
Covalent Inhibition of the Human 20S Proteasome with Homobelactosin C Inquired by QM/MM Studies



Abstract
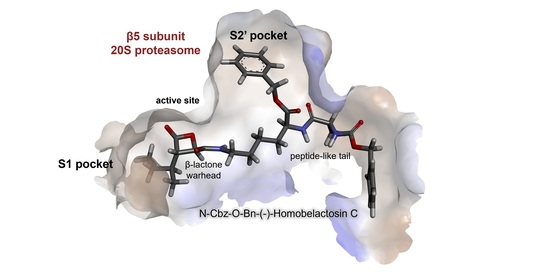
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Recognition Step
2.2. Inactivation Step
3. Computational Methods
3.1. System Setup
3.2. QM/MM Calculations
3.3. Potential Energy Surfaces (PES)
3.4. Free Energy Surfaces (FES)
3.5. Spline Corrections (SP)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyams, J.S. Gastroenterology and Nutrition, 2nd ed.; Elsevier Saunders: Philadelphia, PA, USA, 2001; Volume 13, ISBN 978-1-4377-2603-9. [Google Scholar]
- Etlinger, J.D.; Goldberg, A.L. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. The isolation and purification of a macromolecular protein component from the human erythrocyte ghost. Biochim. Biophys. Acta (BBA)-Protein Struct. 1969, 188, 31–42. [Google Scholar] [CrossRef]
- Hase, J.; Kobashi, K.; Nakai, N.; Mitsui, K.; Iwata, K.; Takadera, T. The quaternary structure of carp muscle alkaline protease. Biochim. Biophys. Acta (BBA)-Enzym. 1980, 611, 205–213. [Google Scholar] [CrossRef]
- Arrigo, A.-P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 1988, 331, 192–194. [Google Scholar] [CrossRef]
- Wilk, S.; Orlowski, M. Cation-Sensitive Neutral Endopeptidase: Isolation and Specificity of the Bovine Pituitary Enzyme. J. Neurochem. 1980, 35, 1172–1182. [Google Scholar] [CrossRef]
- Eytan, E.; Ganoth, D.; Armon, T.; Hershko, A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. USA 1989, 86, 7751–7755. [Google Scholar] [CrossRef]
- The Nobel Prize in Chemistry 2005 NobelPrize.Org. Nobel Prize Outreach AB 2021. Available online: https://www.nobelprize.org/prizes/chemistry/2005/summary/ (accessed on 4 September 2021).
- Hershko, A.; Ciechanover, A.; Heller, H.; Haas, A.L.; Rose, I.A. Proposed role of ATP in protein breakdown: Conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. USA 1980, 77, 1783–1786. [Google Scholar] [CrossRef]
- Ciechanover, A.; Elias, S.; Heller, H.; Ferber, S.; Hershko, A. Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. J. Biol. Chem. 1980, 255, 7525–7528. [Google Scholar] [CrossRef]
- De Martino, G.N.; Proske, R.J.; Moomaw, C.R.; Strong, A.A.; Song, X.; Hisamatsu, H.; Tanaka, K.; Slaughter, C.A. Identification, Purification, and Characterization of a PA700-dependent Activator of the Proteasome. J. Biol. Chem. 1996, 271, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.M.; Crotchett, B.; Slaughter, C.A.; DeMartino, G.N.; Gogol, E.P. Formation of Proteasome−PA700 Complexes Directly Correlates with Activation of Peptidase Activity. Biochemistry 1998, 37, 12927–12932. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.L. An Introduction to Medicinal Chemistry, 6th ed.; Oxford University Press: New York, NY, USA, 1995; ISBN 9780198749691. [Google Scholar]
- Mishra, R.; Upadhyay, A.; Prajapati, V.K.; Mishra, A. Proteasome-mediated proteostasis: Novel medicinal and pharmacological strategies for diseases. Med. Res. Rev. 2018, 38, 1916–1973. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-M. The Anaphase-Promoting Complex: Proteolysis in Mitosis and Beyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 150–162. [Google Scholar] [CrossRef]
- Bonvini, P.; Rosa, H.D.; Vignes, N.; Rosolen, A. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: Role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 2004, 64, 3256–3264. [Google Scholar] [CrossRef]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V Protein of Simian Virus 5 Inhibits Interferon Signalling by Targeting STAT1 for Proteasome-Mediated Degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar] [CrossRef]
- Desai, S.D.; Li, T.-K.; Rodriguez-Bauman, A.; Rubin, E.H.; Liu, L.F. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 2001, 61, 5926–5932. [Google Scholar]
- Ratner, J.N.; Balasubramanian, B.; Corden, J.; Warren, S.L.; Bregman, D.B. Ultraviolet Radiation-induced Ubiquitination and Proteasomal Degradation of the Large Subunit of RNA Polymerase II. J. Biol. Chem. 1998, 273, 5184–5189. [Google Scholar] [CrossRef]
