
Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners

 ,
,  ,
,
Abstract
: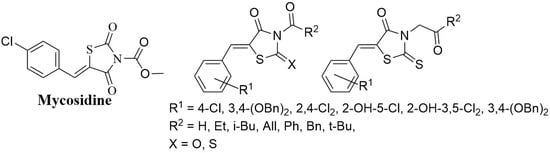
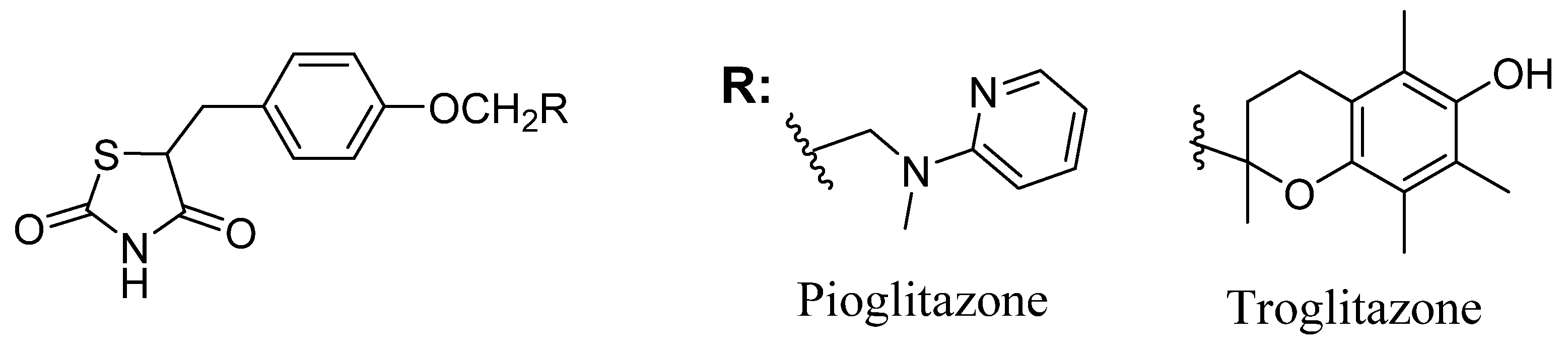
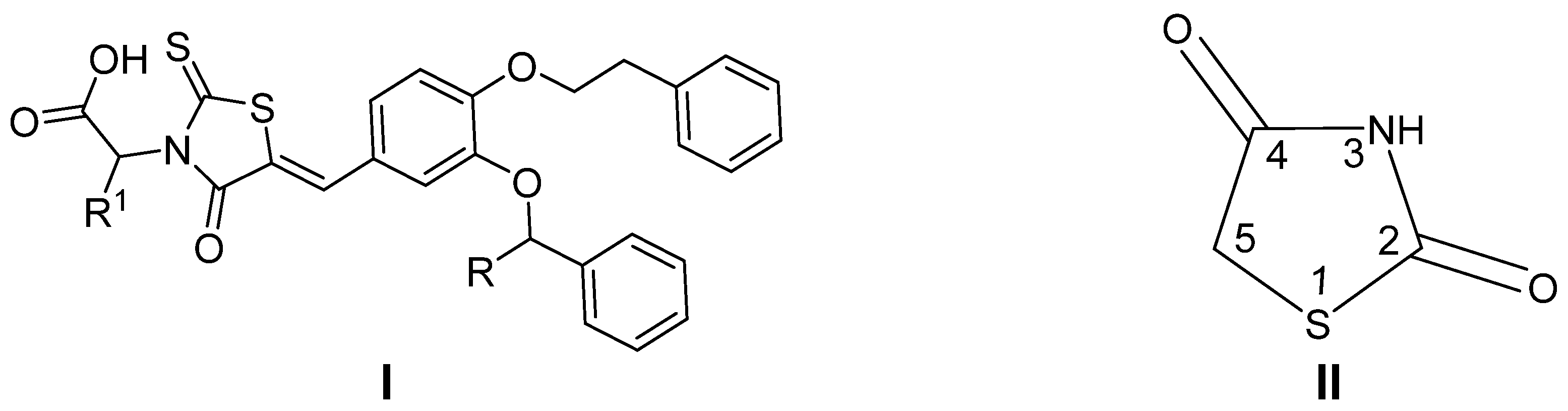
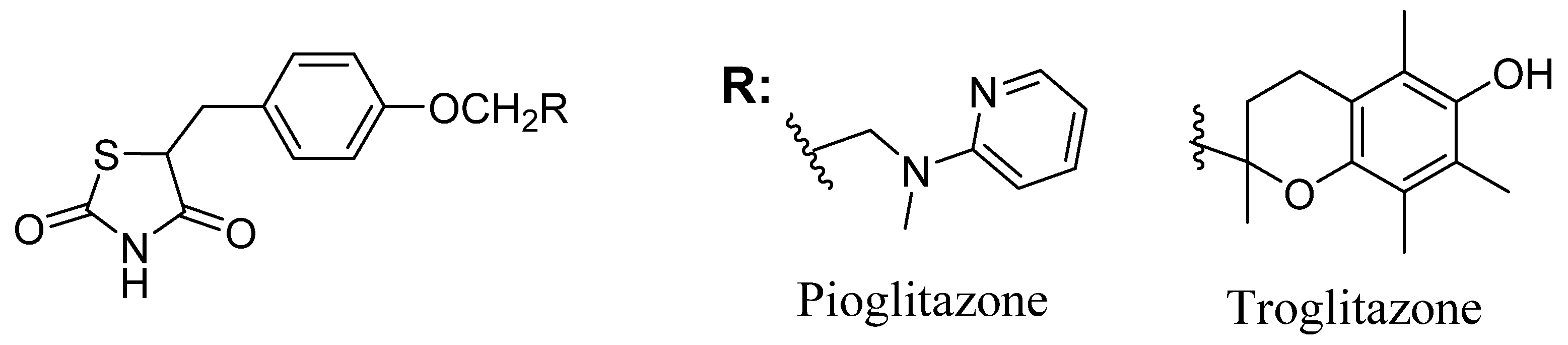
1. Introduction
2. Results
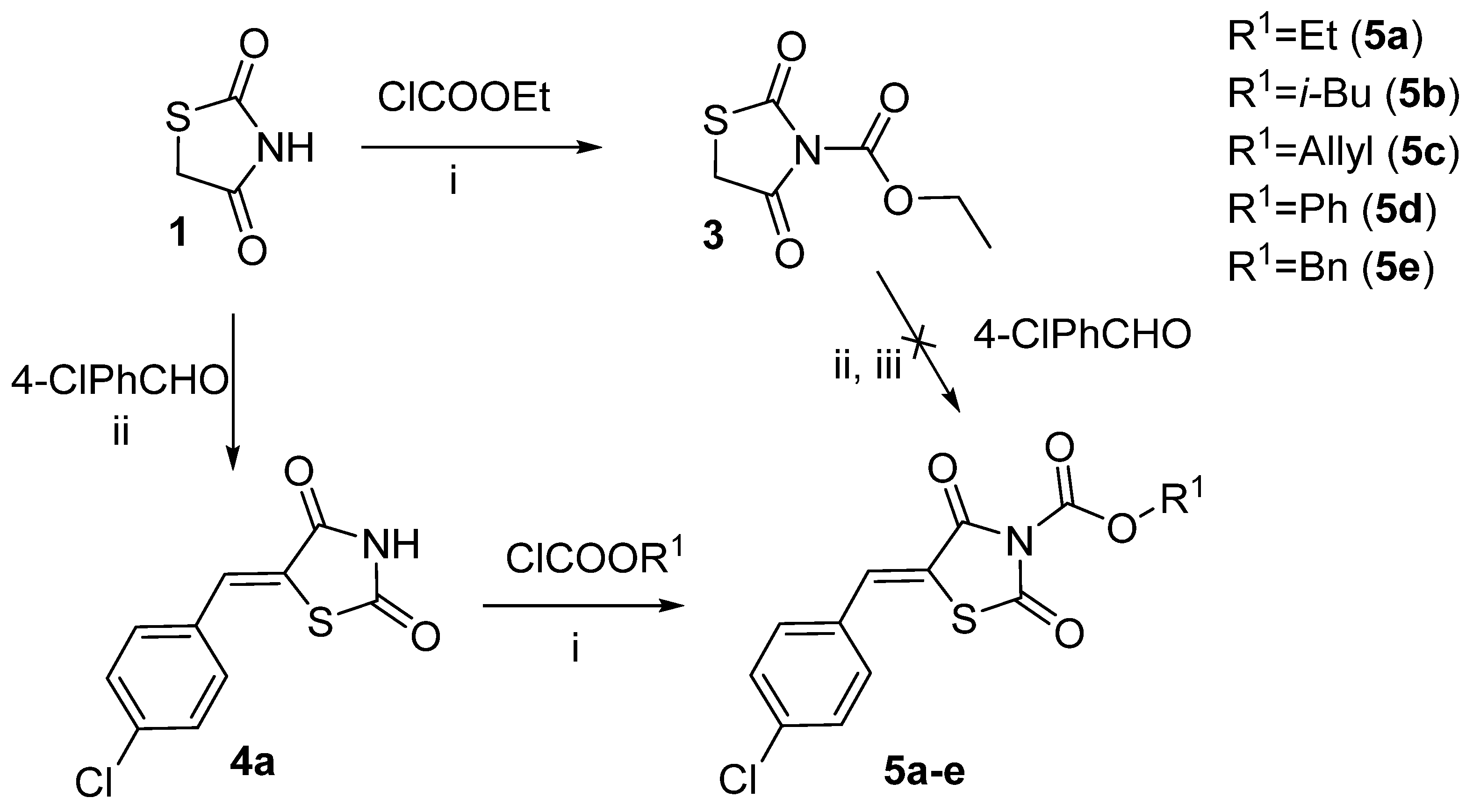
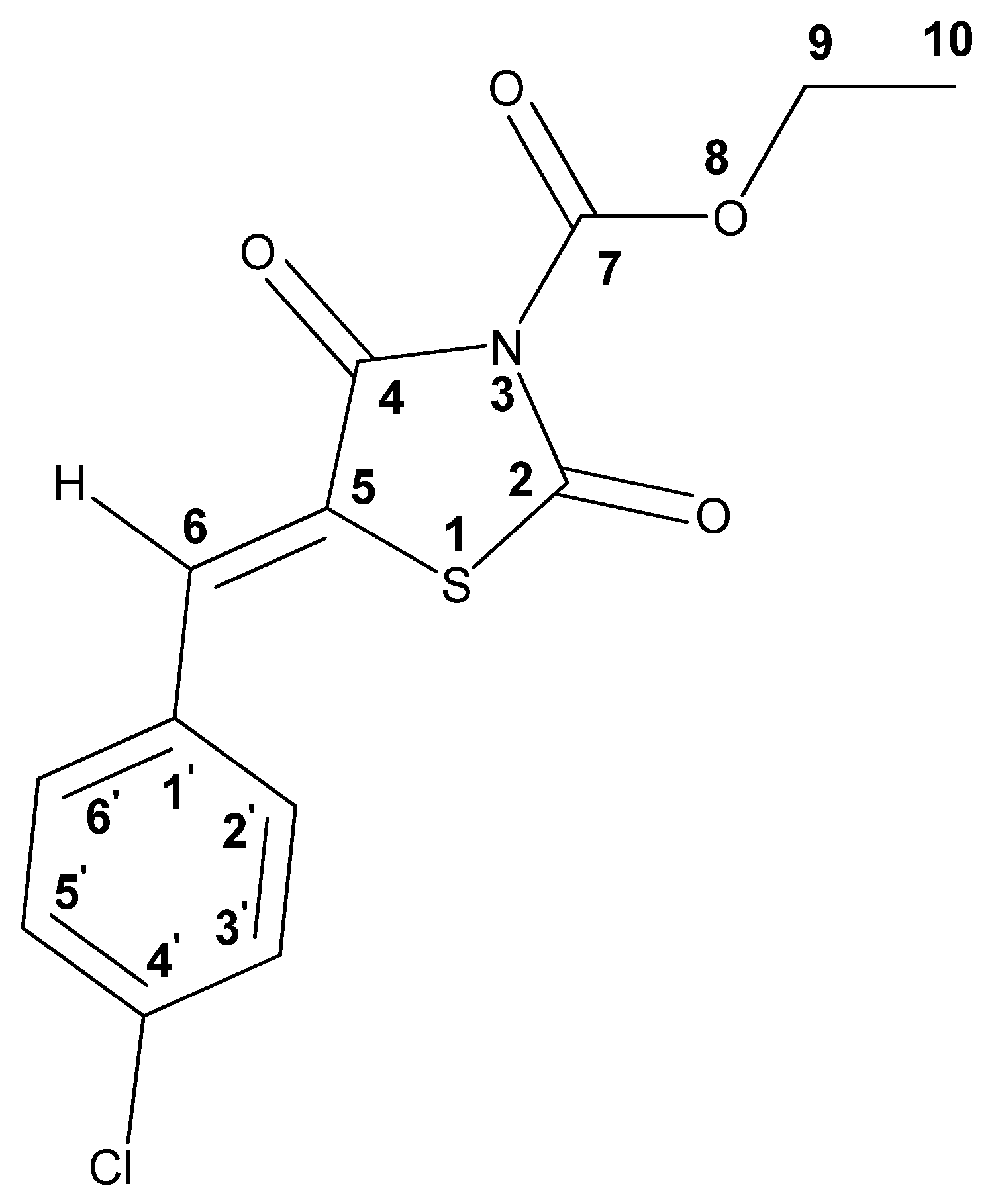
2.1. Ethyl 2,4-Dioxothiazolidine-3-Carboxylate and Other Thiazolidine-3-Carboxylates
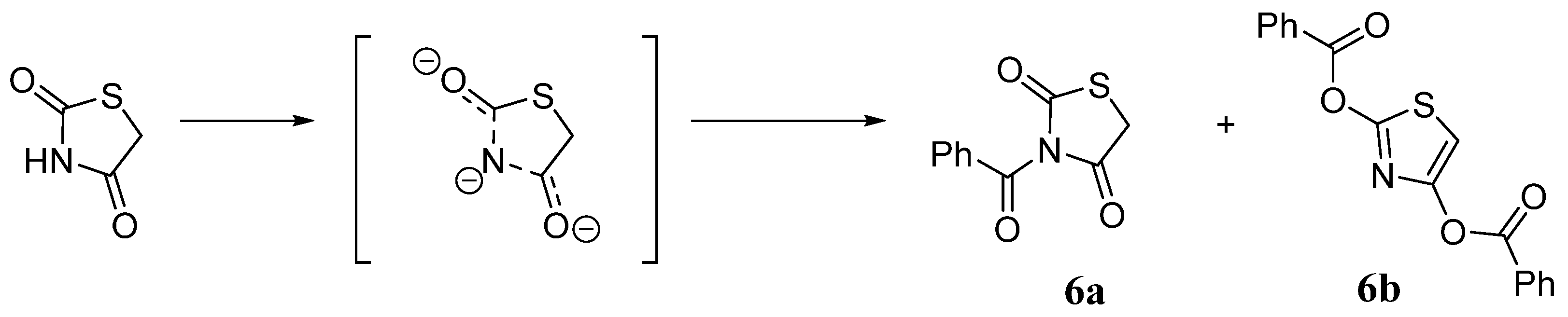
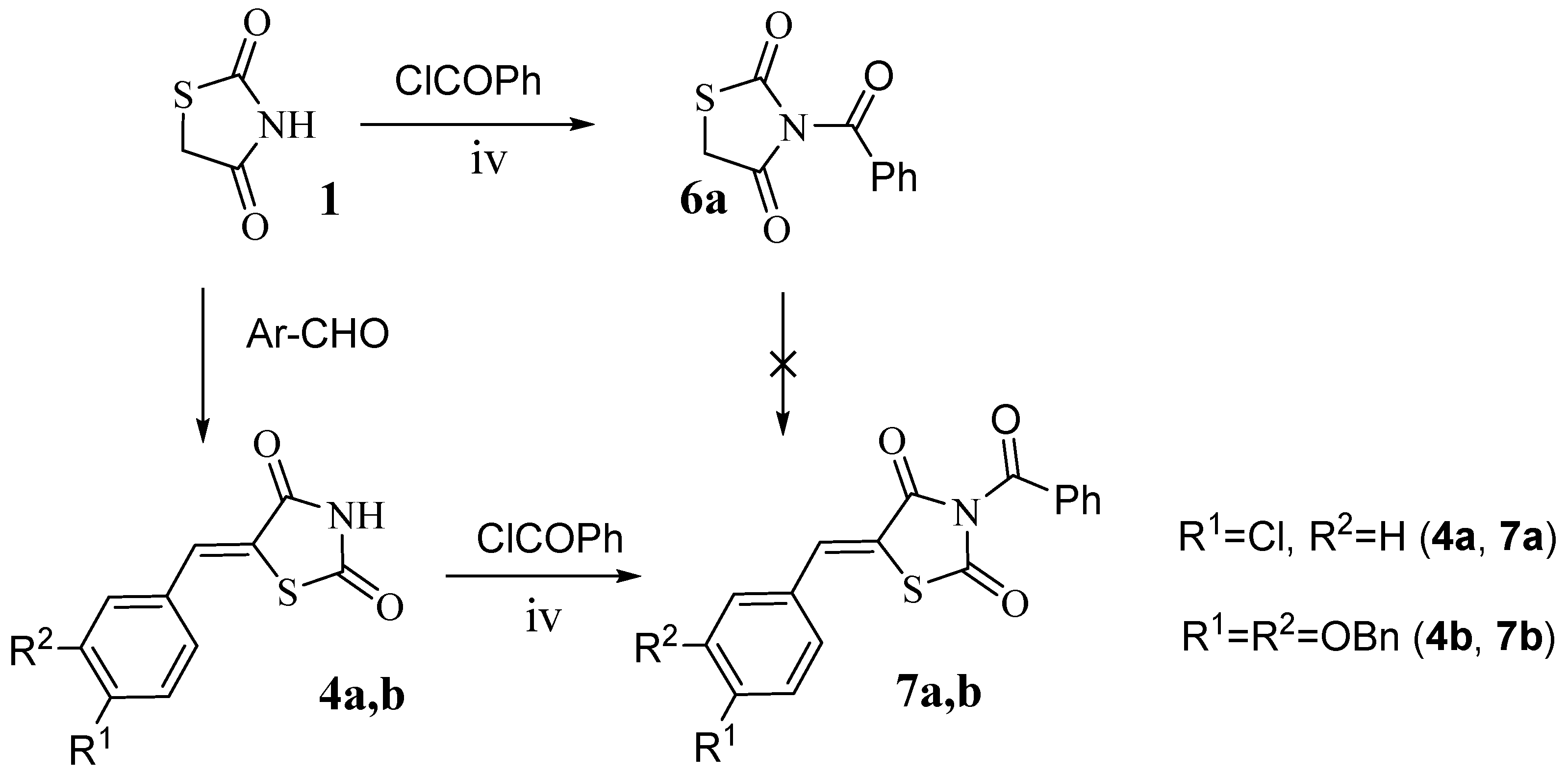
2.2. 5-Arylidene-3-Benzoylthiazolidine-2,4-Dione Derivatives
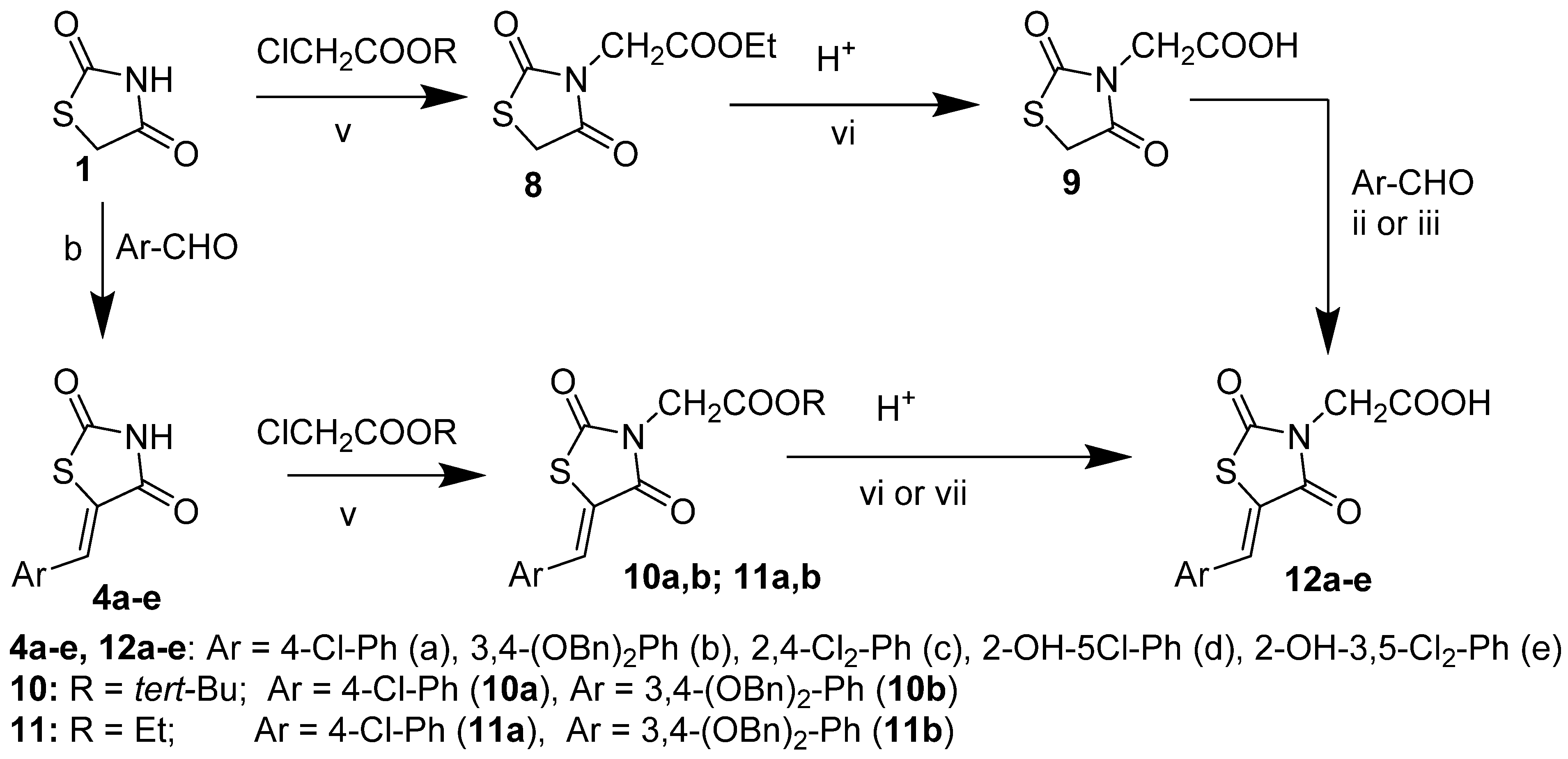
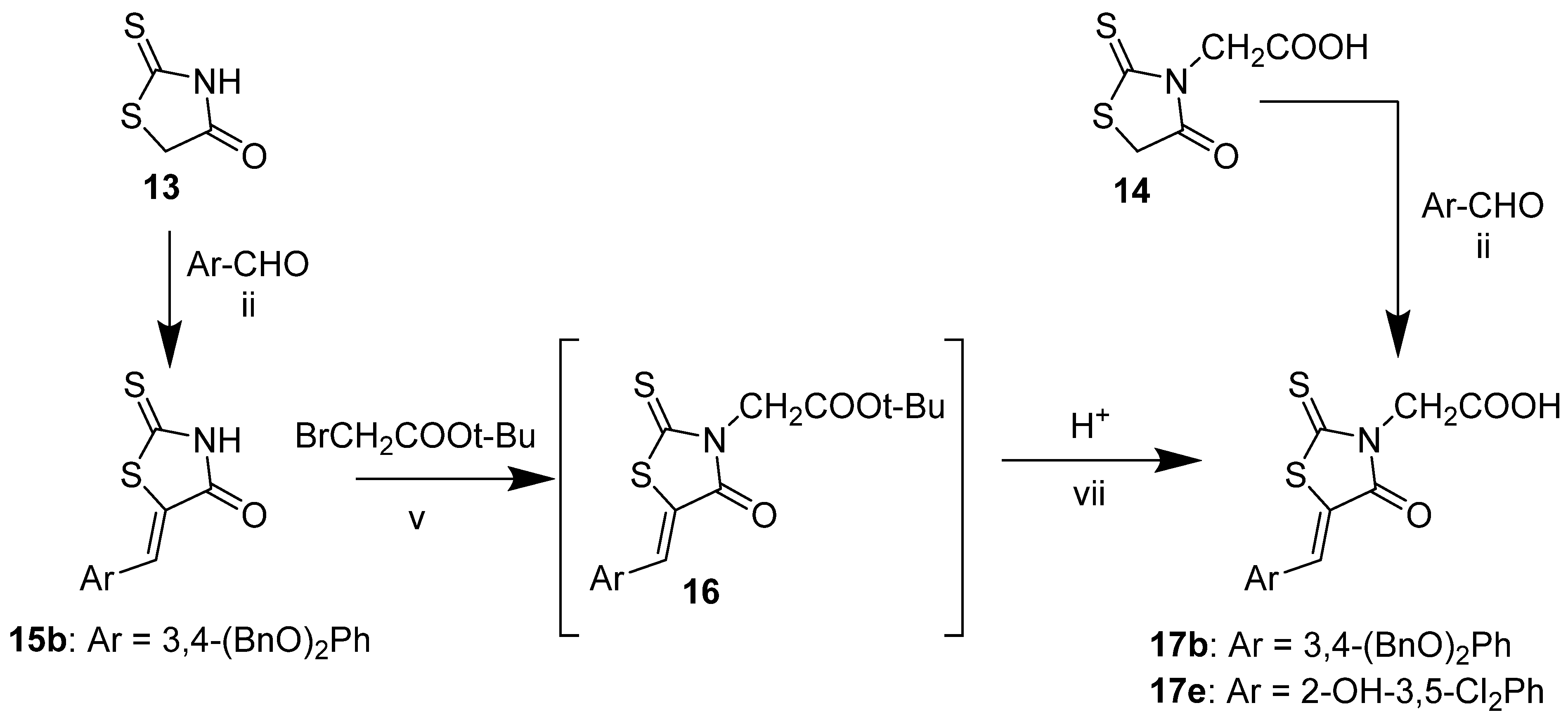
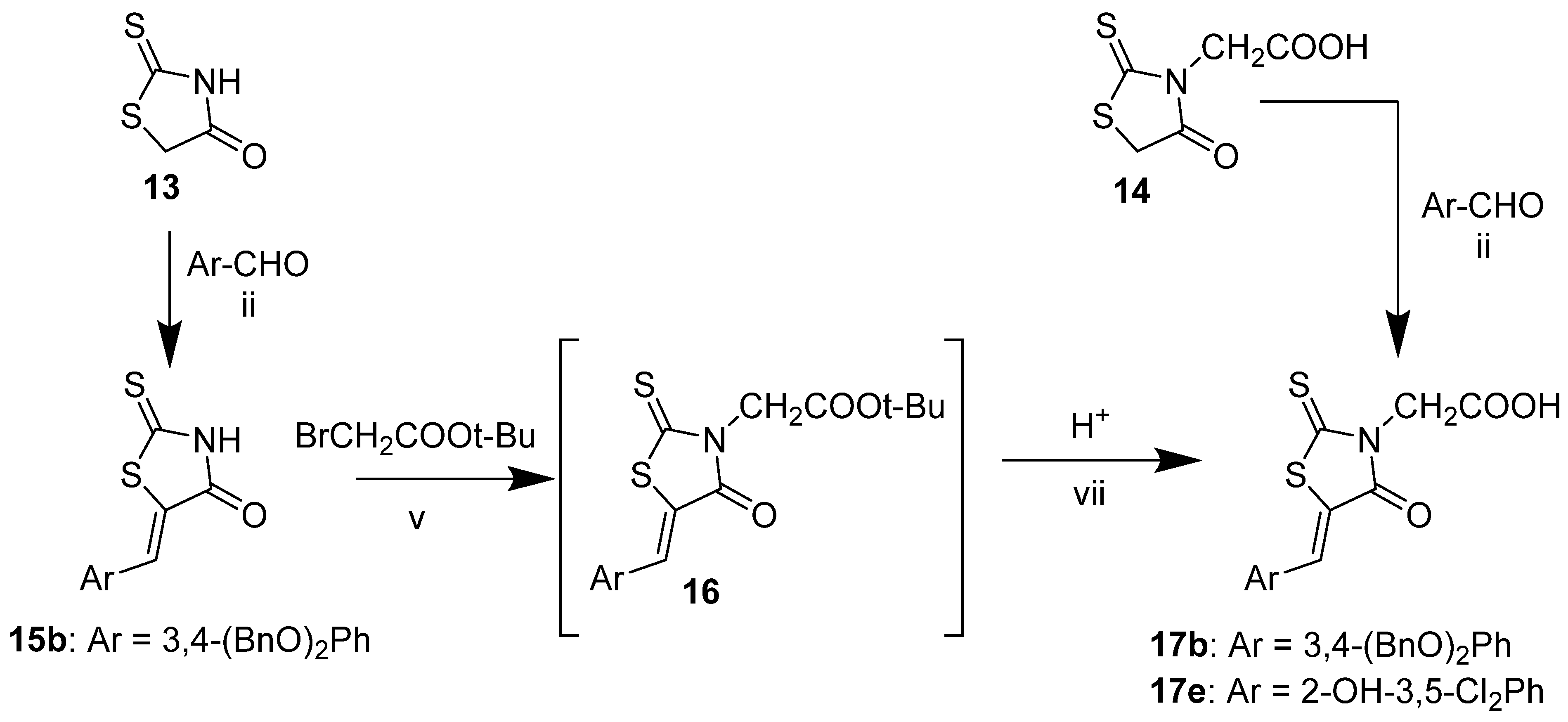
2.3. 2-(2,4-Dioxothiazolidin-3-yl)Acetic Acid and Its Derivatives
2.4. Evaluation of the Biological Activity of Synthesized Compounds
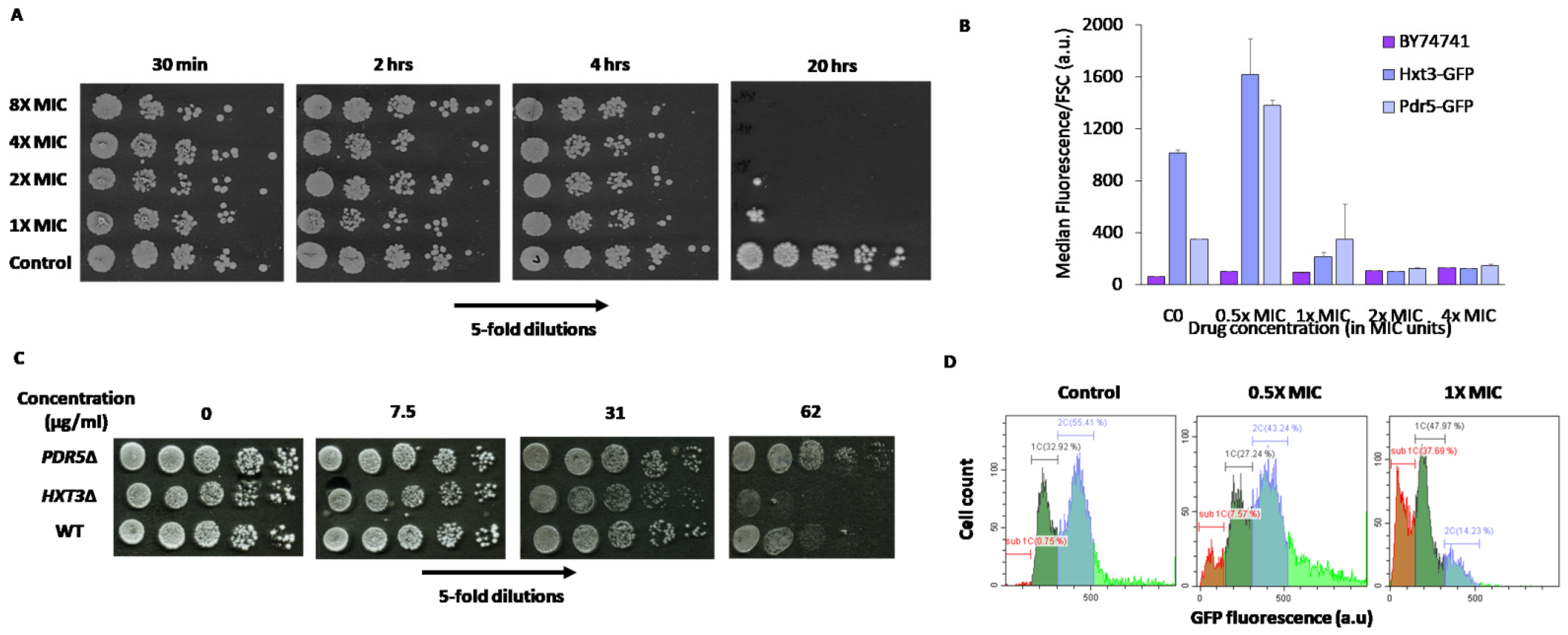
2.5. Cellular Response to Mycosidine
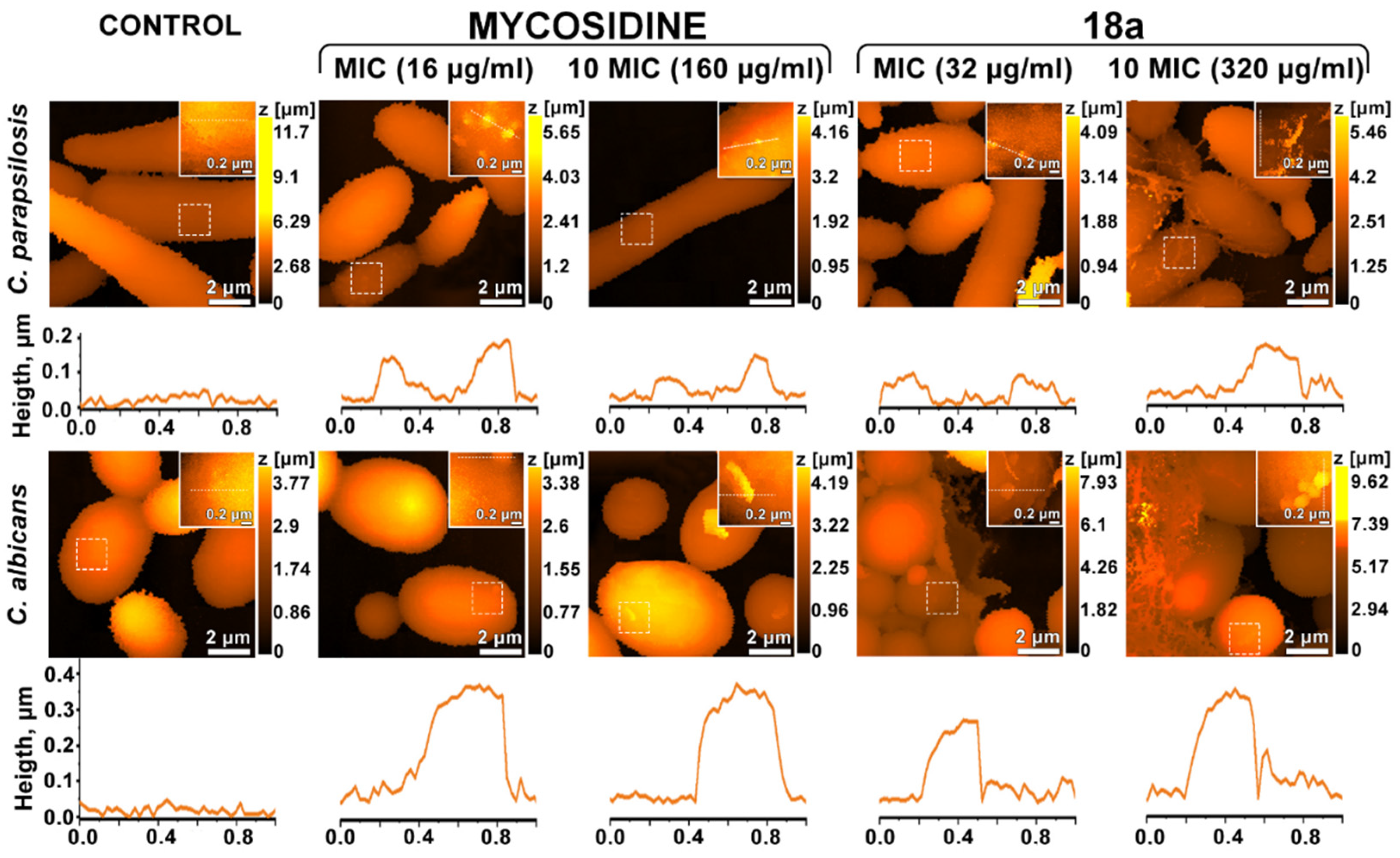
2.6. Effects on the Cell Wall
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and General Methods
3.1.2. Synthesis of the Starting Materials
3.1.3. Synthesis of the Target Compounds
3.2. Biochemistry and Microbiology
3.2.1. MIC Determination
3.2.2. Growth Assays and Cell Death Detection
3.2.3. Flow Cytometric Analysis of Changes in GFP-Tagged Protein Abundance
3.2.4. Sample Preparation for Cell Wall Investigation
3.2.5. Scanning Ion-Conductance Microscopy (SICM)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tripathi, K.D. Essentials of Medical Pharmacology, 3rd ed.; Jaypee Brothers Medical Publishers (P) Ltd.: New Delhi, India, 1994; pp. 707–716. [Google Scholar]
- Denyer, S.P.; Hodges, N.; Gorman, S.P.; Gilmore, B.F. Hugo and Russell’s Pharmaceutical Microbiology, 8th ed.; A John Wiley & Sons, Ltd. Publication: Hoboken, NJ, USA, 2011; pp. 191–193. [Google Scholar]
- Clark, M.A.; Finkel, R.; Rey, J.A.; Whalen, K. Lippincott’s Illustrated Reviews: Pharmacology, 5th ed.; Lippincott Williams & Wilkins, a Wolters Kluwer Business: Philadelphia, PA, USA, 2012; pp. 429–440. [Google Scholar]
- Ghannoum, M.; Arendrup, M.C.; Chaturvedi, V.P.; Lockhart, S.R.; McCormick, T.S.; Chaturvedi, S.; Berkow, E.L.; Juneja, D.; Tarai, B.; Azie, N.; et al. Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida Auris Infections. Antibiotics 2020, 9, 539. [Google Scholar] [CrossRef]
- Georgopapadakou, N.H.; Tkacz, J.S. The Fungal Cell Wall as a Drug Target. Trends Microbiol. 1995, 3, 98–104. [Google Scholar] [CrossRef]
- Georgopapadakou, N.H. Update on Antifungals Targeted to the Cell Wall: Focus on Β-1,3-Glucan Synthase Inhibitors. Expert Opin. Investig. Drugs 2001, 10, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.C.G.; Curto, M.Á.; Carvalho, V.S.; Perez, P.; Ribas, J.C. The Fungal Cell Wall as a Target for the Development of New Antifungal Therapies. Biotechnol. Adv. 2019, 37, 107352. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Cell Wall-Modifying Antifungal Drugs. Curr. Top. Microbiol. Immunol. 2020, 425, 255–275. [Google Scholar] [CrossRef]
