Abstract
Deciphering the drug/virus/host interactions at infected cell reservoirs is a key leading to HIV-1 remission for which positron emission tomography (PET) imaging using radiolabeled antiretroviral (ARV) drugs is a powerful asset. Dolutegravir (DTG) is one of the preferred therapeutic options to treat HIV and can be isotopically labeled with fluorine-18. [18F]DTG was synthesized via a three-step approach of radiofluorination/nitrile reduction/peptide coupling with optimization for each step. Radiofluorination was performed on 2-fluoro-4-nitrobenzonitrile in 90% conversion followed by nitrile reduction using sodium borohydride and aqueous nickel(II) chloride with 72% conversion. Final peptide coupling reaction followed by HPLC purification and formulation afforded ready-to-inject [18F]DTG in 5.1 ± 0.8% (n = 10) decay-corrected radiochemical yield within 95 min. The whole process was automatized using a TRACERlab® FX NPro module, and quality control performed by analytical HPLC showed that [18F]DTG was suitable for in vivo injection with >99% chemical and radiochemical purity and a molar activity of 83 ± 18 GBq/µmol (n = 10). Whole-body distribution of [18F]DTG was performed by PET imaging on a healthy macaque and highlighted the elimination routes of the tracer. This study demonstrated the feasibility of in vivo [18F]DTG PET imaging and paved the way to explore drug/virus/tissues interactions in animals and humans.
1. Introduction
Increasing evidence makes remission an achievable goal for new human immunodeficiency virus (HIV-1) therapies [1,2,3]. Combined antiretroviral therapies (cART) suppress viral replication and drastically reduce morbidity and mortality [4]. However, despite the existence of a few patients capable of controlling HIV replication to undetectable levels (i.e., HIV controllers, <1% of the HIV-infected population) [2], cART do not eradicate infected cells, and viral reservoirs still remain [5]. Early control of these reservoirs together with the limitation of pharmacological sanctuaries where the exposure to cART is suboptimal [6] are among the key scientific challenges to make remission a reality for a majority of patients.
The whole-body distribution of the virus is a key parameter to predict the virus/host/drugs interactions. Indeed, localization of infected cell reservoirs and their interactions with both drugs and the tissue micro-environment remain poorly understood [7]. In this context, in vivo tomographic imaging technologies may be of considerable help to tackle anti-HIV therapy challenges [8,9,10]. In particular, positron emission tomography (PET) imaging is a sensitive and quantitative technique for in vivo kinetic modeling of drug distribution in tissues and accurate detection of molecular targets. Kinetics of drug diffusion and body distribution can theoretically be addressed in relevant animal models of the disease, in healthy volunteers, and in patients. New approaches to directly visualize metabolic pathways or viral replication in the host are rapidly emerging and have a strong potential for being translated in the near future into human clinical studies. Fluorine-18-labeled fluoro-D-deoxyglucose ([18F]FDG) is by far the most common radiotracer for PET imaging and has been used to monitor HIV infection before and after cART in the context of HIV-related inflammation [11]. However, [18F]FDG reacts to immune activation and is not specific for HIV reservoirs. More recently, 18F-labeled nucleoside derivatives have been developed aiming at imaging T-cell trafficking [12,13], but this strategy, which has never been applied to HIV imaging, is only an indirect visualization of immune activation. ImmunoPET imaging using radiolabeled antibodies offers promising perspectives in the path towards higher specificity for infection biomarkers [14]. First-in-human studies using antibodies labeled with copper-64 [15] and zirconium-89 [16] were conducted with HIV patients on and off ART treatment and demonstrated the feasibility and the relevance of PET imaging for HIV patient follow-up. From a therapeutic perspective, it is important to decipher the biodistribution of cART at remaining sanctuary sites and potential HIV-1 reservoirs. This could be achieved with PET imaging using radiolabeled antiretroviral drugs. This approach is currently in an early phase 1 clinical trial (NCT03174977) using fluorine-18-labeled raltegravir (MK-0518), the first integrase strand transfer inhibitor (INSTI) approved by the FDA. Dolutegravir (DTG, Tivicay®, ViiV Healthcare Limited, Brentford, UK) is another INSTI used in combination with other antiretroviral drugs to inhibit the integration of HIV proviral DNA and, hence, the production of new viral particles [17,18]. DTG offers better virological control than raltegravir [19] and is one of the preferred therapeutic options to treat HIV as recommended by the World Health Organization [20]. With two fluorine atoms on its structure, DTG is an excellent candidate for isotopic labeling with fluorine-18, i.e., radiolabeling without modification of the chemical structure and, therefore, without modification of biological properties. To assess the delivery of DTG to various critical tissue compartments and to understand the persistence of HIV-1 reservoirs to prolonged treatments, preclinical and clinical positron emission tomography (PET) using fluorine-18-radiolabeled DTG are of interest.
Herein we describe the different approaches that we explored to radiolabel DTG with fluorine-18 to finally afford a robust and reproducible automatized process to synthesize ready-to-inject [18F]DTG. We also report the first whole-body PET images and biodistribution of [18F]DTG in a healthy non-human primate.
2. Results and Discussion
2.1. Late-Stage Radiolabeling Approach
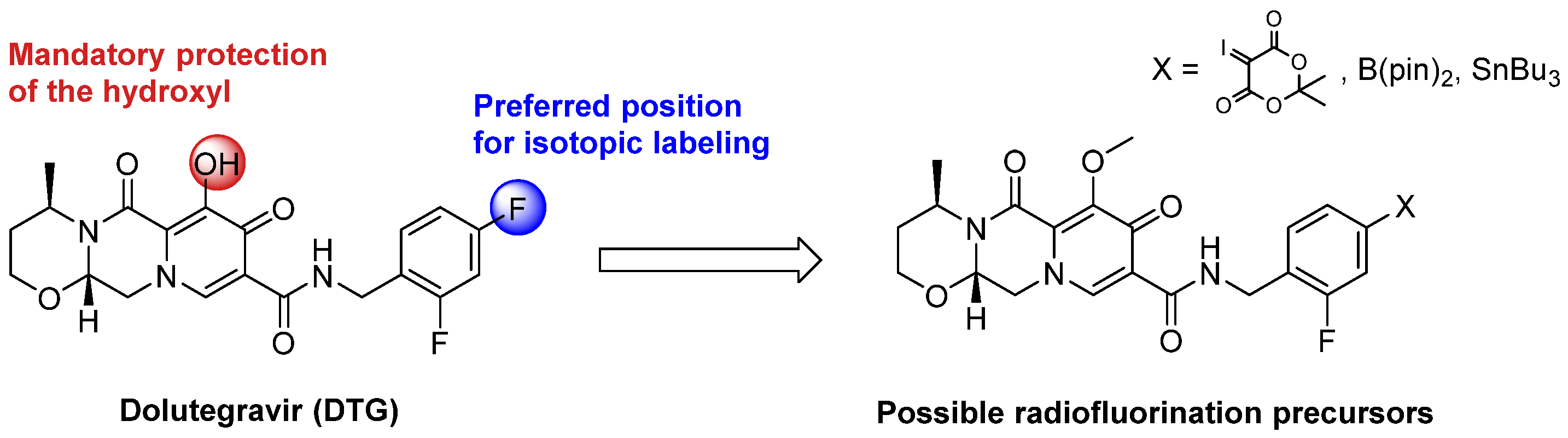
The most efficient strategy to radiolabel DTG with fluorine-18 would be a late-stage approach consisting of the incorporation of fluorine-18 at the final step of the synthesis on the aromatic core of DTG bearing two fluorine atoms (Scheme 1). However, this aromatic core is electron-rich, hampering radiofluorination by standard aromatic nucleophilic substitution (SnAr). Alternative approaches have been described using iodonium salts [21,22,23] or ylides [24,25,26] as precursors or by copper-catalyzed Cham-Lam-like fluorination using boronic acid [27,28], boronic ester [29,30,31,32], or trialkyltin precursors [33,34]. Late-stage isotopic radiolabeling of DTG was considered at the para position of the aromatic core (compared to the methyl amino group), as better radiochemical yields were depicted on this position compared to ortho [27,29,31,33]. Limitations of these fluorination methods were also described in the presence of unprotected alcohol or amine [29,31]. Therefore, the hydroxyl function of DTG was protected with a methyl moiety, the cleavage of such function being well described for DTG [35].
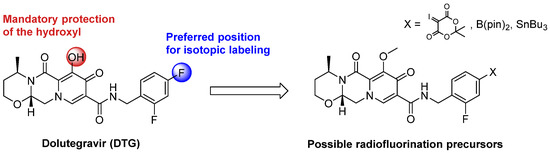
Scheme 1.
Possible precursors for late-stage isotopic radiofluorination of DTG with fluorine-18.
