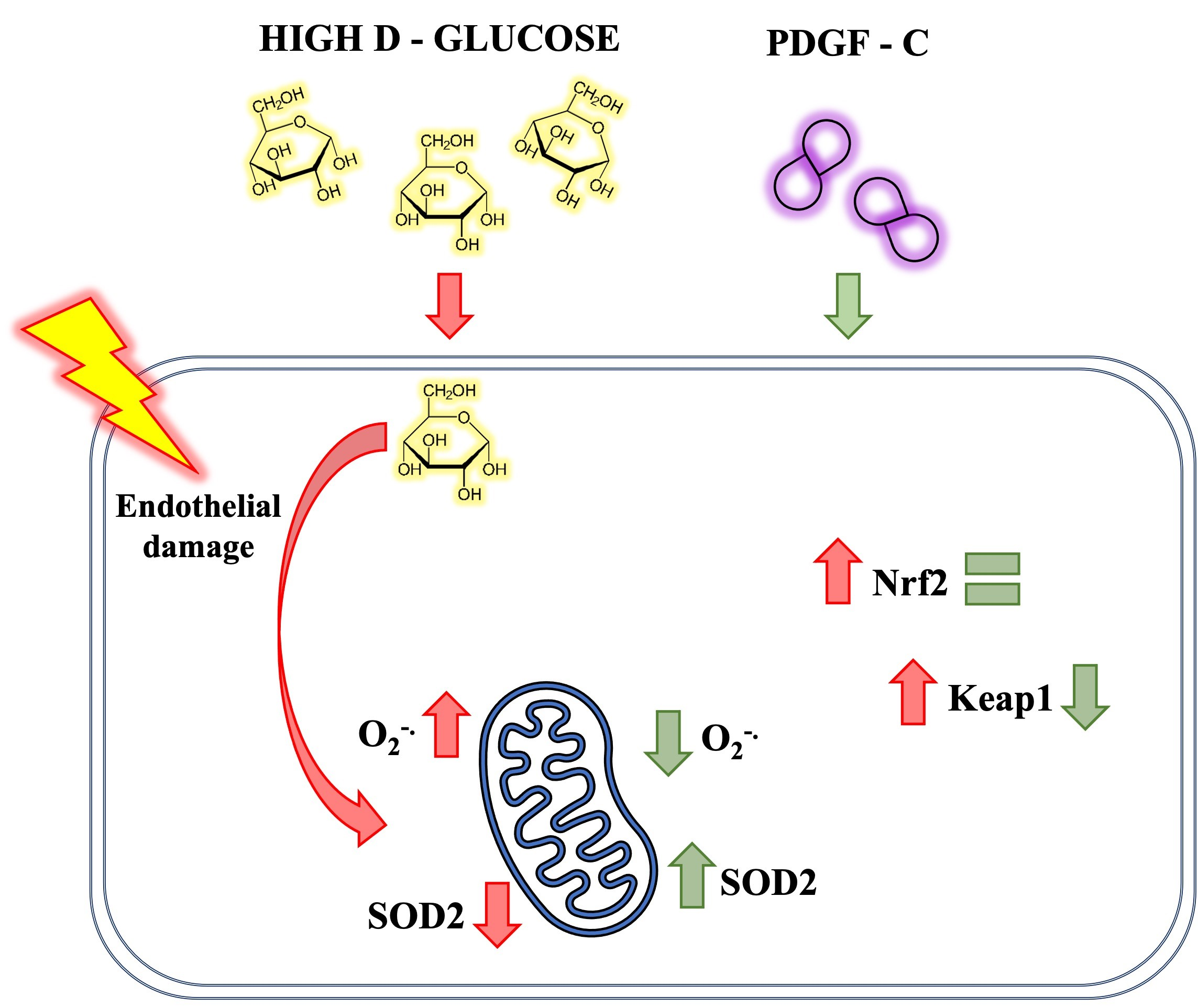
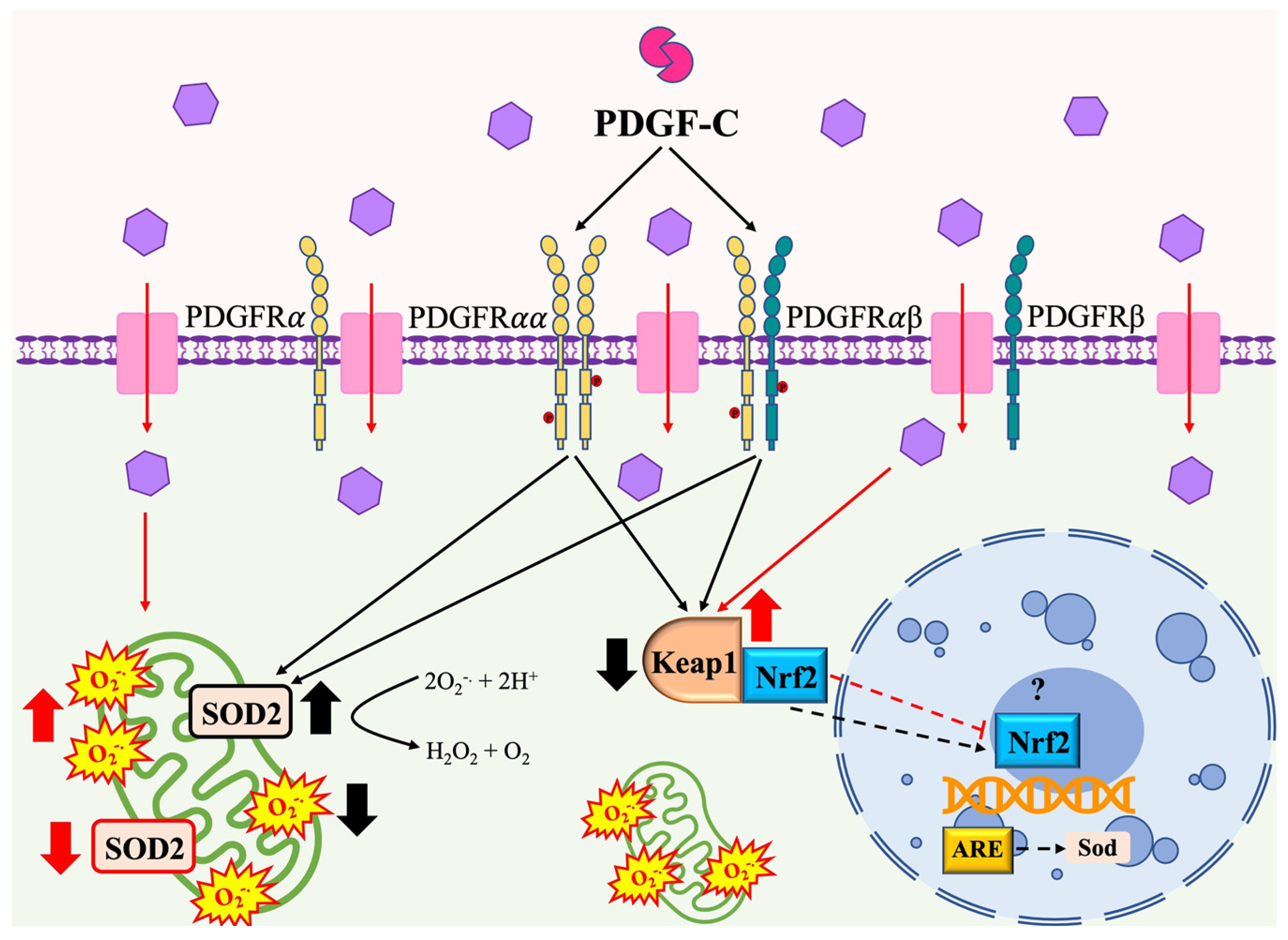
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Cell Culture and Treatments
4.3. Reactive Oxygen Species (ROS) Production
4.4. PDGF α and β Receptor Expressions by Flow Cytometry
4.5. Determination of PDGF-C in Cell Supernatants
4.6. Antioxidant Protein Expression by Western Blot
4.7. SOD Activity
4.8. Gene Expression Assay
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Dal Canto, E.; Ceriello, A.; Rydén, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. S2), 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44 (Suppl. S1), S15–S33. [Google Scholar] [CrossRef]
- Giglio, R.V.; Stoian, A.P.; Haluzik, M.; Pafili, K.; Patti, A.M.; Rizvi, A.A.; Ciaccio, M.; Papanas, N.; Rizzo, M. Novel molecular markers of cardiovascular disease risk in type 2 diabetes mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166148. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Tang, X.; Luo, Y.X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Domingueti, C.P.; Dusse, L.M.S.; Carvalho, M.D.G.; Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Patel, H.; Chen, J.; Das, K.C.; Kavdia, M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc. Diabetol. 2013, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial Dysfunction in Diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. S2), S285–S290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashi, R.; Patel, B.M. NRF2 in Cardiovascular Diseases: A Ray of Hope! J. Cardiovasc. Transl. Res. 2021, 14, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Larose, L. Activation of the PDGFRα-Nrf2 pathway mediates impaired adipocyte differentiation in bone marrow mesenchymal stem cells lacking Nck1. Cell Commun. Signal. 2020, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Rizky, L.; Stefanovic, N.; Tate, M.; Ritchie, R.H.; Ward, K.W.; de Haan, J.B. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activator dh404 protects against diabetes-induced endothelial dysfunction. Cardiovasc. Diabetol. 