- Ibe, C.; Munro, C.A. Fungal cell wall: An Underexploited Target for Antifungal Therapies. PLoS Pathog. 2021, 17, e1009470. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Latge, J.P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis and Function. Microbiol. Spectr. 2017, 5, 1–25. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, E.V.; Polvi, E.J.; Veri, A.O.; Prive, G.G.; Cowen, L.E. Structure-guided Approaches to Targeting Stress Responses in Human Fungal Pathogens. J. Biol. Chem. 2020, 295, 14458–14472. [Google Scholar] [CrossRef]
- Galley, C.M.S.; Neuss, J.C.; Orchard, M.G. Benzylidene thiazolidinediones and their use as antimycotic agents. Patent WO2002017915 A1, 7 March 2002. [Google Scholar]
- Orchard, M.G.; Neuss, J.C.; Galley, C.M.S.; Carr, A.; Porter, D.W.; Smith, P.; Scopes, D.I.C.; Haydon, D.; Vousden, K.; Stubberfield, C.R.; et al. Rhodanine-3-acetic acid Derivatives as Inhibitors of Fungal Protein Mannosyl Transferase 1 (PMT1). Bioorg. Med. Chem. Lett. 2004, 14, 3975–3978. [Google Scholar] [CrossRef]
- Brown, F.C. 4-Thiazolidinones. Chem. Rev. 1961, 61, 463–521. [Google Scholar] [CrossRef]
- Singh, S.P.; Parimar, S.S.; Stenberg, V.I. Chemistry and biological activity of thiazolidinones. Chem. Rev. 1981, 81, 175–203. [Google Scholar] [CrossRef]
- Lesik, R.B.; Zimenkovsky, B.S. 4-Thiazolidones: Centenarian History, Current Status and Perspectives for Modern Organic and Medicinal Chemistry. Curr. Org. Chem. 2004, 8, 1547–1577. [Google Scholar] [CrossRef]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent Developments and Biological Activities of Thiazolidinone Derivatives: A Review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef]
- Jain, V.S.; Vora, D.K.; Ramaa, C.S. Thiazolidine-2,4-Diones: Progress Towards Multifarious Applications. Bioorg. Med. Chem. 2013, 21, 1599–1620. [Google Scholar] [CrossRef]
- Desai, N.C.; Pandit, U.P.; Dodiya, A. Thiazolidinedione Compounds: A Patent Review (2010 present). ExpertOpin. Ther. Pat. 2015, 25, 479–488. [Google Scholar] [CrossRef]
- Chadha, N.; Bahia, M.S.; Kaur, M.; Silakari, O. Thiazolidine-2,4-Dione Derivatives: Programmed Chemical Weapons for Key Protein Targets of Various Pathological Conditions. Bioorg. Med. Chem. 2015, 23, 2953–2974. [Google Scholar] [CrossRef]
- Manjal, S.K.; Kaur, R.; Bhatia, R.; Kumar, K.; Singh, V.; Shanka, R.; Kaur, R.; Rawal, R.K. Synthetic and Medicinal Perspective of Thiazolidinones: A Review. Bioorg. Chem. 2017, 75, 406–423. [Google Scholar] [CrossRef]
- Mounika, P.; Aishwarya, M.N.L.; Sikdar, P.; Prathima, S.; Babu, M.N. A Review on Thiazolidinedione. Asian J. Pharm. Res. 2017, 7, 124–135. [Google Scholar] [CrossRef]
- Tahlan, S.S.; Verma, P.K. Biological Potential of Thiazolidinedione Derivatives of Synthetic Origin. Chem. Cent. J. 2017, 11, 130. [Google Scholar] [CrossRef]
- Naim, M.J.; Alam, M.J.; Ahmad, S.; Nawaz, F.; Shrivastava, N.; Sahu, M.; Alam, O. Therapeutic Journey of 2,4-Thiazolidinediones as a Versatile Scaffold: An Insight Into Structure Activity Relationship. Eur. J. Med. Chem. 2017, 129, 218–250. [Google Scholar] [CrossRef]