Labeling precursors for late-stage radiofluorination of DTG, i.e., iodonium ylide, boronic acid, and ester and tributyltin derivatives were synthesized in two steps (Scheme S1 in Supplementary Materials). Unfortunately, even if the boronic ester precursor was observed by liquid chromatography coupled with mass spectroscopy (LC-MS), it was too unstable to be isolated. The ylide precursor was synthesized but could not be isolated from its starting material and was unstable at 120 °C, whereas a minimal temperature of 150 °C is required to perform radiofluorination with this type of precursor [24,25]. Several attempts of copper-catalyzed radiofluorination using the tributyltin precursor were realized under the standard conditions described in the literature using the Cu(OTf)4Py4 complex [33], but incorporation of fluorine-18 was never observed (Scheme S2 in Supplementary Materials).
2.2. Three-Step Radiolabeling Approach
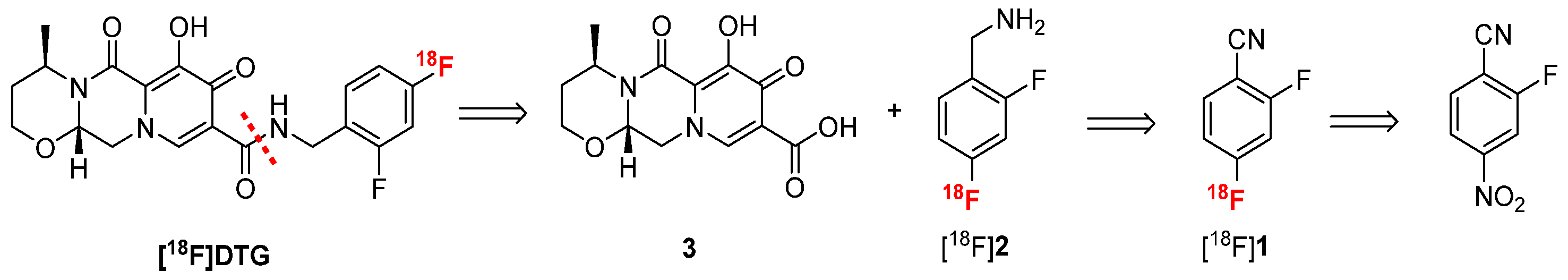
Considering the difficulties encountered for the late-stage labeling of DTG, the strategy described for raltegravir radiosynthesis was explored [36,37]. Mimicking the recent synthesis of dolutegravir described by Dietz et al. [38], [18F]DTG can be synthesized by peptide coupling between carboxylic acid 3 and radiolabeled difluorobenzylamine [18F]2 (Scheme 2). Although the radiosynthesis of [18F]2 has never been reported, p-[18F]fluorobenzylamine has been extensively described in the literature [39]. Following a similar approach, [18F]2 could be synthesized by reduction of a nitrile moiety from [18F]1 which could be obtained by radiofluorination of 2-fluoro-4-nitrobenzonitrile. This approach will benefit from the poor electron density of 2-fluoro-4-nitrobenzonitrile due to the nitrile moiety at the para position of the nitro group, which allows for the introduction of the fluorine-18 ion via a standard SnAr reaction.
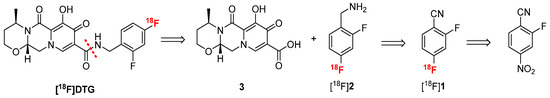
Scheme 2.
Retroradiosynthesis of [18F]DTG following the approach described for [18F]Raltegravir [36].
2.2.1. Radiofluorination Optimization
Radiofluorination of 2-fluoro-4-nitrobenzonitrile was realized by SnAr under standard conditions using the K [18F]F/K222 complex in acetonitrile (Scheme 3) [39]. A kinetic monitoring of the reaction using thin layer chromatography (TLC) analysis on silica gel (see Figure S12 in Supplementary Materials) revealed that performing the radiofluorination in acetonitrile at 95 °C for 5 min was the best compromise to reach a high conversion into [18F]1 (96%) in a short time (Table 1, entry 1). However, acetonitrile, even at trace quantities, could be deleterious for the following step of nitrile reduction, and DMSO is usually preferred [40]. Radiofluorination in DMSO afforded [18F]1 in 90% conversion (Table 1, entry 4), comparable with acetonitrile under the same conditions.
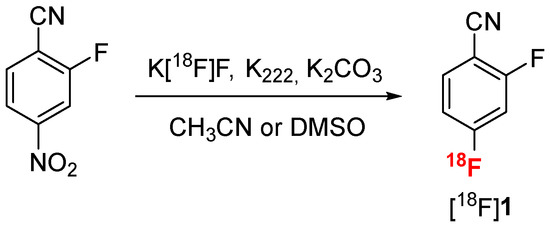
Scheme 3.
Radiofluorination of 2-fluoro-4-nitrobenzonitrile by SnAr.

Table 1.
Reaction conditions for the radiofluorination of 2-fluoro-4-nitrobenzonitrile.
2.2.2. Nitrile Reduction Optimization
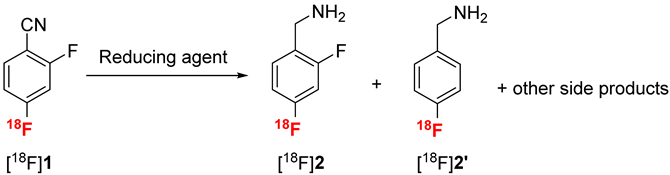
Different approaches were explored for the nitrile reduction of compound [18F]1 into [18F]2 (Table 2). Lithium aluminium hydride (LAH) has been commonly used to reduce [18F]fluorobenzonitrile into [18F]fluorobenzylamine [39]. In our case, using LAH in THF afforded the desired [18F]2 in 15 to 50% conversion depending on the reaction temperature (Table 2, entries 1 and 2) together with different side products. In particular, defluorination of [18F]2 into p-[18F]fluorobenzylamine [18F]2′ was observed. Indeed, hydrodefluorination of arenes in the ortho position of an electron-withdrawing group such as the nitrile moiety has been reported in the literature using metal hydrides [41,42]. Because of their structure similarity, [18F]2 and [18F]2′ are difficult to separate by HPLC (see Figure S13 in Supplementary Materials for radio HPLC examples). Moreover, [18F]2′ will be able to react in the next step of peptide coupling in the same way as [18F]2. Therefore, a compromise has to be found between maximizing the conversion into [18F]2 and minimizing [18F]2′ formation. In addition, results with LAH were not reproducible, and ratios observed between [18F]2 and [18F]2′ were not constant. Therefore, other reducing agents were explored. Borane complexes are also standard reagents for nitrile reduction [43] which have been used for the reduction of [18F]fluorobenzonitrile [39]. In our case, reduction of [18F]1 at either room temperature or 65 °C resulted in only 5% of [18F]2 and mostly degradation of the starting material (Table 2, entries 3 and 4, respectively). Reduction of nitrile moieties by hydrogenation in the presence of palladium on charcoal has also been extensively described in the literature [44], although it has never been used for radiochemical reactions, due to the heterogeneity of the medium. In our case, hydrogenation of compound [18F]1 in methanol at room temperature only resulted in the formation of compound [18F]2′ with 30% conversion (Table 2, entry 5). Indeed, palladium-catalyzed hydrodefluorination has been reported in the case of poly-fluoro arenes [45]. Considering the sensitivity of [18F]1 to defluorination and the significant proportion of side products formed using standard reducing agents, milder conditions were explored. Werkmeister et al. have described an original method to reduce nitrile by hydrogenation under mild conditions using [RuCl2(pCymene)2]2 and 3-bis(diphenylphosphino)propane (DPPP) as the catalytic system and isopropanol as the hydrogen donor [46]. Under those conditions, no side reactions were observed, but [18F]2 was obtained with only 10% conversion (Table 2, entry 6). The in situ generation of metal boride using NaBH4 in the presence of transition metal (Co, Ni) and water is an efficient reducing agent to promote nitrile reduction [47]. This catalytic system has already been described for the reduction of [18F]fluorobenzonitrile to [18F]fluorobenzylamine [40,48,49]. Using excess amounts of NaBH4 and hexahydrated nickel chloride in methanol, [18F]2 was obtained with 42% conversion, together with 22% of side products, mainly corresponding to the defluorinated compound [18F]2′ (Table 2, entry 7). Way and Wuest described a supported version of this nitrile reduction using a borohydride exchange resin (BER) for the automated radiosynthesis of [18F]fluorobenzylamine [48]. In our hands, almost no reaction was observed (Table 2, entry 8).
Table 2.
Different reducing agents explored for the nitrile reduction of compound [18F]1 into [18F]2.