- Rui, L.; Fisher, T.L.; Thomas, J.; White, M.F. Regulation of Insulin/Insulin-like Growth Factor-1 Signaling by Proteasome-mediated Degradation of Insulin Receptor Substrate-2. J. Biol. Chem. 2001, 276, 40362–40367. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Goldberg, J.L.; Qiao, L.Y.; Mitchell, J.J. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes 1999, 48, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.R.; El-Deiry, W.S. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc. Natl. Acad. Sci. USA 2004, 101, 6170–6175. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Nakabayashi, Y.; Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 8662–8667. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Hann, S.R. c-Myc Proteolysis by the Ubiquitin-Proteasome Pathway: Stabilization of c-Myc in Burkitt’s Lymphoma Cells. Mol. Cell. Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular Metabolism and Disease: What Do Metabolic Outliers Teach Us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Kelly, J.M.; Summers, M.; Park, H.S.; Milligan, L.P.; McBride, B.W. Cellular Energy Metabolism and Regulation; Academic Press: New York, NY, USA, 1991; Volume 74, ISBN 978-0-12-066150-3. [Google Scholar]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Zhao, X.; Zhao, K.; Meng, H.; Zhao, W.; Gao, C. TRAF-interacting protein (TRIP) negatively regulates IFN-β production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J. Exp. Med. 2012, 209, 1703–1711. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M. Ubiquitin-dependent proteolysis: Its role in human diseases and the design of therapeutic strategies. Mol. Genet. Metab. 2002, 77, 44–56. [Google Scholar] [CrossRef]
- Layfield, R.; Lowe, J.; Bedford, L. The ubiquitin–proteasome system and neurodegenerative disorders. Essays Biochem. 2005, 41, 157–171. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Tabrizi, S. The Ubiquitin-Proteasome System in Neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef]
- Dou, Q.P.; Smith, D.M.; Daniel, K.G.; Kazi, A. Interruption of tumor cell cycle progression through proteasome inhibition: Implications for cancer therapy. Prog. Cell Cycle Res. 2003, 5, 441–446. [Google Scholar]
- Papandreou, C.N. The Proteasome as a Target for Cancer Treatment. Am. J. Cancer 2005, 4, 359–372. [Google Scholar] [CrossRef][Green Version]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Zmuda, F.; Sastry, L.; Shepherd, S.M.; Jones, D.; Scott, A.; Craggs, P.D.; Cortes, A.; Gray, D.W.; Torrie, L.S.; De Rycker, M. Identification of Novel Trypanosoma cruzi Proteasome Inhibitors Using a Luminescence-Based High-Throughput Screening Assay. Antimicrob. Agents Chemother. 2019, 63, e00309-19. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Central Sci. 2017, 3, 830–838. [Google Scholar] [CrossRef]
- Aminake, M.N.; Arndt, H.-D.; Pradel, G. The proteasome of malaria parasites: A multi-stage drug target for chemotherapeutic intervention? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Le Chapelain, C.; Groll, M. Rational Design of Proteasome Inhibitors as Antimalarial Drugs. Angew. Chem. Int. Ed. 2016, 55, 6370–6372. [Google Scholar] [CrossRef] [PubMed]
- Gandotra, S.; Schnappinger, D.; Monteleone, M.; Hillen, W.; Ehrt, S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 2007, 13, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Ditzel, L.; Groll, M.; Huber, R. Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 6070–6074. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C.; Hill, C.P. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Mol. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Dick, T.P.; Nussbaum, A.K.; Deeg, M.; Heinemeyer, W.; Groll, M.; Schirle, M.; Keilholz, W.; Stevanović, S.; Wolf, D.H.; Huber, R.; et al. Contribution of Proteasomal β-Subunits to the Cleavage of Peptide Substrates Analyzed with Yeast Mutants. J. Biol. Chem. 1998, 273, 25637–25646. [Google Scholar] [CrossRef]
- Huber, E.M. Introduction. In Structural and Functional Characterization of the Immunoproteasome; Huber, E.M., Ed.; Springer Theses TS; Springer International Publishing: Cham, Switzerland, 2013; pp. 1–18. ISBN 978-3-319-01555-2. [Google Scholar]
- Dou, Q.P.; Goldfarb, R.H. Bortezomib (millennium pharmaceuticals). IDrugs Investig. Drugs J. 2002, 5, 828–834. [Google Scholar]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA Approval for the Treatment of Multiple Myeloma Progressing on Prior Therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [Google Scholar] [CrossRef]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Asai, A.; Yamashita, Y.; Katahira, R.; Hasegawa, A.; Ochiai, K.; Akinaga, S. UCK 14 Compounds. U.S. Patent 5,663, 298, 1997. Available online: https://patentimages.storage.googleapis.com/53/ba/fc/d9b04aa7e971dd/US5663298.pdf (accessed on 1 March 2022).