- Sethi, N.S.; Prasad, D.N.; Singh, R.K. An Insight into the Synthesis and SAR of 2,4-Thiazolidinediones (2,4-TZD) as Multifunctional Scaffold: A Review. Mini Rev. Med. Chem. 2020, 20, 308–330. [Google Scholar] [CrossRef] [PubMed]
- Bireddy, S.R.; Konkala, V.S.; Godugu, C.; Dubey, P.K. A Review on the Synthesis and Biological Studies of 2,4-Thiazolidinedione Derivatives. Mini Rev. Org. Chem. 2020, 17, 958–974. [Google Scholar] [CrossRef]
- Agrawal, N. Synthetic and Therapeutic Potential of 4-Thiazolidinone and its Analogs. Curr. Chem. Lett. 2021, 10, 119–138. [Google Scholar] [CrossRef]
- Wiesenberg, I.; Chiesi, M.; Missbach, M.; Spanka, C.; Pignat, W.; Carlberg, C. Specific activation of the nuclear receptors PPARgamma and RORA by the antidiabetic thiazolidinedione BRL 49653 and the antiarthritic thiazolidinedione derivative CGP 52608. Mol. Pharmacol. 1998, 53, 1131. [Google Scholar]
- Tomašic, T.; Zidar, N.; Mueller-Premru, M.; Kikelj, D.L.; Peterlin, M.L. Discovery of Novel 5-Benzylidenerhodanine and 5-Benzylidenethiazolidine-2,4-dione Inhibitors of MurD Ligase. Eur. J. Med. Chem. 2010, 53, 6584–6594. [Google Scholar] [CrossRef]
- Zidar, N.; Tomašić, T.; Šink, R.; Kovač, A.; Patin, D.; Blanot, D.; Contreras-Martel, C.; Dessen, A.; Premru, M.; Zega, A.; et al. New 5-Benzylidenethiazolidin-4-one Inhibitors of Bacterial Murd Ligase: Design, Synthesis, Crystal Structures, and Biological Evaluation. Eur. J. Med. Chem. 2011, 11, 5512–5523. [Google Scholar] [CrossRef]
- Zvarec, O.; Polyak, S.W.; Tieu, W.; Kuan, K.; Dai, H.; Pedersen, D.S.; Morona, R.; Zhang, L.; Booker, G.W.; Abell, A.D. 5-Benzylidenerhodanine and 5-Benzylidene-2,4-thiazolidinedione Based Antibacterials. Bioorg. Med. Chem. Lett. 2012, 22, 2720. [Google Scholar] [CrossRef]
- Sortino, M.; Delgado, P.; Juarez, S.; Quiroga, J.; Abonia, R.; Insuasty, B.; Nogueras, M.; Rodero, L.; Garibotto, F.M.; Enriz, R.D.; et al. Synthesis and antifungal activity of (Z)-5-arylidenerhodanines. Bioorg. Med. Chem. 2007, 15, 484–494. [Google Scholar] [CrossRef]
- Ma, L.A.; Xie, C.F.; Ma, Y.H.; Liu, J.A.; Xiang, M.L.; Ye, X.; Zheng, H.; Chen, Z.Z.; Xu, Q.Y.; Chen, T.; et al. Synthesis and Biological Evaluation of Novel 5-Benzylidenethiazolidine-Derivatives 2,4-Dione Derivatives for the Treatment of Infammatory Diseases. J. Med. Chem. 2011, 54, 2060–2068. [Google Scholar] [CrossRef]
- Rekha, S.; Shanthram, U.; Vineet, C. Synthesis and Evaluation of Novel Thiazolidinedionesforanti Inflammatory Activity. IRJP 2011, 2, 81–84. [Google Scholar]
- Azam, F.; Prasad, M.V.; Thangavel, V. Targeting Oxidative Stress Component in the Therapeutics of Epilepsy. Curr. Top. Med. Chem. 2012, 12, 994–1007. [Google Scholar] [CrossRef]
- Salamone, S.; Colin, C.; Grillier-Vuissoz, I.; Kuntz, S.; Mazerbourg, S.; Flament, S.; Martin, H.; Richert, L.; Chapleur, Y.; Boisbrun, M. Synthesis of New Troglitazone Derivatives: Anti-Proliferative Activity in Breast Cancer Cell Lines and Preliminary Toxicological Study. Eur. J. Med. Chem. 2012, 51, 206–215. [Google Scholar] [CrossRef]
- Long, N.; Le Gresley, A.; Wren, S.P. Thiazolidinediones: An In–Depth Study of Their Synthesis and Application to Medicinal Chemistry in the Treatment of Diabetes Mellitus. ChemMedChem 2021, 16, 1717–1736. [Google Scholar] [CrossRef]