Clearly the homogeneous nitrile reduction using NaBH4 and hexahydrated NiCl2 was the best compromise to achieve a moderate conversion into the desired benzylamine [18F]2 while minimizing the formation of side products. In order to further improve the conversion into [18F]2, different parameters (quantity of reagents, solvent, time, and temperature) were studied (Table 3). THF was also explored to realize this reduction, but conversion into [18F]2 was always lower than in methanol (data not shown). Decreasing the quantity of NaBH4 resulted in a total conversion of the starting material [18F]1 but significantly increased the proportion of side products compared to the desired compound [18F]2 (Table 3, entry 2). Increasing the time of reaction to 10 min (Table 3, entry 3) or the temperature to 50 °C (Table 3, entry 4) did not significantly increase the conversion into [18F]2. Additional water (100 µL) was introduced to the reaction to promote the formation of the Ni2B reducing agent and improve the homogeneity of the reaction (Table 3, entry 5). In those conditions, the conversion of [18F]2 rose to 45%, with 55% of side products formed. To further improve the conversion of [18F]2, the influence of the quantity of NiCl2·6H2O was studied. Increasing the quantity to 20 µmol (Table 3, entry 6) or 40 µmol (Table 3, entry 7) did not result in significant improvement, whereas using 10 µmol of NiCl2·6H2O (Table 3, entry 8) afforded a conversion of 72% of [18F]2 with only 28% of side products. In those conditions, increasing the reaction time to 10 min (Table 3, entry 9) did not yield any significant improvement of the conversion rate, whereas increasing the temperature to 50 °C (Table 3, entry 10) resulted in an increase of side product formation. Finally, the conversion of [18F]2 dropped to 46% when increasing the quantity of NaBH4 while using 100 µmol of NiCl2·6H2O and additional water with unreacted [18F]1 remaining (Table 3, entry 11). Overall, the best compromise to optimize the conversion into [18F]2 (72%) while minimizing the formation of deleterious side products (28%) for the next reaction step was found when using a rather low quantity of NaBH4 (40 µmol) and a large excess of hexahydrated nickel chloride with additional water (Table 3, entry 8).
Table 3.
Optimization of the nitrile reduction using NaBH4 (2M) and NiCl2·6H2O as reducing agents.
2.2.3. Peptide Coupling Optimization
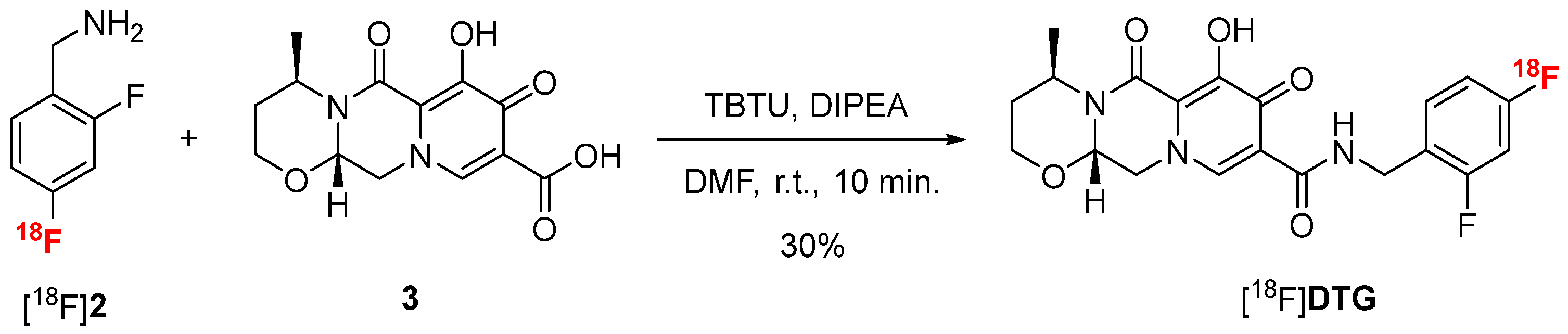
Inspired by the recent work of Dietz and colleagues [38], [18F]DTG was finally obtained in a last step of peptide coupling between [18F]2 and carboxylic acid 3 using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) as coupling agent and diisopropylethylamine (DIPEA) in DMF (Scheme 4). Compound 3 was synthesized in one-step from compound GSK3210346A [50] (see Scheme S3 in Supplementary Materials). In those conditions, [18F]DTG was obtained in 30% conversion after 10 min of reaction at room temperature. Increasing the temperature or the reaction time did not result in any significant improvement of the conversion.

Scheme 4.
Peptide coupling reaction between [18F]2 and compound 3 to afford [18F]DTG.
2.3. Automated Radiosynthesis of [18F]DTG
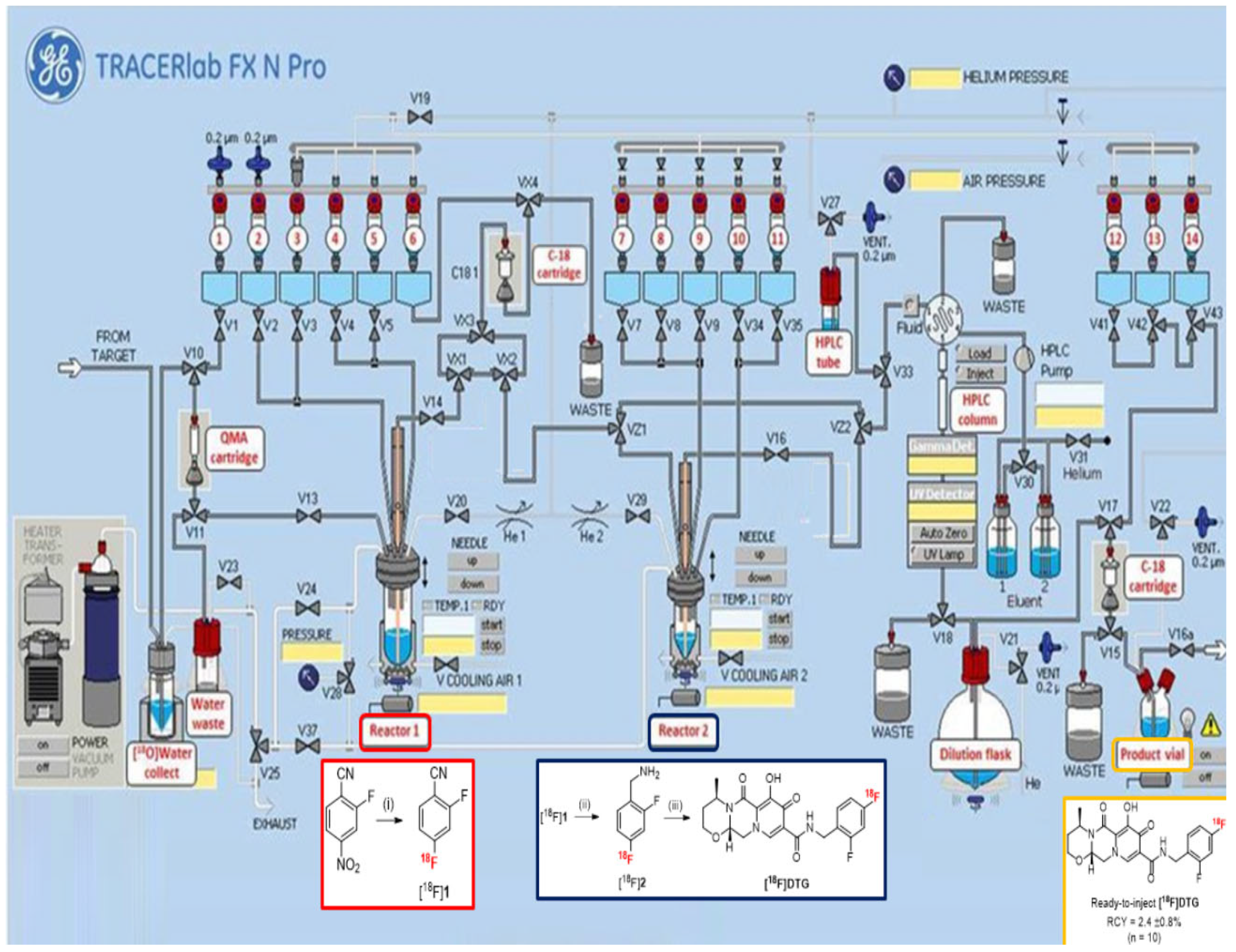
For in vivo PET imaging applications, the radiosynthesis of [18F]DTG has to be automated for reproducibility and radioprotection reasons. With optimized conditions for each step in hand, the automation was carried out using a TRACERlab® FX N Pro (GE Healthcare, Chicago, IL, USA) module (Figure 1). The presence of two separated reactors on the module was mandatory, as the reaction conditions, the solvents in particular, between the fluorination and the reduction steps are not compatible. Moreover, unreacted fluoride ions could promote the decomposition of sodium borohydride [51]. In contrast, the reduction and the peptide coupling reaction could be realized in one pot in the second reactor. After using a standard procedure for the preparation of the K [18F]F/K222 complex with azeotropic drying, the radiofluorination step was realized in reactor 1 following the optimized conditions depicted above. In order to remove unreacted fluoride ions before the reduction step, the crude mixture from radiofluorination was pre-purified by solid-phase extraction (SPE) on a C18 cartridge. This step also achieved a solvent exchange from DMSO to methanol for the reduction step. The crude [18F]1 was eluted with methanol from the C18 cartridge to the reactor 2 to avoid any contamination for the reduction step from possible residual DMSO or fluoride ions in reactor 1. To realize the reduction step, the sodium borohydride solution in methanol and the nickel chloride hexahydrate solution in water were added from separate vials into the reactor 2. Mixing the two solutions beforehand resulted in the degradation of the formed Ni2B reducing agent and a poor conversion into [18F]2. The coupling reaction was then performed in one pot by adding a mixture of compound 3, TBTU, and DIPEA in DMF directly into reactor 2 at the end of the reduction step. At the end of the radiosynthesis, purification of the crude reaction mixture was realized by semi-preparative HPLC on a Symmetry C18 column (Waters) using a mixture of acetonitrile, water, and trifluoroacetic acid (TFA) as eluent. [18F]DTG was isolated with a retention time of about 17 min (see Figure S17 in Supplementary Materials). Final formulation by SPE on a C18 cartridge afforded ready-to-inject [18F]DTG in 95 min and 5.1 ± 0.8% (n = 10) decay-corrected radiochemical yield. This yield is comparable to the similar radiosynthesis of [18F]Raltegravir (4.61 ± 0.3% within 135 min) described by Blecha et al. [37]. The quantity of [18F]DTG produced was in the range 500–700 MBq, consistent with administration to humans (typically in the range 200–400 MBq).
