
Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
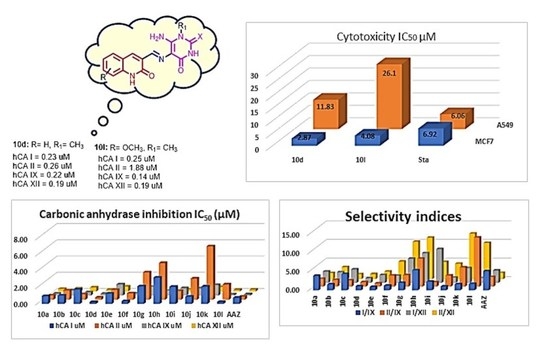
2.2. Biology
2.2.1. Carbonic Anhydrase Inhibition
2.2.2. Antiproliferative Activity
2.2.3. Assessment of Apoptotic Marker Levels
2.3. In Silico Study
2.3.1. Physicochemical and Pharmacokinetic Parameters
2.3.2. Molecular Docking Study
2.4. SAR Study
- -
- Both uracil and thiouracil had CA inhibitory activity.
- -
- Substitution on uracil N-1 with a bulky group (benzyl 10b and 10i) decreases activity.
- -
- Substitution on the quinoline ring has tolerable activity, but greatly improves the selectivity, particularly when in combination with thiouracil (10l).
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedures for the Preparation of 10a–l
(E)-6-Amino-5-(((2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino)pyrimidine-2,4(1H,3H)-dione (10a)
(E)-6-Amino-1-benzyl-5-(((2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino) pyrimidine-2,4(1H,3H)-dione (10b)
(E)-6-Amino-1-ethyl-5-(((2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino)pyrimidine-2,4(1H,3H)-dione (10c)
(E)-6-Amino-1-methyl-5-(((2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino) pyrimidine-2,4(1H,3H)-dione (10d)
(E)-3-(((6-Amino-1-methyl-4-Oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)imino)methyl) quinolin-2(1H)-one (10e)
(E)-6-Amino-1-ethyl-5-(((8-methyl-2-oxo-1,2-dihydroquinolin-3-yl)methylene) amino)pyrimidine-2,4(1H,3H)-dione (10f)
(E)-3-(((6-Amino-1-methyl-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl) imino)methyl)-8-methylquinolin-2(1H)-one (10g)
(E)-6-Amino-5-(((6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino) pyrimidine-2,4(1H,3H)-dione (10h)
(E)-6-Amino-1-benzyl-5-(((6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)methylene) amino) pyrimidine-2,4(1H,3H)-dione (10i)
(E)-6-Amino-1-ethyl-5-(((6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino)pyrimidine-2,4(1H,3H)-dione (10j)
(E)-6-Amino-5-(((6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)methylene)amino)-1-methyl pyrimidine-2,4(1H,3H)-dione (10k)
(E)-3-(((6-Amino-1-methyl-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)imino)methyl)-6-methoxyquinolin-2(1H)-one (10l)
3.2. Biology
3.3. Computational Studies
3.3.1. Molecular Modeling Study
3.3.2. Prediction of Pharmacokinetics Properties and Drug Likeliness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Supuran, C. Carbonic Anhydrases An Overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T.; Capasso, C. An overview on the recently discovered iota-carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibition/Activation: Trip of a Scientist Around the World in the Search of Novel Chemotypes and Drug Targets. Curr. Pharm. Des. 2010, 16, 3233–3245. [Google Scholar] [CrossRef] [PubMed]
- Strapcova, S.; Takacova, M.; Csaderova, L.; Martinelli, P.; Lukacikova, L.; Gal, V.; Kopacek, J.; Svastova, E. Clinical and Pre-Clinical Evidence of Carbonic Anhydrase IX in Pancreatic Cancer and Its High Expression in Pre-Cancerous Lesions. Cancers 2020, 12, 2005. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: A patent review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef]
- Krasavin, M.; Kalinin, S.; Sharonova, T.; Supuran, C.T. Inhibitory activity against carbonic anhydrase IX and XII as a candidate selection criterion in the development of new anticancer agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1555–1561. [Google Scholar] [CrossRef]
- Supuran, C.T. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J. Clin. Oncol. 2012, 3, 98. [Google Scholar] [CrossRef]