- Besstina, E.L.; Tunde, O.A.; Moss, P.E.; Stewart, L.V. Thiazolidinediones Regulate Expression of Cell Cycle Proteins in Human Prostate Cancer Cells via Pparγ-Dependent and Pparγ-Independent Pathways. Cell Cycle 2009, 8, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Yang, J.; Lee, S.L.; Kulp, S.K.; Chen, C.S. PPARgamma-Independent Antitumor Effects of Thiazolidinediones. Cancer Lett. 2009, 276, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Bhanushali, U.; Rajendran, S.; Sarma, K.; Kulkarni, P.; Chatti, K.; Chatterjee, S.; Ramaa, C.S. 5-Benzylidene-2,4-thiazolidenedione Derivatives: Design, Synthesis and Evaluation as Inhibitors of Angiogenesis Targeting VEGR-2. Bioorg. Chem. 2016, 67, 139–147. [Google Scholar] [CrossRef]
- Joshi, H.; Pal, T.; Ramaa, C.S. A New Dawn for the Use of Thiazolidinediones in Cancer Therapy. Expert Opin. Investig. Drugs 2014, 23, 501–510. [Google Scholar] [CrossRef]
- Shah, D.K.; Menon, K.M.J.; Cabrera, L.M.; Vahratian, A.; Kavoussi, S.K.; Lebovic, D.I. Thiazolidinediones Decrease Vascular Endothelial Growth Factor (VEGF) Production b Human Luteinized Granulosa Cells In Vitro. Fertil. Steril. 2010, 93, 2042–2047. [Google Scholar] [CrossRef]
- Joseph, A.; Shah, C.S.; Kumar, S.S.; Alex, A.T.; Maliyakkal, N.; Moorkoth, S.; Mathew, J.E. Synthesis, In Vitro Anticancer and Antioxidant Activity of Thiadiazole Substituted Thiazolidin-4-Ones. Acta Pharm. 2013, 63, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef]
- Levshin, I.B.; Glushkov, R.G.; Silin, V.A.; Padejskaja, E.N. Benzylidenethiazolidine-2,4-Diones Showing Antimicrobial Activity. SU1417436A1, 10 December 1986. [Google Scholar]
- Glushkov, R.G.; Levchuk, T.A.; Shilova, I.B.; Gus’kova, T.A. Pharmaceutical Composition For Treatment of Fungal Disease and Method For Its Preparing. RU Patent 2295958 C1, 27 March 2007. [Google Scholar]
- Puchkina, T.V.; Shilova, I.B.; Sharova, S.A.; Chistykov, V.V.; Stepanova, E.V.; Novoselov, A.U.; Koshichkina, N.V.; Kozukova, O.A. Experimental and Clinical Study of A New Thiazolidine-2,4-Dione-Mycosidine Derivative Ointment in Treatment of Smooth Skin Dermatomycoses. Adv. Med. Mycol. 2007, 10, 94–95. [Google Scholar]
- Levshin, I.B.; Stovbun, S.V.; Gomberg, M.A.; Bibikova, M.V. Study of the Mechanism of Action of an Original Domestic Drug From A New Class of Antimycotics-Thiazolidin-2,4-dione Derivatives. Adv. Med. Mycol. 2014, 12, 415–417. [Google Scholar]
- Shilova, I.B. Study of a Thiazolidine-2,4-Dione Derivative (Mycosidine) as a Potential Antifungal Agent. Ph.D. Thesis, TsHLS-VNIHFI, Moscow, Russia, 30 May 2007. (In Russian). [Google Scholar]
- Nawale, S.L.; Dhake, A.S. Synthesis and Evaluation of Novel Thiazolidinedione Derivatives for Antibacterial Activity. Der Pharma Chem. 2012, 4, 2270–2277. [Google Scholar]