2017, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Bailey-Downs, L.; Gautam, T.; Jimenez, R.; Losonczy, G.; Zhang, C.; Ballabh, P.; Recchia, F.A.; Wilkerson, D.C.; Sonntag, W.E.; et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1133–H1140. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.M.; Ziyad, S.; Briot, A.; Der, A.; Iruela-Arispe, M.L. A Ligand-Independent VEGFR2 Signaling Pathway Limits Angiogenic Responses in Diabetes. Sci. Signal. 2014, 7, ra1. [Google Scholar] [CrossRef] [Green Version]
- Moriya, J.; Ferrara, N. Inhibiting the Response to VEGF in Diabetes. Sci. Signal. 2014, 7, pe1. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, A.; Simons, M. VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 2012, 24, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Abid, M.R.; Schoots, I.G.; Spokes, K.C.; Wu, S.-Q.; Mawhinney, C.; Aird, W.C. Vascular Endothelial Growth Factor-mediated Induction of Manganese Superoxide Dismutase Occurs through Redox-dependent Regulation of Forkhead and IκB/NF-κB. J. Biol. Chem. 2004, 279, 44030–44038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltenberger, J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem. Soc. Trans. 2009, 37 Pt 6, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kumar, A.; Zhang, F.; Lee, C.; Li, Y.; Tang, Z.; Arjunan, P. VEGF-independent angiogenic pathways induced by PDGF-C. Oncotarget 2010, 1, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S. Platelet-Derived Growth Factor CC—A Clinically Useful Angiogenic Factor at Last? N. Engl. J. Med. 2005, 352, 1815–1816. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Folestad, E.; Kunath, A.; Wågsäter, D. PDGF-C and PDGF-D signaling in vascular diseases and animal models. Mol. Asp. Med. 2018, 62, 1–11. [Google Scholar] [CrossRef]
- Lee, C.; Zhang, F.; Tang, Z.; Liu, Y.; Li, X. PDGF-C: A new performer in the neurovascular interplay. Trends Mol. Med. 2013, 19, 474–486. [Google Scholar] [CrossRef]