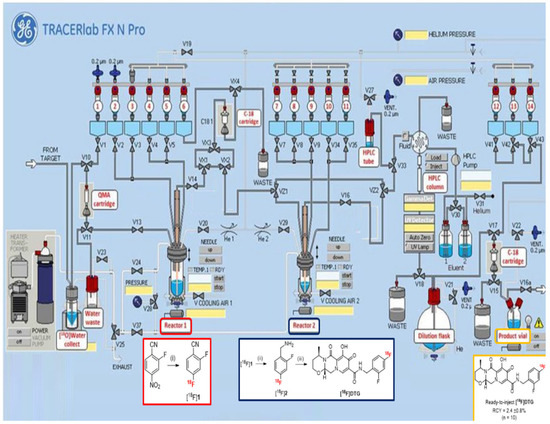
Figure 1.
Automated radiosynthesis of [18F]DTG on a TRACERlab® FX N Pro module. Radiofluorination (red square) takes place in reactor 1, whereas the nitrile reduction and peptide coupling reactions (blue square) take place in reactor 2. Ready-to-inject [18F]DTG (orange square) is obtained after semi-preparative HPLC purification and SPE formulation. Reagents and conditions: (i) K [18F]F, K222, K2CO3, DMSO, 95 °C, 5 min; (ii) NaBH4 2M (20 µL), NiCl2·6H2O (25 mg), MeOH, H2O (100 µL), r.t., 5 min; (iii) 3, TBTU, DIPEA, DMF, r.t., 10 min.
2.4. Quality Control
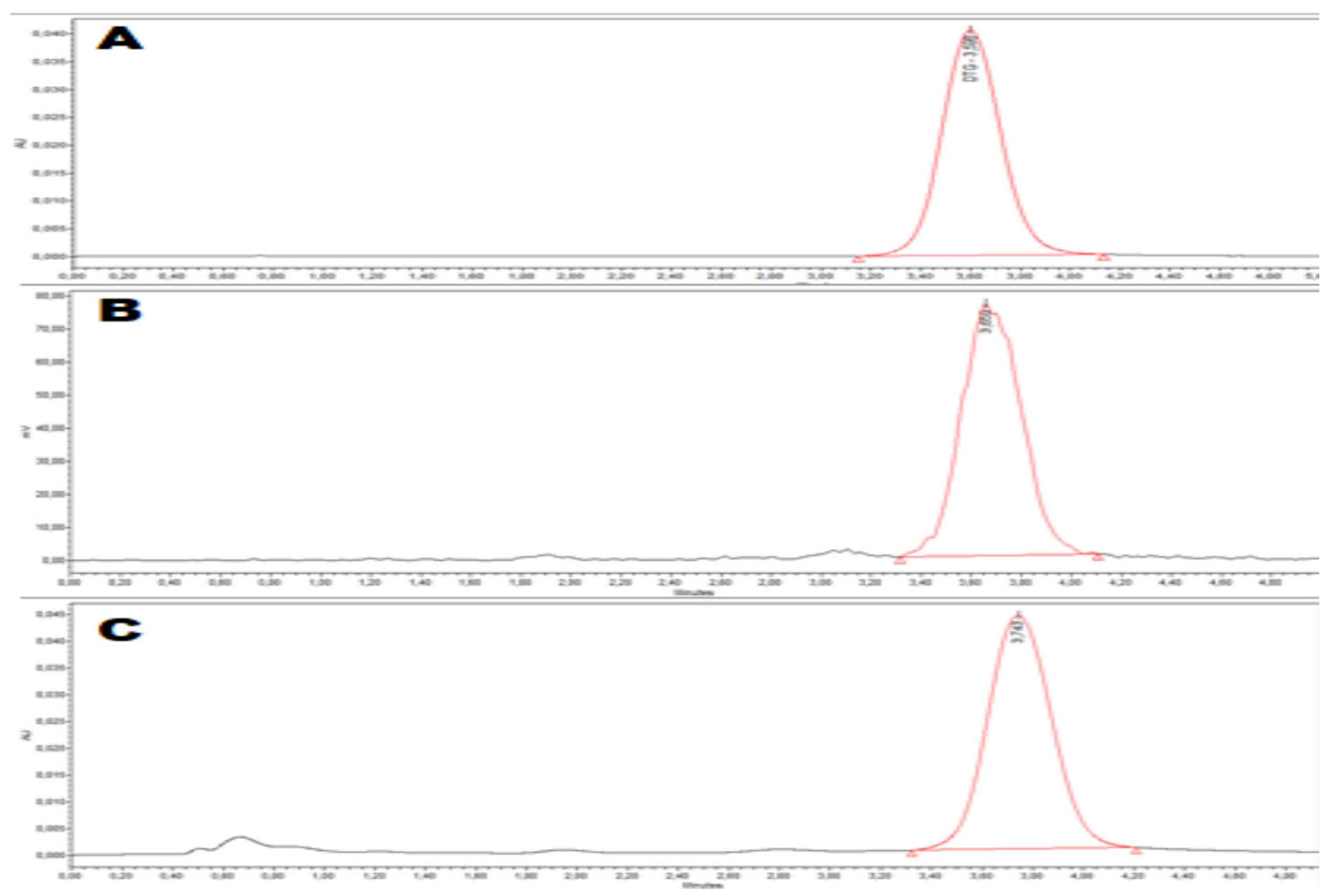
Quality control was performed on [18F]DTG by analytical HPLC with both UV (258 nm) and gamma detection to confirm the identity of the radiotracer and its chemical and radiochemical purity and to assess the molar activity (MA) of the radiotracer (Figure 2). The identity of the radiotracer was confirmed by the retention time of [18F]DTG (tR = 3.74 min, Figure 2C), which was within the range tRref ± 10% compared to the retention time of the DTG reference (tRref = 3.60 min., Figure 2A). [18F]DTG was obtained with both a radiochemical and chemical purity above 99% (Figure 2B and C respectively). [18F]DTG was obtained with a MA of 83 ± 18 GBq/µmol (n = 10) calculated with the help of a calibration curve (see Figure S18 in Supplementary Materials). In summary, the quality control of [18F]DTG demonstrated that the radiotracer was suitable for in vivo injection to perform PET imaging. Pending additional tests, [18F]DTG would be suitable for human injection according to guidance from the European Pharmacopeia [52].
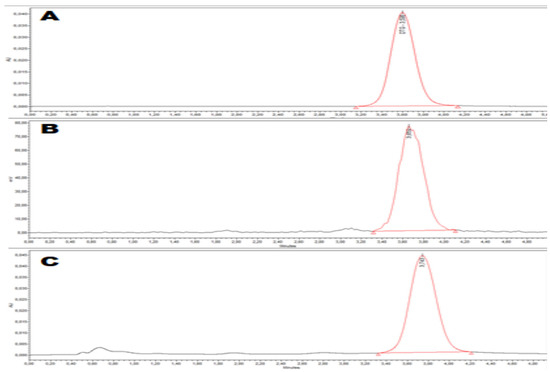
Figure 2.