- Tran, H.Q.; Nguyen, H.C.; Van Nguyen, T.; Nguyen, T.T.; Vo, T.N.; Nguyend, C.T. Synthesis and Evaluation of Cytotoxic Activity on Mcf-7 Cell Line of Some Diesters Derived from 5-(Hydroxybenzylidene)Thiazolidine-2,4-Diones. Acta Chemica IASI 2018, 26, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Mousseron, M. 5-substituted thiazolidine-4-one and process for production thereof. Patent CA897687A, 11 April 1972. [Google Scholar]
- Levshin, I.B.; Tsurkan, A.A.; V’yunov, K.A.; Ginak, A.I. Preparation Method of 5-Arylideneselena- and Thiazolidine-2,4-Diones. Chem. Inf. 1983, 14. [Google Scholar] [CrossRef]
- Popov-Pergal, K.M.; Poleti, D.; Rančić, M.P.; Meden, A.; Pergal, M.V. Synthesis and Structure of New 5-(Arylidene)-3-(4-methylbenzoyl)thiazolidine-2,4-diones. J. Het. Chem. 2010, 47, 224–228. [Google Scholar] [CrossRef]
- Shaikh, F.M.; Navin, B.; Patel, B.; Rajani, D. Synthesis of New Thiazolidine Derivatives and Their Antimicrobial and Antitubercular Activity. Ind. J. Res. Pharm. Biotechnol. 2013, 1, 496–503. [Google Scholar]
- Levshin, I.B.; Rastorgueva, N.A.; Kiselev, A.V.; Vedenkin, A.S.; Stovbun, S.V.; Churakov, A.V.; Saveliev, O.Y.; Polshakov, V.I. Thiazolidine-2,4-dione in Benzoylation Reaction. Chem. Heterocycl. Compd. 2019, 55, 178–183. [Google Scholar] [CrossRef]
- Bhat, B.A.; Ponnala, S.; Sahu, D.P.; Tiwari, P.; Tripathi, B.K.; Srivastava, A.K. Synthesis and Antihyperglycemic Activity Profiles of Novel Thiazolidinedione Derivatives. Bioorg. Med. Chem. 2004, 12, 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Costantino, L.; Curinga, C.; Maccari, R.; Monforte, F.; Nicolo, F.; Ottana, R.; Vigorita, M.G. Synthesis and Aldose Reductase Inhibitory Activity of 5-Arylidene-2,4-thiazolidinediones. Bioorg. Med. Chem. 2002, 10, 1077–1084. [Google Scholar] [CrossRef]
- Sambasivarao, S.V.; Soni, L.K.; Arun, K.; Gupta, A.K.; Hanumantharao, P.; Kaskhedikar, S.G. Quantitative Structure–Activity Analysis of 5-Arylidene-2,4-Thiazolidinediones as Aldose Reductase Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 512–520. [Google Scholar] [CrossRef]
- Maccari, R.; Ottana, R.; Curinga, C.; Vigorita, M.G.; Rakowitz, D.; Steindl, T.; Langer, T. Structure–Activity Relationships and Molecular Modelling of 5-Arylidene-2,4-Thiazolidinediones Active as Aldose Reductase Inhibitors. Bioorg. Med. Chem. 2005, 13, 2809–2823. [Google Scholar] [CrossRef]
- Maccari, R.; Ottana, R.; Ciurleo, R.; Vigorita, M.G.; Rakowitz, D.; Steind, T.; Langer, T. Evaluation of In Vitro Aldose Redutase Inhibitory Activity of 5-Arylidene-2,4-Thiazolidinediones. Bioorg. Med. Chem. Lett. 2007, 17, 3886–3893. [Google Scholar] [CrossRef]
- Maccari, R.; Vitale, R.M.; Ottanà, R.; Rocchiccioli, M.; Marrazzo, A.; Venera, C.V.; Graziano, A.C.E.; Amodeo, P.; Mura, U.; Del Corso, A. Structure-Activity Relationships and Molecular Modelling of New 5-Arylidene-4-Thiazolidinone Derivatives As Aldose Reductase Inhibitors and Potential Anti-Inflammatory Agents. Eur. J. Med. Chem. 2014, 81, 316082. [Google Scholar] [CrossRef]
- Datar, P.A.; Aher, S.B. Design and Synthesis of Novel Thiazolidine-2,4-diones As Hypoglycemic Agents. J. Saudi Chem. Soc. 2012, 12, 196–201. [Google Scholar] [CrossRef] [Green Version]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Eissa, I.H.; El-Hddad, S.S.A.; Shoman, F.M.I.A. Design, Synthesis, Molecular Docking and Anticancer Evaluations of 5-Benzylidenethiazolidine-2,4-Dione Derivatives Targeting VEGFR-2 Enzyme. Bioorg. Chem. 2020, 102, 104059. [Google Scholar] [CrossRef]