- Supuran, C.T. Experimental Carbonic Anhydrase Inhibitors for the Treatment of Hypoxic Tumors. J. Exp. Pharmacol. 2020, 12, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Temiz, E.; Koyuncu, I.; Durgun, M.; Caglayan, M.; Gonel, A.; Güler, E.M.; Kocyigit, A.; Supuran, C.T. Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6098. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L-and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: Stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J. Med. Chem. 2006, 49, 3019–3027. [Google Scholar] [PubMed]
- Supuran, C. Carbonic Anhydrases as Drug Targets—An Overview. Curr. Top. Med. Chem. 2007, 7, 825–833. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 2007, 15, 4336–4350. [Google Scholar] [CrossRef]
- Ye, R.; Tan, C.; Chen, B.; Li, R.; Mao, Z. Zinc-Containing Metalloenzymes: Inhibition by Metal-Based Anticancer Agents. Front. Chem. 2020, 8, 402. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Frühauf, A.; Meyer-Almes, F.-J. Non-Hydroxamate Zinc-Binding Groups as Warheads for Histone Deacetylases. Molecules 2021, 26, 5151. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Dong, M.; Ning, Z.; Newman, M.J.; Xu, J.; Dou, G.; Cao, H.; Shi, Y.; Gingras, M.A.; Lu, X.; Feng, F. Phase I study of chidamide (CS055/HBI-8000), a novel histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. J. Clin. Oncol. 2009, 27, 3529. [Google Scholar] [CrossRef]
- Clawson, G.A. Histone deacetylase inhibitors as cancer therapeutics. Ann. Transl. Med. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kouketsu, A.; Matsuura, A.; Kohara, A.; Ninomiya, S.-I.; Kohda, K.; Miyata, N. Thiol-based SAHA analogues as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3313–3317. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Dul, E.; Sung, C.-M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of Novel Isoform-Selective Inhibitors within Class I Histone Deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones As Lead-Like Natural Products: The Development of Potent and Selective Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef]
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as Novel Zinc Binding Group for Selective Histone Deacetylase Inhibition. J. Med. Chem. 2013, 56, 3492–3506. [Google Scholar] [CrossRef]
- Valente, S.; Conte, M.; Tardugno, M.; Nebbioso, A.; Tinari, G.; Altucci, L.; Mai, A. Developing novel non-hydroxamate histone deacetylase inhibitors: The chelidamic warhead. MedChemComm 2012, 3, 298–304. [Google Scholar] [CrossRef]
- Wang, Y.; Stowe, R.L.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.L.; Wang, Y.; Wang, Y.; et al. Identification of Histone Deacetylase Inhibitors with Benzoylhydrazide Scaffold that Selectively Inhibit Class I Histone Deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Li, Y.; Woster, P.M. Discovery of a new class of histone deacetylase inhibitors with a novel zinc binding group. MedChemComm 2015, 6, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.M.; Wang, Q.; Fuller, J.H.; West, N.; Martinez, N.M.; Morse, E.M.; Weïwer, M.; Schreiber, S.L.; Bradner, J.E.; Koehler, A.N. A novel HDAC inhibitor with a hydroxy-pyrimidine scaffold. Bioorg. Med. Chem. Lett. 2011, 21, 4164–4169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, M.; Chen, N.; Wang, S.; Luo, H.-B.; Zhang, Y.; Wu, R. Computational Design of a Time-Dependent Histone Deacetylase 2 Selective Inhibitor. ACS Chem. Biol. 2015, 10, 687–692. [Google Scholar] [CrossRef]
- Beena; Rawat, D.S. Antituberculosis Drug Research: A Critical Overview. Med. Res. Rev. 2013, 33, 693–764. [Google Scholar] [CrossRef] [PubMed]
- Van de Walle, T.; Cools, L.; Mangelinckx, S.; D’Hooghe, M. Recent contributions of quinolines to antimalarial and anticancer drug discovery research. Eur. J. Med. Chem. 2021, 226, 113865. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kumar, K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem. 2021, 215, 113220. [Google Scholar] [CrossRef] [PubMed]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-virulence, Antiviral and Anti-parasitic Agents. Adv. Microbiol. Infect. Dis. Public Health 2019, 1282, 37–69. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Kumar Jain, P.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef]