Quality control of [18F]DTG by analytical HPLC. (A) UV (258 nm) chromatogram of the reference compound DTG; (B) gamma chromatogram of the radiotracer [18F]DTG; (C) UV (258 nm) chromatogram of the radiotracer [18F]DTG.
2.5. PET Imaging
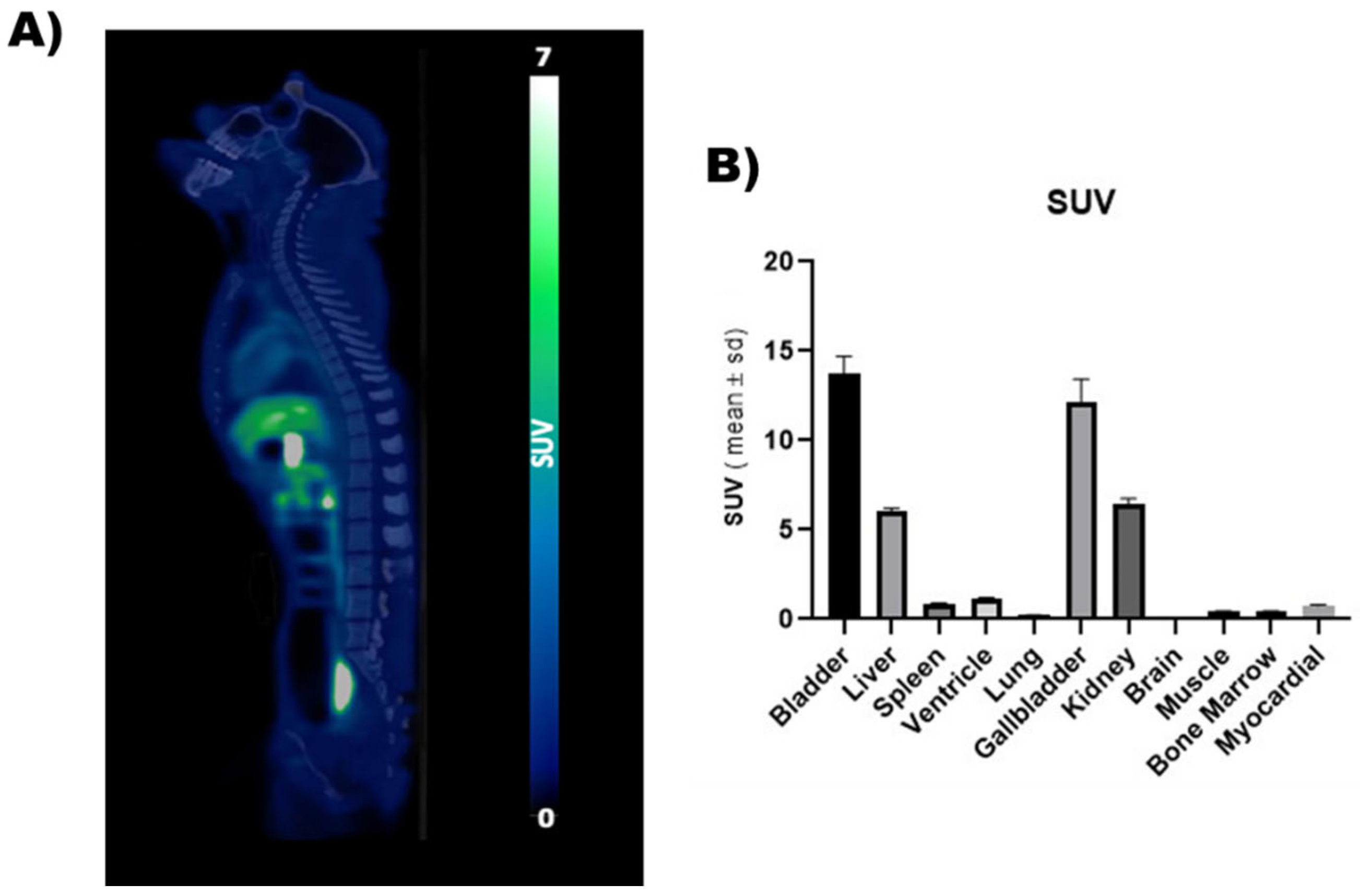
Feasibility of in vivo PET imaging using [18F]DTG was first assessed in a non-human primate. PET images unveiled the whole-body distribution of [18F]DTG (Figure 3). Highest PET signal was observed in the liver (SUV = 6.06 ± 0.14), kidneys (SUV = 6.45 ± 0.31), gallbladder (SUV = 12.16 ± 1.26), and urinary bladder (SUV = 13.73 ± 0.97), consistent with the known elimination routes of DTG [53]. PET signal in the brain was strikingly low (SUV = 0.04 ± 0.001) compared with peripheral organs, suggesting limited brain penetration of [18F]DTG. Minimally invasive [18F]DTG PET, therefore, sheds light on the elimination of DTG and its distribution to tissues. Interestingly, it suggests a minimal distribution to the brain which could correspond to a pharmacological sanctuary. Previous studies suggest a low brain distribution of [18F]DTG in mice [54]. Further experiments with dynamic brain PET imaging and kinetic modelling will however be necessary to correctly estimate the brain distribution of [18F]DTG in macaques.
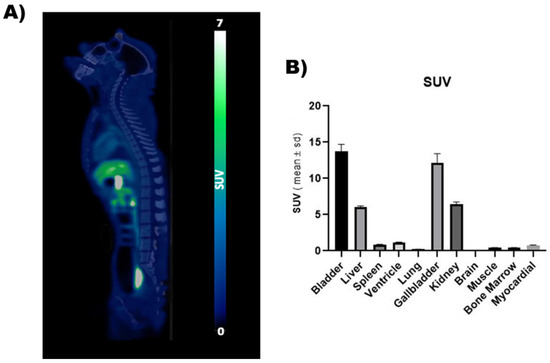
Figure 3.
PET imaging of a non-human primate using [18F]DTG. (A) Representative whole-body [18F]DTG PET-CT image obtained in a healthy macaque. PET signal is expressed in SUV units. (B) SUV (calculated as the mean value at the equilibrium between 150 and 180 min) in the organs of interest.
3. Materials and Methods
3.1. Chemistry
Chemicals were purchased from Aldrich (Saint-Quentin-Fallavier, France) and used as received. 2-Fluoro-4-nitrobenzonitrile was purchased from Fluorochem. Dolutegravir and compound GSK3210346A were kindly provided by GSK. Reactions were monitored by thin layer chromatography (TLC) on aluminum pre-coated plates of silica gel 60F254 (VWR, Rosny-sous-bois, France). The compounds were localized at 254 nm using a UV lamp. 1H and 13C NMR spectra were recorded on a Bruker Advance 400 MHz apparatus using DMF-d4 as solvent. The chemical shifts (δ) are reported in ppm (s, d, t, q, and b for singlet, doublet, triplet, quadruplet, and broad signal, respectively) and referenced with the solvent residual chemical shift. Ultra-performance liquid chromatography-mass spectroscopy (UPLC-MS) was realized on an Ultimate 3000 (Thermo Scientific, Waltham, MA, USA) device equipped with an Acquity BEH 2.1 × 50 mm, 1.7 µm column (Waters, Milford, MA, USA). A gradient of water with 0.1% of formic acid and acetonitrile with 0.1% of formic acid (3% of CH3CN/HCHO for 2 min, then rising to 100% during 7 min, then decreasing to 3% during 1 min, then keeping 3% for 2 min) at a flow rate of 0.3 mL/min was used. Mass spectroscopy was performed with a Linear Trap Quadripole Orbitrap Velos (Thermo Scientific, Waltham, MA, USA) equipped with an electron spray ionization (ESI) chamber. Spectra were recorded between 100 and 1000 m/z. High-resolution mass spectrometry (HRMS) analyses were performed by the Small Molecule Mass Spectrometry platform of ICOA, (Orléans, France) by electrospray with positive (ESI+) ionization mode.