- Alegaon, S.G.; Alagawadi, K.R.; Pawar, S.M.; Vinod, D.; Rajput, U. Synthesis, characterization, and biological evaluation of thiazolidine-2,4-dione derivatives. Med. Chem. Res. 2014, 23, 987–994. [Google Scholar] [CrossRef]
- Froes, T.Q.; Chaves, B.T.; Mendes, M.S.; Ximenes, R.M.; da Silva, I.M.; da Silva, P.B.G.; de Albuquerque, J.F.C.; Castilho, M.S. Synthesis and Biological Evaluation of Thiazolidinedione Derivatives with High Ligand Efficiency to P. Aeruginosa PhzS. J. Enzyme Inhib. Med. Chem. 2021, 36, 1217–1229. [Google Scholar] [CrossRef]
- Kumar, B.R.P.; Karvekar, M.D.; Adhikary, L.; Nanjan, M.J.; Suresh, D. Microwave Induced Synthesis of the Thiazolidine-2,4-dione Motif and the Efficient Solvent Free-Solid Phase Parallel Syntheses of 5-Benzylidene-thiazolidine-2,4-dione and 5-Benzylidene-2-thioxo-thiazolidine-4-one Compounds. J. Heterocycl. Chem. 2006, 43, 897–903. [Google Scholar] [CrossRef]
- Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z.M.; McInnes, C.; Wang, W.; Kraft, A.; Smith, C.D. Synthesis and Evaluation of Novel Inhibitors of Pim-1 and Pim-2 Protein Kinases. J. Med. Chem. 2009, 52, 74–86. [Google Scholar] [CrossRef] [Green Version]
- da Silva, I.M.; da Silva Filho, J.; Santiago, P.B.; do Egito, M.S.; de Souza, C.A.; Gouveia, F.L.; Ximenes, R.M.; de Sena, K.X.; de Faria, A.R.; Brondani, D.J.; et al. Synthesis and Antimicrobial Activities of 5-Arylidene-thiazolidine-2,4-dione Derivatives. BioMed Res. Int. 2014, 316082. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Santiago, P.; Froes, T.Q.; da Silva Filho, J.; da Silva, S.G.; Ximenes, R.M.; de Faria, A.R.; Brondani, D.J.; de Albuquerque, J.; Castilho, M.S. Structure-guided discovery of thiazolidine-2,4-dione derivatives as a novel class of Leishmania major pteridine reductase 1 inhibitors. Eur. J. Med. Chem. 2016, 123, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Naeem, F.; Nadeem, H.; Muhammad, A.; Muhammad, Z.M.A.; Saeed, A. Synthesis, α-Amylase Inhibitory Activity and Molecular Docking Studies of 2,4-Thiazolidinedione Derivatives. Open Chem. J. 2018, 5, 134–144. [Google Scholar] [CrossRef]
- Tiekat, K.; Upadhyay, N.; Schweipert, M.; Hess, J.D.; Macias, L.H.; Mrowka, P.; Meyer-Almes, F.-J.; Aguilera, R.J.; Iancu, C.V.; Choe, J.-Y.; et al. Permuted 2,4-Thiazolidinedione (TZD) Analogs As GLUT Inhibitors and Their In-Vitro Evaluation in Leukemic Cells. Eur. J. Pharm. Sci. 2020, 154, 105512. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing for Yeasts, 4th ed.; CLSI Standard M27; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi, 2nd ed.; Approved Standard. Document M-38A2; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- Soste, M.; Hrabakova, R.; Wanka, S.; Melnik, A.; Boersema, P.; Maiolica, A.; Wernas, T.; Tognetti, M.; von Mering, C.; Picotti, P.A. Sentinel Protein Assay for Simultaneously Quantifying Cellular Processes. Nat. Methods 2014, 11, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B. Functional Profiling of the Saccharomyces Cerevisiae Genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Galkina, K.V.; Finkelberg, J.M.; Markova, O.V.; Azbarova, A.V.; Banerjee, A.; Kumari, S.; Sokolov, S.S.; Severin, F.F.; Prasad, R.; Knorre, D.A. Protonophore FCCP Provides Fitness Advantage To PDR-Deficient Yeast Cells. J. Bioenerg. Biomembr. 