- Asai, A.; Hasegawa, A.; Ochiai, K.; Yamashita, Y.; Mizukami, T. Belactosin A, a Novel Antitumor Antibiotic Acting on Cyclin/CDK Mediated Cell Cycle Regulation, Produced by Streptomyces sp. J. Antibiot. 2000, 53, 81–83. [Google Scholar] [CrossRef]
- Asai, A.; Tsujita, T.; Sharma, S.V.; Yamashita, Y.; Akinaga, S.; Funakoshi, M.; Kobayashi, H.; Mizukami, T. A new structural class of proteasome inhibitors identified by microbial screening using yeast-based assay. Biochem. Pharmacol. 2004, 67, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Harshbarger, W.D.; Robles, O.; Krysiak, J.; Hull, K.G.; Cho, S.W.; Richardson, R.D.; Yang, Y.; Garcia, A.; Spiegelman, L.; et al. A strategy for dual inhibition of the proteasome and fatty acid synthase with belactosin C-orlistat hybrids. Bioorganic Med. Chem. 2017, 25, 2901–2916. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Groettrup, M.; Cerny, T. The Proteasome, a New Target for Cancer Therapy. Onkologie 2002, 25, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Almond, J.B.; Cohen, G.M. The Proteasome: A Target Novel for Cancer Chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef]
- Elliott, P.J.; Zollner, T.M.; Boehncke, W.-H. Proteasome inhibition: A new anti-inflammatory strategy. Klin. Wochenschr. 2003, 81, 235–245. [Google Scholar] [CrossRef]
- Armstrong, A.; Scutt, J.N. Total synthesis of (+)-belactosin A. Chem. Commun. 2004, 4, 510–511. [Google Scholar] [CrossRef]
- Groll, M.; Larionov, O.V.; Huber, R.; de Meijere, A. Inhibitor-binding mode of homobelactosin C to proteasomes: New insights into class I MHC ligand generation. Proc. Natl. Acad. Sci. USA 2006, 103, 4576–4579. [Google Scholar] [CrossRef]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Aparicio, N.S.; Świderek, K.; Moliner, V. Theoretical study of the inhibition mechanism of human 20S proteasome by dihydroeponemycin. Eur. J. Med. Chem. 2019, 164, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Aparicio, N.; Moliner, V.; Świderek, K. Nature of Irreversible Inhibition of Human 20S Proteasome by Salinosporamide A. The Critical Role of Lys–Asp Dyad Revealed from Electrostatic Effects Analysis. ACS Catal. 2021, 11, 3575–3589. [Google Scholar] [CrossRef]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of β-Lactone Ring Opening and a Mechanism for Irreversible Binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Serrano-Aparicio, N.; Moliner, V.; Świderek, K. On the Origin of the Different Reversible Characters of Salinosporamide A and Homosalinosporamide A in the Covalent Inhibition of the Human 20S Proteasome. ACS Catal. 2021, 11, 11806–11819. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, J.-I.; Chung, S.J.; Park, J.D.; Park, N.-K.; Han, J.H. Cleavage of β-lactone ring by serine protease. Mechanistic implications. Bioorg. Med. Chem. 2002, 10, 2553–2560. [Google Scholar] [CrossRef]
- Galmés, M.A.; García-Junceda, E.; Świderek, K.; Moliner, V. Exploring the Origin of Amidase Substrate Promiscuity in CALB by a Computational Approach. ACS Catal. 2020, 10, 1938–1946. [Google Scholar] [CrossRef]
- Burgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Burgi, H.B.; Dunitz, J.D.; Lehn, J.; Wipff, G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions, Student Edition; John Wiley & Sons: Hoboken NJ, USA, 2011; pp. 214–215. ISBN 9780470746608. [Google Scholar]