- Moriya, J.; Wu, X.; Zavala-Solorio, J.; Ross, J.; Liang, X.H.; Ferrara, N. Platelet-derived growth factor C promotes revascularization in ischemic limbs of diabetic mice. J. Vasc. Surg. 2014, 59, 1402–1409.e1–e4. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tjwa, M.; Moons, L.; Fons, P.; Noel, A.; Ny, A.; Zhou, J.M.; Lennartsson, J.; Li, H.; Luttun, A.; et al. Revascularization of ischemic tissues by PDGF-CC via effects on endothelial cells and their progenitors. J. Clin. Investig. 2005, 115, 118–127. [Google Scholar] [CrossRef]
- He, C.; Zhao, C.; Kumar, A.; Lee, C.; Chen, M.; Huang, L.; Wang, J.; Ren, X.; Jiang, Y.; Chen, W.; et al. Vasoprotective effect of PDGF-CC mediated by HMOX1 rescues retinal degeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 14806–14811. [Google Scholar] [CrossRef] [Green Version]
- Moriya, J.; Ferrara, N. Inhibition of protein kinase C enhances angiogenesis induced by platelet-derived growth factor C in hyperglycemic endothelial cells. Cardiovasc. Diabetol. 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas, R.; Vega-Vela, N.E.; González-Sanmiguel, J.; González, J.; Esquinas, P.; Echeverria, V.; Barreto, G.E. PDGF-BB Preserves Mitochondrial Morphology, Attenuates ROS Production, and Upregulates Neuroglobin in an Astrocytic Model Under Rotenone Insult. Mol. Neurobiol. 2018, 55, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hasegawa, G.; Ishii, M.; Yamasaki, M.; Fukui, M.; Nakamura, N.; Yoshikawa, T. Short-term exposure of high glucose concentration induces generation of reactive oxygen species in endothelial cells: Implication for the oxidative stress associated with postprandial hyperglycemia. Redox Rep. 2004, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Benzie, I.F.; Ma, S.-W.; Strain, J.J.; Hannigan, B.M. Acute hyperglycemia and oxidative stress: Direct cause and effect? Free Radic. Biol. Med. 2008, 44, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ma, D.; Li, A. SENP3 regulates high glucose-induced endothelial dysfunction via ROS dependent signaling. Diabetes Vasc. Dis. Res. 2020, 17, 1479164120970895. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, X.; Zhang, Z.; Li, B.; Yang, G. Vildagliptin protects endothelial cells against high glucose-induced damage. Biomed. Pharmacother. 2018, 108, 1790–1796. [Google Scholar] [CrossRef]
- Ke, S.-Y.; Yu, S.-J.; Liu, D.-H.; Shi, G.-Y.; Wang, M.; Zhou, B.; Wu, L.; Song, Z.-M.; Zhu, J.-M.; Wu, C.-D.; et al. Ginsenoside Rb1 Protects Human Umbilical Vein Endothelial Cells against High Glucose-Induced Mitochondria-Related Apoptosis through Activating SIRT3 Signalling Pathway. Chin. J. Integr. Med. 2021, 27, 336–344. [Google Scholar] [CrossRef]
- Lin, F.; Yang, Y.; Wei, S.; Huang, X.; Peng, Z.; Ke, X.; Zeng, Z.; Song, Y. Hydrogen Sulfide Protects Against High Glucose-Induced Human Umbilical Vein Endothelial Cell Injury Through Activating PI3K/Akt/eNOS Pathway. Drug Des. Dev. Ther. 2020, 14, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wu, M.; Lu, Y.; Xian, T.; Wang, Y.; Huang, B.; Zeng, G.; Huang, Q. Protective effects of 6-Gingerol on vascular endothelial cell injury induced by high glucose via activation of PI3K-AKT-eNOS pathway in human umbilical vein endothelial cells. Biomed. Pharmacother. 2017, 93, 788–795. [Google Scholar] [CrossRef]
- Qin, W.; Ren, B.; Wang, S.; Liang, S.; He, B.; Shi, X.; Wang, L.; Liang, J.; Wu, F. Apigenin and naringenin ameliorate PKCβII-associated endothelial dysfunction via regulating ROS/caspase-3 and NO pathway in endothelial cells exposed to high glucose. Vasc. Pharmacol. 2016, 85, 39–49. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell. Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gero, D. Hyperglycemia-Induced Endothelial Dysfunction. In Endothelial Dysfunction-Old Concepts and New Challenges; Lenasi, H., Ed.; IntechOpen: London, UK, 2017; Available online: https://www.intechopen.com/chapters/57915 (accessed on 18 February 2022).