2020, 52, 383–395. [Google Scholar] [CrossRef]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Li, C.; Shevchuk, A.I.; Stepanyan, R.; Caldwell, M.; Hughes, S.; Smart, T.G.; Gorelik, J.; Ostanin, V.P.; Lab, M.J.; et al. Nanoscale Live-Cell Imaging Using Hopping Probe Ion Conductance Microscopy. Nat. Methods 2009, 6, 279–281. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, V.S.; Erofeev, A.S.; Woodcock, E.; Efremov, Y.M.; Iakovlev, A.P.; Savin, N.A.; Alova, A.V.; Lavrushkina, S.V.; Kireev, I.I.; Prelovskaya, A.O.; et al. Mapping Mechanical Properties of Living Cells At Nanoscale Using Intrinsic Nanopipette–Sample Force Interactions. Nanoscale 2021, 13, 6558–6568. [Google Scholar] [CrossRef]
- Clarke, R.W.; Novak, P.; Zhukov, A.; Tyler, E.J.; Cano-Jaimez, M.; Drews, A.; Richards, O.; Volynski, K.; Bishop, C.; Klenerman, D. Low Stress Ion Conductance Microscopy of Sub-Cellular Stiffness. Soft Matter 2016, 12, 7953–7958. [Google Scholar] [CrossRef] [Green Version]
- Abd Alhameed, R.; Almarhoon, Z.B.S.I.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Synthesis and Antimicrobial Activity of a New Series of Thiazolidine-2,4-diones Carboxamide and Amino Acid Derivatives. Molecules 2020, 25, 105. [Google Scholar] [CrossRef] [Green Version]

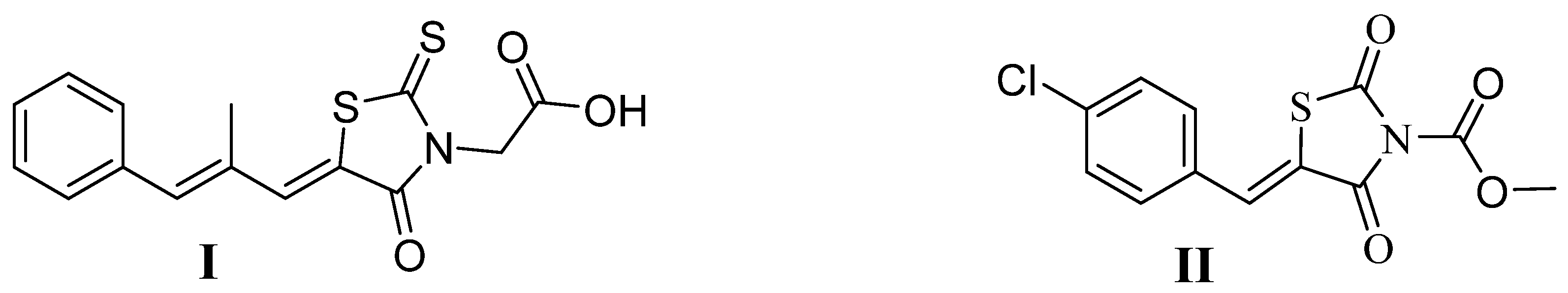

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
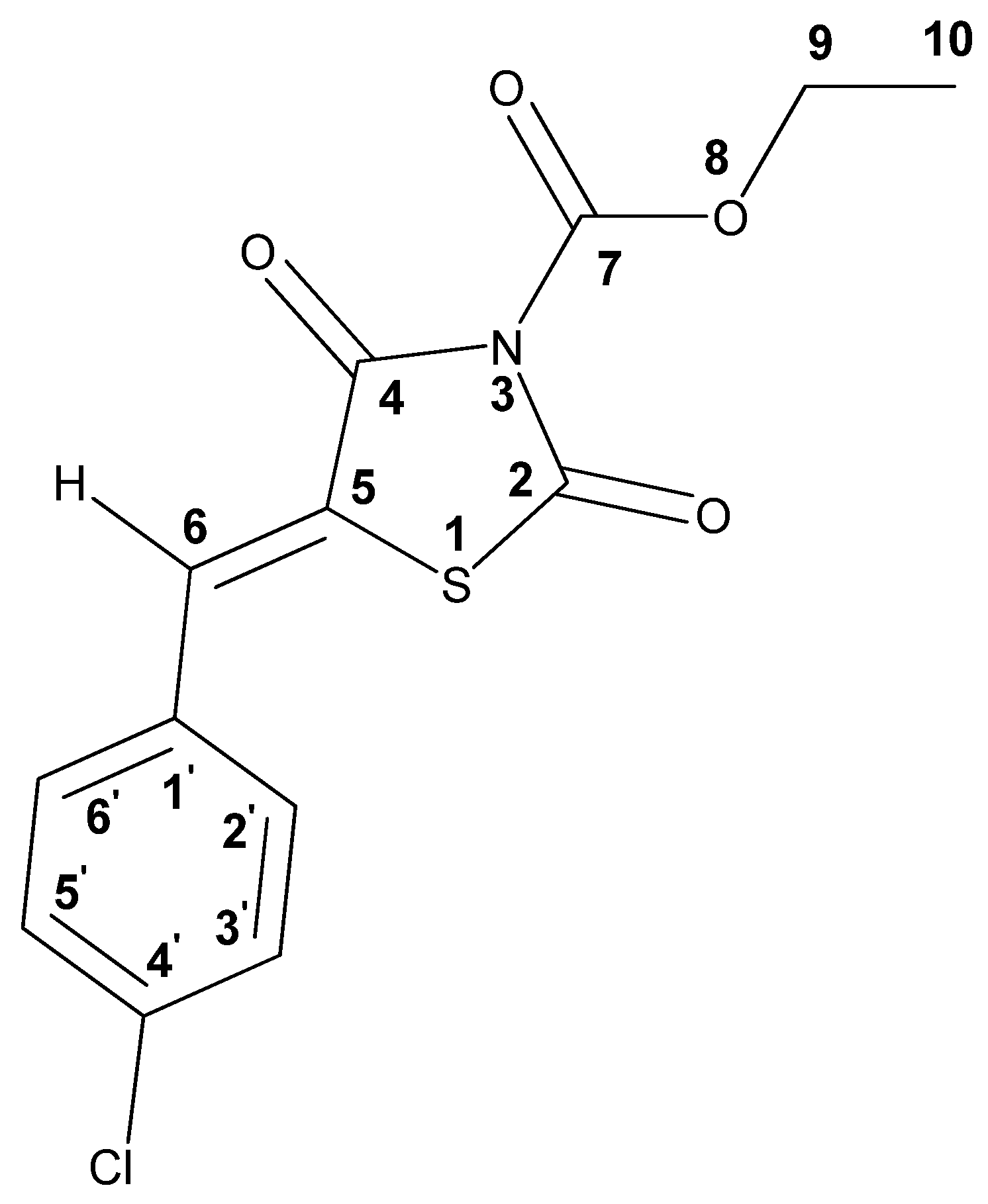
| Solvent | Chemical Shift, ppm | |||
|---|---|---|---|---|
| CH (6) | Har (2′,3′,5′,6′) | CH2 (9) | CH3 (10) | |
| DMSO-d6 | 7.90 | 7.59–7.67 | 4.42 | 1.32 |
| CDCl3 | 7.85 | 7.41–7.46 | 4.50 | 1.43 |
| Atom | C(2) | C(4) | C(5) | C(6) | C(7) | C(9) | C(10) | C(1′) | C(2′) C(6′) | C(3′) C(5′) | C(4′) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| δ (13C) ppm | 163.3 | 162.4 | 120.4 | 134.0 | 147.3 | 66.0 | 13.9 | 137.2 | 131.4 | 129.6 | 131.2 |
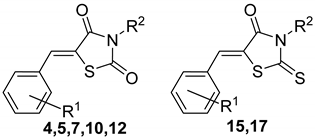 | ||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | C. albicans ATCC 24433 24h 48h | A. niger 37a | A. fumigatus ATCC 46645 | M. canis B-200 | T. rubrum 2002 | ||
| 4a | 4-Cl | H | >128 | >128 | 64 | >64 | 16 | 32 |
| 4b | 3,4-(OBn)2 | H | >128 | >128 | >64 | >64 | >32 | >32 |
| 4c | 2,4-di-Cl | H | >128 | >128 | 64 | >64 | 64 | 128 |
| 4d | 2-OH-5-Cl | H | 64 | 64 | 16 | 32 | 64 | 16 |
| 4e | 2-OH-3,5-di-Cl | H | 8 | 32 | 16 | 32 | 32 | 16 |
| 15b | 3,4-(OBn)2 | H | 32 | >64 | >64 | >64 | >64 | >64 |
| 5a | 4-Cl | COOEt | 8–16 | 16 | 1 | 4 | 8 | 8 |
| 5b | 4-Cl | COOi-Bu | 4 | 8 | 8–16 | 16 | 4–8 | 8 |
| 5c | 4-Cl | COOAll | 8 | 8 | 8 | 16 | 8 | 4 |
| 5d | 4-Cl | COOPh | 16 | 64 | 32 | 64 | 4 | 32 |
| 5e | 4-Cl | COOBn | >64 | >64 | 64 | >64 | 16 | 32 |
| 7a | 4-Cl | COPh | 8 | 32 | 32 | 64 | 4 | 8–16 |