- Musiol, R.; Serda, M.; Hensel-Bielowka, S.; Polanski, J. Quinoline-Based Antifungals. Curr. Med. Chem. 2010, 17, 1960–1973. [Google Scholar] [CrossRef]
- Razzaghi-Asl, N.; Sepehri, S.; Ebadi, A.; Karami, P.; Nejatkhah, N.; Johari-Ahar, M. Insights into the current status of privileged N-heterocycles as antileishmanial agents. Mol. Divers. 2019, 24, 525–569. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pal, M. Quinolines: A new hope against inflammation. Drug Discov. Today 2013, 18, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.M.; Pawara, R.; Surana, S.J. Chapter 1—Introduction. In Third Generation EGFR Inhibitors; Patel, H.M., Pawara, R., Surana, S.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–24. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Wang, D.; Campone, M.; Calles, A.; Leip, E.; Turnbull, K.; Bardy-Bouxin, N.; Duvillié, L.; Calvo, E. Bosutinib plus capecitabine for selected advanced solid tumours: Results of a phase 1 dose-escalation study. Br. J. Cancer 2014, 111, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Bronte, E.; Galvano, A.; Novo, G.; Russo, A. Chapter 5—Cardiotoxic Effects of Anti-VEGFR Tyrosine Kinase Inhibitors. In Cardio-Oncology; Gottlieb, R.A., Mehta, P.K., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 69–89. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Mross, K.; Jakob, A.; Kanz, L.; Bokemeyer, C. Topotecan—A Novel Topoisomerase I Inhibitor: Pharmacology and Clinical Experience. Oncology 1999, 56, 1–12. [Google Scholar] [CrossRef]
- El-Sayed, M.A.A.; El-Husseiny, W.M.; Abdel-Aziz, N.I.; El-Azab, A.S.; Abuelizz, H.A.; Abdel-Aziz, A.A.M. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: Molecular docking study. J. Enzym. Inhib. Med. Chem. 2017, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- George, R.F.; Samir, E.M.; Abdelhamed, M.N.; Abdel-Aziz, H.A.; Abbas, S.E.S. Synthesis and anti-proliferative activity of some new quinoline based 4,5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019, 83, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, R.; Elseginy, S.; Ziedan, N.; Jones, A.; Westwell, A. New Quinoline-Based Heterocycles as Anticancer Agents Targeting Bcl-2. Molecules 2019, 24, 1274. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaset, M.S.; Abuo-Rahma, G.E.-D.A.; Abdelrahman, M.H.; Ramadan, M.; Youssif, B.G.M.; Bukhari, S.N.A.; Mohamed, M.F.A.; Abdel-Aziz, M. Novel pyrrol-2(3H)-ones and pyridazin-3(2H)-ones carrying quinoline scaffold as anti-proliferative tubulin polymerization inhibitors. Bioorg. Chem. 2018, 80, 151–163. [Google Scholar] [CrossRef]
- Kundu, B.; Das, S.K.; Paul Chowdhuri, S.; Pal, S.; Sarkar, D.; Ghosh, A.; Mukherjee, A.; Bhattacharya, D.; Das, B.B.; Talukdar, A. Discovery and Mechanistic Study of Tailor-Made Quinoline Derivatives as Topoisomerase 1 Poison with Potent Anticancer Activity. J. Med. Chem. 2019, 62, 3428–3446. [Google Scholar] [CrossRef]
- Rabal, O.; Sánchez-Arias, J.A.; San José-Enériz, E.; Agirre, X.; de Miguel, I.; Garate, L.; Miranda, E.; Sáez, E.; Roa, S.; Martínez-Climent, J.A.; et al. Detailed Exploration around 4-Aminoquinolines Chemical Space to Navigate the Lysine Methyltransferase G9a and DNA Methyltransferase Biological Spaces. J. Med. Chem. 2018, 61, 6546–6573. [Google Scholar] [CrossRef]