(4R,12aS)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxylic acid (3). To a solution of GSK3210346A (500 mg, 1.0 equiv.) in DMF (20 mL) was added lithium chloride (690 mg, 10.0 equiv.), and the mixture was heated for 5 h at 120 °C. Upon cooling to room temperature, an aqueous solution of HCl (1 M, 10 mL) was added to reach a pH of 1. The aqueous phase was extracted with ethyl acetate (2 × 10 mL), and the combined organic phases were washed with a saturated solution of sodium chloride (2 × 10 mL), dried over sodium sulfate, and the solvent was evaporated. The residue was dried under vacuum to afford compound 3 (360 mg, 76%) as a white powder. 1H-NMR (DMF-d4, 400 MHz) δ 15.39 (s, 1H), 12.94 (bs, 1H), 8.70 (s, 1H), 5.68 (q, J = 4 Hz, 1H), 4.92–4.84 (b, 2H), 4.57 (dd, J2 = 14 Hz, J3 = 6 Hz, 1H), 4.17 (td, J2 = 12 Hz, J3 = 2 Hz, 1H), 4.00 (dq, J2 = 12 Hz, J3 = 2 Hz, 1H), 2.22–2.12 (b, 1H), 1.69–1.65 (b, 1H), 1.43 (d, J = 7 Hz, 3H) ppm. 13C-NMR (DMF-d4, 100 MHz) δ 172.9, 165.6, 162.0, 154.6, 141.2, 118.6, 113.6, 76.6, 62.4, 52.2, 45.3, 29.5, 14.9 ppm. HR-ESI(+)-MS m/z calcd for C13H15N2O6: 295.0925 [M + H]+, found 295.0923. NMR and mass spectra are presented in Suppporting Information (Figure S14, S15 and S16 respectively).
3.2. Radiochemistry
All reactions were carried out using a TRACERlab® FX N Pro module (GE Healthcare).
No carrier-added [18F]fluoride ion was produced via the 18O(p, n)18F nuclear reaction by irradiation of a 2 mL [18O]water (>97% enriched, Rotem) target with an IBA Cyclone-18/9 (IBA) cyclotron.
Analytical HPLC was performed using a 717plus Autosampler system, a 1525 binary pump, a 2996 photodiode array detector (Waters, Milford, MA, USA), and a Flowstar LB 513 (Berthold, Thoiry, France) gamma detector. The system was operated with the Empower 3 (Waters, Milford, MA, USA) software. HPLC were realized on a reverse phase analytical Symmetry® C18 (150 × 3.9 mm, 5 μm, Waters) column using a mixture of H2O/CH3CN/PicB7® as eluent. The chemical identification of the peak was assessed by comparing the retention time of the radiotracer with the retention time of the non-radioactive reference (tRref). For acceptance, the retention time must be within the tRref ± 10% range. Radiochemical and chemical purities were calculated as the ratio of the AUC of the radiotracer peak over the sum of the AUCs of all other peaks on gamma and UV chromatograms, respectively. The conversion of the reaction was calculated as the ratio of the decay-corrected activity of the radiotracer at the end of the synthesis over the activity of the starting material, both measured in an ionization chamber (Capintec®, Berthold, Thoiry, France).
2-Fluoro-4-([18F]fluoro)benzonitrile ([18F]1). [18F]F− (2–3 GBq) was trapped on an ion exchange resin QMA light (Waters, Milford, MA, USA) and eluted with a solution of K2CO3 (2 mg) in a mixture of CH3CN (800 µL) and H2O (200 µL) containing Kryptofix-222 (12–15 mg). The resulting complex was dried upon heating at 60 °C for 7 min under vacuum and a stream of helium followed by heating at 120 °C for 5 min under vacuum only. Upon cooling to 70 °C, a solution of 2-fluoro-4-nitrobenzonitrile (5 mg) in the solvent (CH3CN or DMSO, 700 µL) was added and the mixture was heated at 95 °C for 5, 10, or 15 min. Upon cooling to room temperature, the mixture was diluted with CH3CN (2 mL) and analyzed by TLC on aluminum pre-coated plates of silica gel 60F254 (VWR, Rosny-sous-bois, France).) using a mixture of dichloromethane/methanol 9/1 v/v as eluent. TLC plates were analyzed with a Mini-scan TLC imaging scanner equipped with a FC-3600 beta probe (Bioscan Inc., Washington, DC, WA, USA). TLC chromatograms were recorded using the ChromeleonTM (ThermoFischer, Waltham, MA, USA) software.
2-Fluoro-4-([18F]fluoro)benzylamine ([18F]2). To a solution of [18F]1 (tracer quantity) in the appropriate solvent (1 mL) were added the reduction agents, and the reaction took place under the conditions depicted in Table 2 and Table 3. The reaction was quenched using a mixture of CH3CN/H2O 1/1 v/v (1 mL), and the crude was analyzed by HPLC according to the general procedure. A mixture of H2O/CH3CN/Et3N (80/20/0.1 v/v/v, 2 mL/min) was used as eluent, and UV detection was performed at 254 nm.
[18F]Dolutegravir ([18F]DTG). To a solution of [18F]2 (tracer quantity) in DMF (500 µL) were added compound 3 (12 mg), TBTU (13 mg), and DIPEA (15 µL). The reaction was stirred at room temperature for 10 min and quenched with a mixture of CH3CN/H2O 1/1 v/v (1 mL). The crude was analyzed by HPLC according to the general procedure. A mixture of H2O/CH3CN/PicB7® (70/30/0.2, 2 mL/min) was used as eluent, and UV detection was performed at 258 nm.
Fully automated radiosynthesis of [18F]Dolutegravir. [18F]F− (20–30 GBq) was trapped on an ion exchange resin QMA light (Waters) and eluted in reactor 1 with a solution of K2CO3 (2 mg) in a mixture of CH3CN (800 µL) and H2O (200 µL) containing Kryptofix-222 (12–15 mg). The resulting complex was dried upon heating at 60 °C for 7 min under vacuum and a stream of helium followed by heating at 120 °C for 5 min under vacuum only. Upon cooling to 70 °C, a solution of 2-fluoro-4-nitrobenzonitrile (5 mg) in DMSO (700 µL) was added, and the mixture was heated at 95 °C for 5 min. After cooling to room temperature, the mixture was diluted with water (10 mL) and passed through a C18 Sep Pak® cartridge (Waters). The cartridge was rinsed with water (5 mL) and eluted with methanol (1.5 mL) to reactor 2. A solution of NaBH4 2M (20 µL) in THF (100µL) was added, followed by the addition of a solution of NiCl2·6H2O (25 mg) in water (100 µL). The mixture was stirred at room temperature for 5 min. A solution of compound 3 (12 mg), TBTU (13 mg), and DIPEA (15 µL) in DMF (500 µL) was added, and the mixture was stirred at room temperature for 10 min. The reaction was diluted with a mixture of CH3CN/H2O/TFA 30/70/0.1 v/v/v (1.5 mL), and the crude was purified by reverse phase semi-preparative HPLC (Waters Symmetry® C18 7.8 × 300 mm, 7 μm) with a 515 HPLC Pump (Waters, Milford, MA, USA) using a mixture of H2O/CH3CN/TFA (70/30/0.1 v/v/v, 5 mL/min) as eluent. UV detection (K2501, Knauer, Berlin, Germany) was performed at 254 nm. The purified compound was diluted with water (20 mL) and passed through a Sep-Pak® C18 cartridge (Waters, Milford, MA, USA). The cartridge was rinsed with water (10 mL) and eluted with ethanol (2 mL), and the final compound was diluted with saline (0.9% w/v, 8 mL). Ready-to-inject [18F]DTG (0.5–0.7 GBq) was obtained in 5.1 ± 0.8% (n = 10) non-decay corrected radiochemical yield within 95 min.
Quality control. Quality control was performed by reverse phase analytical HPLC according to the general procedure described above. A mixture of H2O/CH3CN/PicB7® (70/30/0.2 v/v/v, 2 mL/min) was used as eluent, and UV detection was performed at 258 nm. Molar activity was calculated as the ratio of the activity of the collected peak of [18F]DTG measured in an ionization chamber (Capintec®, Berthold, Thoiry, France) over the molar quantity of DTG determined using a calibration curve (see Figure S18 in Supplementary Materials). Molar activity is calculated as the mean value of three consecutive runs.
3.3. PET Imaging
Whole-body distribution of [18F]DTG was assessed using whole-body (3-bed step) PET acquisition in a healthy male macaque (Macaca fascicularis, 8.35 kg, 10 years). Animal was i.v. injected with 181 MBq of 18F-DTG followed by PET acquisition performed using a Siemens Biograph PET-CT scanner (Siemens Healthineers, Knoxville, TN, USA) as previously described [55]. Mean PET signal acquired from 150 to 180 min after injection of [18F]DTG was normalized by injected dose of [18F]DTG and animal weight (standardized uptake value, SUV). Representative PET image was co-registered to the anatomical CT scan.