| 7b | 3,4-(OBn)2 | COPh | >128 | >125 | 32 | >64 | >64 | >64 |
| 10a | 4-Cl | CH2COOt-Bu | 32–64 | 64 | >64 | >64 | >64 | >64 |
| 10b | 3,4-(OBn)2 | CH2COOt-Bu | >32 | >64 | >64 | >64 | >64 | >64 |
| 11a | 4-Cl | CH2COOEt | 32–64 | >64 | >64 | >64 | >64 | >64 |
| 11b | 3,4-(OBn)2 | CH2COOEt | >64 | >64 | >64 | >64 | >64 | >64 |
| 12a | 4-Cl | CH2COOH | 32 | >64 | 64 | >64 | 32–64 | 32 |
| 12b | 3,4-(OBn)2 | CH2COOH | 64 | >64 | >64 | >64 | >64 | >64 |
| 12c | 2,4-Cl2 | CH2COOH | 32 | >32 | 16 | 2 | 16–32 | 32 |
| 12d | 2-OH-5-Cl | CH2COOH | 32 | >64 | 4–8 | 16 | 16 | 16 |
| 12e | 2-OH-3,5-di-Cl | CH2COOH | 0.125 | 0.5 | 8 | 16 | 16 | 16 |
| 17b | 3,4-(OBn)2 | CH2COOH | 64 | >64 | >64 | >64 | >64 | >64 |
| 17e | 2-OH-3,5-di-Cl | CH2COOH | 8 | 16 | 8 | 16 | 2 | 8 |
| Mycosidine (R1 = 4-Cl, R2 = COOMe) | 16 | 32 | 4 | 8 | 4 | 4 | ||
| Fluconazole | 4 | 32 | >64 | >64 | 16 | 64 | ||
| Microorganism | MIC (µg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Flc | 2 | 5a | 7a | 12e | |||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | ||
| C. parapsilosis ATCC 22019 | 4 | 16 | 32 | 16 | 32 | 32 | 64 | <0.125 | 0.5 |
| C. albicans ATCC 24433 | 4 | 8 | 16 | 8 | 16 | 8 | 32 | 0.125 | 0.25 |
| C. tropicalis 3019 | 2 | 32 | 64 | 32 | 32 | 64 | 64 | 64 | 64 |
| C. kefyr 77 | n.d. | 8 | 16 | 8 | 16 | 16 | 32 | 64 | 64 |
| C. famata 312 | n.d. | 32 | 64 | 32 | 32 | 64 | >64 | >64 | >64 |
| C. guilliermondii 355 | n.d. | 64 | 64 | n.d. | n.d. | 64 | 64 | 64 | 64 |
| C. parapsilosis 58N | n.d. | 64 | 64 | n.d. | n.d. | 64 | 64 | >64 | >64 |
| C. krusei 432M | >32 | 16 | 32 | 16 | 16 | 32 | 64 | 64 | 64 |
| C. glabrata 61L | 32 | 8 | 16 | 8 | 16 | 16 | 32 | 64 | 64 |
| C. albicans 604M | >32 | 32 | 64 | 32 | 32 | 64 | 64 | >64 | >64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levshin, I.B.; Simonov, A.Y.; Lavrenov, S.N.; Panov, A.A.; Grammatikova, N.E.; Alexandrov, A.A.; Ghazy, E.S.M.O.; Savin, N.A.; Gorelkin, P.V.; Erofeev, A.S.; et al. Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners. Pharmaceuticals 2022, 15, 563. https://doi.org/10.3390/ph15050563
Levshin IB, Simonov AY, Lavrenov SN, Panov AA, Grammatikova NE, Alexandrov AA, Ghazy ESMO, Savin NA, Gorelkin PV, Erofeev AS, et al. Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners. Pharmaceuticals. 2022; 15(5):563. https://doi.org/10.3390/ph15050563
Chicago/Turabian StyleLevshin, Igor B., Alexander Y. Simonov, Sergey N. Lavrenov, Alexey A. Panov, Natalia E. Grammatikova, Alexander A. Alexandrov, Eslam S. M. O. Ghazy, Nikita A. Savin, Peter V. Gorelkin, Alexander S. Erofeev, and et al. 2022. "Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners" Pharmaceuticals 15, no. 5: 563. https://doi.org/10.3390/ph15050563
APA StyleLevshin, I. B., Simonov, A. Y., Lavrenov, S. N., Panov, A. A., Grammatikova, N. E., Alexandrov, A. A., Ghazy, E. S. M. O., Savin, N. A., Gorelkin, P. V., Erofeev, A. S., & Polshakov, V. I. (2022). Antifungal Thiazolidines: Synthesis and Biological Evaluation of Mycosidine Congeners. Pharmaceuticals, 15(5), 563. https://doi.org/10.3390/ph15050563