- Brownlee, M. The Pathobiology of Diabetic Complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Chen, J.; Brodsky, S.V.; Huang, H.; Adler, S.; Lee, J.H.; Dhadwal, N.; Cohen-Gould, L.; Gross, S.S.; Goligorsky, M.S. Docking of Endothelial Nitric Oxide Synthase (eNOS) to the Mitochondrial Outer Membrane: A pentabasic amino acid sequence in the autoinhibitory domain of eNOS targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J. Biol. Chem. 2004, 279, 15968–15974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, V.; Elfering, S.L.; Squires, R.J.; Traaseth, N.; Solien, J.; Ettl, A.; Giulivi, C. Mitochondrial Nitric-oxide Synthase: Role in Pathophysiology. IUBMB Life 2003, 55, 599–603. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The role of Nrf2 in cardiovascular function and disease. Oxid. Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, Q.; Karlstetter, M.; Xu, Z.; Yang, J.; Zhou, L.; Eilken, H.M.; Terjung, C.; Cho, H.; Gong, J.; Lai, M.J.; et al. Inhibition of the Keap1-Nrf2 protein-protein interaction protects retinal cells and ameliorates retinal ischemia-reperfusion injury. Free Radic. Biol. Med. 2020, 146, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.G.; Winkles, J.A. The half-lives of platelet-derived growth factor A- and B-chain mRNAS are similar in endothelial cells and unaffected by heparin-binding growth factor-1 or cycloheximide. J. Cell. Physiol. 1991, 147, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen | Forward Sequence | Reverse Sequence |
|---|---|---|
| Nrf2 | TCCAGTCAGAAACCAGTGGAT | GAATGTCTGCGCCAAAAGCTG |
| Keap1 | CAACTTCGCTGAGCAGATTGGC | TGATGAGGGTCACCAGTTGGCA |
| Rpl27 | ATCGCCAAGAGATCAAAGATAA | TCTGAAGACATCCTTATTGACG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grismaldo Rodríguez, A.; Zamudio Rodríguez, J.A.; Mendieta, C.V.; Quijano Gómez, S.; Sanabria Barrera, S.; Morales Álvarez, L. Effect of Platelet-Derived Growth Factor C on Mitochondrial Oxidative Stress Induced by High d-Glucose in Human Aortic Endothelial Cells. Pharmaceuticals 2022, 15, 639. https://doi.org/10.3390/ph15050639
Grismaldo Rodríguez A, Zamudio Rodríguez JA, Mendieta CV, Quijano Gómez S, Sanabria Barrera S, Morales Álvarez L. Effect of Platelet-Derived Growth Factor C on Mitochondrial Oxidative Stress Induced by High d-Glucose in Human Aortic Endothelial Cells. Pharmaceuticals. 2022; 15(5):639. https://doi.org/10.3390/ph15050639
Chicago/Turabian StyleGrismaldo Rodríguez, Adriana, Jairo A. Zamudio Rodríguez, Cindy V. Mendieta, Sandra Quijano Gómez, Sandra Sanabria Barrera, and Ludis Morales Álvarez. 2022. "Effect of Platelet-Derived Growth Factor C on Mitochondrial Oxidative Stress Induced by High d-Glucose in Human Aortic Endothelial Cells" Pharmaceuticals 15, no. 5: 639. https://doi.org/10.3390/ph15050639
APA StyleGrismaldo Rodríguez, A., Zamudio Rodríguez, J. A., Mendieta, C. V., Quijano Gómez, S., Sanabria Barrera, S., & Morales Álvarez, L. (2022). Effect of Platelet-Derived Growth Factor C on Mitochondrial Oxidative Stress Induced by High d-Glucose in Human Aortic Endothelial Cells. Pharmaceuticals, 15(5), 639. https://doi.org/10.3390/ph15050639