4. Conclusions
We have developed the first fluorine-18 isotopic radiolabeling of the antiretroviral drug dolutegravir. The three steps of the radiosynthesis, i.e., radiofluorination, reduction, and peptide coupling reactions, have been optimized to maximize the quantity of [18F]DTG produced. The whole process has been automated for reproducibility and radioprotection purposes to ensure the feasibility of preclinical PET imaging studies. For that matter, quality control has been set up, and 500–700 MBq of ready-to-inject [18F]DTG was produced, a quantity compatible with injection into primates or humans. As a proof of concept, the first in vivo injection of [18F]DTG was carried out in a macaque, and PET imaging was performed to observe the biodistribution of the tracer. [18F]DTG is currently being used in PET studies with healthy and simian immunodeficiency virus (SIV)-positive non-human primates to explore the distribution of cART at remaining SIV sanctuaries and to understand their resistance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15050587/s1, Scheme S1: Synthesis of late-stage labelling precursors of DTG.; Scheme S2: Radiolabelling of the tributyltin precursor s5 according to the conditions of the literature; Figure S1: 1H NMR of compound s1; Figure S2: 13C NMR of compound s1; Figure S3: HRMS analysis of compound s1; Figure S4: 1H NMR of compound s2; Figure S5: 13C NMR of compound s2; Figure S6: HRMS analysis of compound s2; Figure S7: LC-MS− analysis of crude compound s3; Figure S8: LC-MS+ analysis of crude compound s4; Figure S9: 1H NMR of compound s5; Figure S10: 13C NMR of compound s5; Figure S11: HRMS analysis of compound s5; Figure S12: TLC analysis of the radiofluorination step; Figure S13: HPLC analysis of a mixture of compounds 1, 2 and 2′; Scheme S3: Synthesis of compound 3 from GSK3210346A; Figure S14: 1H NMR of compound 3; Figure S15: 13C NMR of compound 3; Figure S16: HRMS analysis of compound 3; Figure S17: Semi-preparative HPLC purification of [18F]DTG; Figure S18: Calibration curve of DTG for molar activity calculation.
Author Contributions
Conceptualization, R.L.G., C.M.P., N.T., B.K. and T.B.; methodology, M.T., S.G., M.G., B.K. and F.C.; writing—original draft preparation, M.T. and F.C.; writing—review and editing, F.C.; supervision, D.D. and R.L.G.; project administration, D.D.; funding acquisition, R.L.G. and C.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the ANRS RHIVIERA and ViiV Healthcare.
Institutional Review Board Statement
All animal use procedures were in accordance with the recommendations of the European Community for the care and use of laboratory animals (2010/63/UE) and the French National Committees (French Decret 2013-118). Experimental protocols were validated by a local ethics committee for animal use (CETEA/A15-002 and A18-065) and approved by the French government.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within article and Supplementary Materials.
Acknowledgments
The authors thank Sarah Leterrier for her contribution to the quantitative PET data determination. This work was performed on a platform member of the France Life Imaging network (ANR-11-INBS-0006). The Infectious Disease Models and Innovative Therapies (IDMIT) research infrastructure is supported by the “Programme Investissements d’Avenir” managed by the ANR under reference ANR-11-INBS-0008.
Conflicts of Interest
Thomas Bonasera is employed by GSK. Chris M. Parry is employed by ViiV Healthcare and has stock/shares in GSK.
References
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-Treatment HIV-1 Controllers with a Long-Term Virological Remission after the Interruption of Early Initiated Antiretroviral Therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Pancino, G. HIV controllers: A genetically determined or inducible phenotype? Immunol. Rev. 2013, 254, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Passaes, C.P.; Sáez-Cirión, A. HIV cure research: Advances and prospects. Virology 2014, 454, 340–352. [Google Scholar] [CrossRef] [PubMed]
- The Antiretroviral Therapy Cohort Collaboration. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: A collaborative analysis of 14 cohort studies. Lancet 2008, 372, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Cory, T.J.; Schacker, T.W.; Stevenson, M.; Fletcher, C.V. Overcoming pharmacologic sanctuaries. Curr. Opin. HIV AIDS 2013, 8, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.-H.; Lichterfeld, M. Recent progress in understanding HIV reservoirs. Curr. Opin. HIV AIDS 2018, 13, 137–142. [Google Scholar] [CrossRef]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral tissue kinetics: In vivo imaging using positron emission tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Di Mascio, M.; Paik, C.H.; Carrasquillo, J.A.; Maeng, J.-S.; Jang, B.-S.; Shin, I.S.; Srinivasula, S.; Byrum, R.; Neria, A.; Kopp, W.; et al. Antiretroviral Tissue Kinetics: In Vivo Imaging Using Positron Emission Tomography. Blood 2009, 114, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, P.J.; Rogers, K.A.; Zurla, C.; Blanchard, E.L.; Gumber, S.; Strait, K.; Connor-Stroud, F.; Schuster, D.M.; Amancha, P.K.; Hong, J.J.; et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat. Methods 2015, 12, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Henrich, T.J.; Hsue, P.Y.; VanBrocklin, H. Seeing Is Believing: Nuclear Imaging of HIV Persistence. Immunology 2019, 10, 2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronald, J.A.; Kim, B.-S.; Gowrishankar, G.; Namavari, M.; Alam, I.S.; D’Souza, A.; Nishikii, H.; Chuang, H.-Y.; Ilovich, O.; Lin, C.-F.; et al. A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant. Cancer Res. 2017, 77, 2893–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namavari, M.; Chang, Y.-F.; Kusler, B.; Yaghoubi, S.; Mitchell, B.S.; Gambhir, S.S. Synthesis of 2′-Deoxy-2′-[18F]Fluoro-9-β-D-Arabinofuranosylguanine: A Novel Agent for Imaging T-Cell Activation with PET. Mol. Imaging Biol. 2011, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.-Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef]
- McMahon, J.H.; Zerbato, J.M.; Lau, J.S.Y.; Lange, J.L.; Roche, M.; Tumpach, C.; Dantanarayana, A.; Rhodes, A.; Chang, J.; Rasmussen, T.A.; et al. A clinical trial of non-invasive imaging with an anti-HIV antibody labelled with copper-64 in people living with HIV and uninfected controls. EBioMedicine 2021, 65, 103252. [Google Scholar] [CrossRef]
- Vera, D.B.; Schulte, B.; Henrich, T.; Flavell, R.; Seo, Y.; Abdelhafez, Y.; Badawi, R.; Cherry, S.; VanBrocklin, H. First-in-human total-body PET imaging of HIV with 89Zr-VRC01 on the EXPLORER. J. Nucl. Med. 2020, 61, 545. [Google Scholar]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Sax, P.E.; Pozniak, A.; Montes, M.L.; Koenig, E.; DeJesus, E.; Stellbrink, H.-J.; Antinori, A.; Workowski, K.; Slim, J.; Reynes, J.; et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380–1490): A randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet 2017, 390, 2073–2082. [Google Scholar]
- Cahn, P.; Pozniak, A.L.; Mingrone, H.; Shuldyakov, A.; Brites, C.; Andrade-Villanueva, J.F.; Richmond, G.; Buendia, C.B.; Fourie, J.; Ramgopal, M.; et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: Week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013, 382, 700–708. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news/item/22-07-2019-who-recommends-dolutegravir-as-preferred-hiv-treatment-option-in-all-population (accessed on 10 January 2022).