- Heathcock, C.H. Understanding and Controlling Diastereofacial Selectivity in Carbon-Carbon Bond-Forming Reactions. Aldrichimica Acta 1990, 23, 94–111. [Google Scholar]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20 S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Schafmeister, C.E.A.; Ross, W.S.; Romanovski, V. LEAP; University of California: San Francisco, CA, USA, 1995. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, T.; Lafiosca, P.; Cappelli, C. A General Route to Include Pauli Repulsion and Quantum Dispersion Effects in QM/MM Approaches. J. Chem. Theory Comput. 2017, 13, 4854–4870. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, T.; Lafiosca, P.; Chandramouli, B.; Barone, V.; Cappelli, C. Effective yet reliable computation of hyperfine coupling constants in solution by a QM/MM approach: Interplay between electrostatics and non-electrostatic effects. J. Chem. Phys. 2019, 150, 124102. [Google Scholar] [CrossRef] [PubMed]
- Lennard-Jones, J.E. Cohesion. Proc. Phys. Soc. 1931, 43, 461–482. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Field, M.J.; Albe, M.; Bret, C.; Martin, F.P.-D.; Thomas, A. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J. Comput. Chem. 2000, 21, 1088–1100. [Google Scholar] [CrossRef]
- Krzemińska, A.; Paneth, P.; Moliner, V.; Świderek, K. Binding Isotope Effects as a Tool for Distinguishing Hydrophobic and Hydrophilic Binding Sites of HIV-1 RT. J. Phys. Chem. B 2014, 119, 917–927. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Baker, J. An algorithm for the location of transition states. J. Comput. Chem. 1986, 7, 385–395. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.; Kollman, P.A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Ruiz-Pernía, J.J.; Silla, E.; Tuñón, I.; Martí, S. Hybrid Quantum Mechanics/Molecular Mechanics Simulations with Two-Dimensional Interpolated Corrections: Application to Enzymatic Processes. J. Phys. Chem. B 2006, 110, 17663–17670. [Google Scholar] [CrossRef]
- Chuang, Y.-Y.; Corchado, A.J.C.; Truhlar, D.G. Mapped Interpolation Scheme for Single-Point Energy Corrections in Reaction Rate Calculations and a Critical Evaluation of Dual-Level Reaction Path Dynamics Methods. J. Phys. Chem. A 1999, 103, 1140–1149. [Google Scholar] [CrossRef]
- Świderek, K.; Tuñón, I.; Martí, S.; Moliner, V. Protein Conformational Landscapes and Catalysis. Influence of Active Site Conformations in the Reaction Catalyzed by L-Lactate Dehydrogenase. ACS Catal. 2015, 5, 1172–1185. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: https://gaussian.com/glossary/g09/ (accessed on 1 March 2022).












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano-Aparicio, N.; Ferrer, S.; Świderek, K. Covalent Inhibition of the Human 20S Proteasome with Homobelactosin C Inquired by QM/MM Studies. Pharmaceuticals 2022, 15, 531. https://doi.org/10.3390/ph15050531
Serrano-Aparicio N, Ferrer S, Świderek K. Covalent Inhibition of the Human 20S Proteasome with Homobelactosin C Inquired by QM/MM Studies. Pharmaceuticals. 2022; 15(5):531. https://doi.org/10.3390/ph15050531
Chicago/Turabian StyleSerrano-Aparicio, Natalia, Silvia Ferrer, and Katarzyna Świderek. 2022. "Covalent Inhibition of the Human 20S Proteasome with Homobelactosin C Inquired by QM/MM Studies" Pharmaceuticals 15, no. 5: 531. https://doi.org/10.3390/ph15050531
APA StyleSerrano-Aparicio, N., Ferrer, S., & Świderek, K. (2022). Covalent Inhibition of the Human 20S Proteasome with Homobelactosin C Inquired by QM/MM Studies. Pharmaceuticals, 15(5), 531. https://doi.org/10.3390/ph15050531