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [Green Version]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cheng, R.; Chen, P.; Liu, G.; Liang, S.H. Novel Path to Aryl(isoquinoline)iodonium(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem. Int. Ed. 2016, 55, 11882–11886. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B.H.; Stephenson, N.A.; Vasdev, N.; Liang, S.H. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 2014, 5, 4365. [Google Scholar] [CrossRef] [Green Version]
- Rotstein, B.H.; Wang, L.; Liu, R.Y.; Patteson, J.; Kwan, E.E.; Vasdev, N.; Liang, S.H. Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem. Sci. 2016, 7, 4407–4417. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, J.E.; Grønnevik, G.; Riss, P.J. Organocatalyst-assisted Ar–18F bond formation: A universal procedure for direct aromatic radiofluorination. Chem. Commun. 2017, 53, 12906–12909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef] [PubMed]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Krasikova, R. Effect of Pyridine Addition on the Efficiency of Copper-Mediated Radiofluorination of Aryl Pinacol Boronates. ChemistrySelect 2017, 2, 7909–7912. [Google Scholar] [CrossRef]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Zhang, X.; Basuli, F.; Swenson, R.E. Positron Emission Tomography Imaging with 2-[18F]F-p-Aminobenzoic Acid Detects Staphylococcus aureus Infections and Monitors Drug Response. J. Label. Compd. Radiopharm. 2019, 62, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef] [PubMed]
- Lahdenpohja, S.; Keller, T.; Rajander, J.; Kirjavainen, A.K. Radiosynthesis of the norepinephrine transporter tracer [18F]NS12137 via copper-mediated 18F-labelling. J. Label. Compd. Radiopharm. 2019, 62, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Goodman, S.N.; Mans, D.; Kowalski, M. Process for Preparing Carbamoylpyridone Derivatives and Intermediates. EP3260457A1, 27 December 2017. [Google Scholar]
- Li, W.; Thompson, W.; Fisher, T.; Wai, J.S.; Hazuda, D.; Burns, H.D.; Hamill, T.G. Radiosynthesis of the HIV integrase inhibitor [18F]MK-0518 (Isentress). J. Label. Compd. Radiopharm. 2010, 53, 517–520. [Google Scholar] [CrossRef]
- Blecha, J.; Neumann, K.; VanBrocklin, H. Automated synthesis of [18F]Raltegravir through [18F]fluorobenzylamine. J. Nucl. Med. 2018, 59, 669. [Google Scholar]
- Dietz, J.-P.; Lucas, T.; Groß, J.; Seitel, S.; Brauer, J.; Ferenc, D.; Gupton, B.F.; Opatz, T. Six-Step Gram-Scale Synthesis of the Human Immunodeficiency Virus Integrase Inhibitor Dolutegravir Sodium. Org. Process Res. Dev. 2021, 25, 1898–1910. [Google Scholar] [CrossRef]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [Green Version]
- Koslowsky, I.; Mercer, J.; Wuest, F. Synthesis and application of 4-[18F]fluorobenzylamine: A versatile building block for the preparation of PET radiotracers. Org. Biomol. Chem. 2010, 8, 4730–4735. [Google Scholar] [CrossRef]
- Clot, E.; Eisenstein, O.; Jasim, N.; Macgregor, S.A.; McGrady, J.E.; Perutz, R.N. C−F and C−H Bond Activation of Fluorobenzenes and Fluoropyridines at Transition Metal Centers: How Fluorine Tips the Scales. Acc. Chem. Res. 2011, 44, 333–348. [Google Scholar] [CrossRef]
- Lv, H.; Cai, Y.-B.; Zhang, J.-L. Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chem. Int. Ed. 2013, 52, 3203–3207. [Google Scholar] [CrossRef]
- Burkhardt, E.R.; Matos, K. Boron Reagents in Process Chemistry: Excellent Tools for Selective Reductions. Chem. Rev. 2006, 106, 2617–2650. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, L.; Máthé, T. Selective heterogeneous catalytic hydrogenation of nitriles to primary amines in liquid phase: Part I. Hydrogenation of benzonitrile over palladium. Appl. Catal. Gen. 2005, 296, 209–215. [Google Scholar] [CrossRef]
- Gair, J.J.; Grey, R.L.; Giroux, S.; Brodney, M.A. Palladium Catalyzed Hydrodefluorination of Fluoro-(hetero)arenes. Org. Lett. 2019, 21, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Werkmeister, S.; Bornschein, C.; Junge, K.; Beller, M. Selective Ruthenium-Catalyzed Transfer Hydrogenations of Nitriles to Amines with 2-Butanol. Chem. Eur. J. 2013, 19, 4437–4440. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Judd, D.B.; Lewis, A.K.D.K.; Reich, M.T.; Williams, M.R.V. A generic approach for the catalytic reduction of nitriles. Tetrahedron 2003, 59, 5417–5423. [Google Scholar] [CrossRef]
- Way, J.; Wuest, F. Fully automated synthesis of 4-[18F]fluorobenzylamine based on borohydride/NiCl2 reduction. Nucl. Med. Biol. 2013, 40, 430–436. [Google Scholar] [CrossRef]
- Tietz, O.; Sharma, S.K.; Kaur, J.; Way, J.; Marshall, A.; Wuest, M.; Wuest, F. Synthesis of three 18F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org. Biomol. Chem. 2013, 11, 8052–8064. [Google Scholar] [CrossRef]
- Budidet, S.R.; Dussa, N.; Kaki, G.R.; Yatcherla, S.R.; Sanapureddy, J.M.R.; Danda, S.R.; Katuroju, S.; Meenakshisunderam, S. An Improved Process for the Preparation of Dolutegravir. International Patent Application No. WO2014128545A2, 28 August 2014. [Google Scholar]
- Kalantzopoulos, G.N.; Guzik, M.N.; Deledda, S.; Heyn, R.H.; Muller, J.; Hauback, B.C. Destabilization effect of transition metal fluorides on sodium borohydride. Phys. Chem. Chem. Phys. 2014, 16, 20483–20491. [Google Scholar] [CrossRef]
- General Monograph 0125—Radiopharmaceutical Preparations in European Pharmacopoeia; Council of Europe: Strasbourg, France, 2016.
- Castellino, S.; Moss, L.; Wagner, D.; Borland, J.; Song, I.; Chen, S.; Lou, Y.; Min, S.S.; Goljer, I.; Culp, A.; et al. Metabolism, Excretion, and Mass Balance of the HIV-1 Integrase Inhibitor Dolutegravir in Humans. Antimicrob. Agents Chemother. 2013, 57, 3536–3546. [Google Scholar] [CrossRef] [Green Version]
- Labarthe, L.; Gelé, T.; Gouget, H.; Benzemrane, M.-S.; Le Calvez, P.; Legrand, N.; Lambotte, O.; Le Grand, R.; Bourgeois, C.; Barrail-Tran, A. Pharmacokinetics and tissue distribution of tenofovir, emtricitabine and dolutegravir in mice. J. Antimicrob. Chemother. 2022, 77, 1094–1101. [Google Scholar] [CrossRef]
- Tournier, N.; Saba, W.; Cisternino, S.; Peyronneau, M.-A.; Damont, A.; Goutal, S.; Dubois, A.; Dollé, F.; Scherrmann, J.-M.; Valette, H.; et al. Effects of Selected OATP and/or ABC Transporter Inhibitors on the Brain and Whole-Body Distribution of Glyburide. AAPS J. 2013, 15, 1082–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
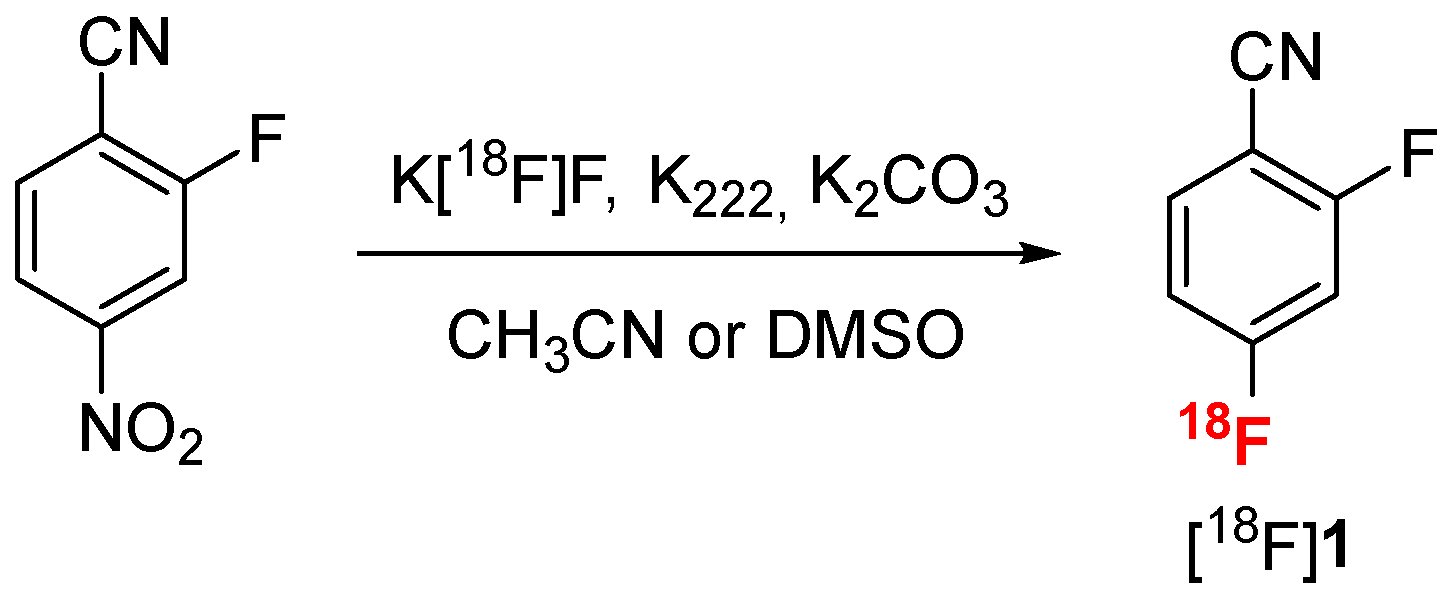
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).