- Chung, P.; Lam, P.; Zhou, Y.; Gasparello, J.; Finotti, A.; Chilin, A.; Marzaro, G.; Gambari, R.; Bian, Z.; Kwok, W.; et al. Targeting DNA Binding for NF-κB as an Anticancer Approach in Hepatocellular Carcinoma. Cells 2018, 7, 177. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Bua, S.; Ha Lee, S.; Abdelgawad, M.A.; Roh, E.J.; Eldehna, W.M.; Supuran, C.T. Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino)benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014, 2014, 202784. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.; Siddiqui, N. A review on biological importance of pyrimidines in the new era. Int. J. Pharm. Pharm. Sci. 2016, 8, 8–21. [Google Scholar]
- Nerkar, A.U. Use of Pyrimidine and Its Derivative in Pharmaceuticals: A Review. J. Adv. Chem. Sci. 2021, 7, 729–732. [Google Scholar] [CrossRef]
- Haouala, A.; Widmer, N.; Duchosal, M.A.; Montemurro, M.; Buclin, T.; Decosterd, L.A. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood J. Am. Soc. Hematol. 2011, 117, e75–e87. [Google Scholar] [CrossRef] [PubMed]
- Bartzatt, R. Potential antineoplastic structural variations of uracil mustard (uramustine) retaining cytotoxic activity and drug-likeness suitable for oral administration. J. Cancer Tumor Int. 2015, 2, 50. [Google Scholar] [CrossRef][Green Version]
- Pullarkat, S.T.; Stoehlmacher, J.; Ghaderi, V.; Xiong, Y.P.; Ingles, S.A.; Sherrod, A.; Warren, R.; Tsao-Wei, D.; Groshen, S.; Lenz, H.J. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharm. J. 2001, 1, 65–70. [Google Scholar] [CrossRef]
- Ariav, Y.; Ch’ng, J.H.; Christofk, H.R.; Ron-Harel, N.; Erez, A. Targeting nucleotide metabolism as the nexus of viral infections, cancer, and the immune response. Sci. Adv. 2021, 7, eabg6165. [Google Scholar] [CrossRef]
- Grem, J.L.; Chabner, B.A.; Chu, E.; Johnson, P.; Yeh, G.C.; Allegra, C.J. Antimetabolites. Cancer Chemother. Biol. Response Modif. 1991, 12, 1–25. [Google Scholar]
- Rider, B.J. Cytarabine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef]
- Si, J.; Boumber, Y.A.; Shu, J.; Qin, T.; Ahmed, S.; He, R.; Jelinek, J.; Issa, J.-P.J. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res. 2010, 70, 6968–6977. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, M.; Momparler, L.F.; Raynal, N.J.M.; Bernstein, M.L.; Momparler, R.L. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2008, 63, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Shaldam, M.; Nocentini, A.; Elsayed, Z.M.; Ibrahim, T.M.; Salem, R.; El-Domany, R.A.; Capasso, C.; Supuran, C.T.; Eldehna, W.M. Development of Novel Quinoline-Based Sulfonamides as Selective Cancer-Associated Carbonic Anhydrase Isoform IX Inhibitors. Int. J. Mol. Sci. 2021, 22, 11119. [Google Scholar] [CrossRef]
- Nemr, M.T.M.; AboulMagd, A.M.; Hassan, H.M.; Hamed, A.A.; Hamed, M.I.A.; Elsaadi, M.T. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agents via carbonic anhydrase IX inhibition. RSC Adv. 2021, 11, 26241–26257. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A. Carbonic Anhydrase Inhibitors: Aromatic Sulfonamides and Disulfonamides Act as Efficient Tumor Growth Inhibitors. J. Enzym. Inhib. 2008, 15, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Briganti, F.; Tilli, S.; Chegwidden, W.R.; Scozzafava, A. Carbonic anhydrase inhibitors: Sulfonamides as antitumor agents? Bioorg. Med. Chem. 2001, 9, 703–714. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.-Y.; Qin, H.-L. Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef]
- Dorn, J.M.; Alpern, M.; McNulty, C.; Volcheck, G.W. Sulfonamide Drug Allergy. Curr. Allergy Asthma Rep. 2018, 18, 38. [Google Scholar] [CrossRef]
- Giles, A.; Foushee, J.; Lantz, E.; Gumina, G. Sulfonamide Allergies. Pharmacy 2019, 7, 132. [Google Scholar] [CrossRef]
- Alper Türkoğlu, E.; Şentürk, M.; Supuran, C.T.; Ekinci, D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J. Enzym. Inhib. Med. Chem. 2017, 32, 74–77. [Google Scholar] [CrossRef]
- Yiğit, B.; Yiğit, M.; Taslimi, P.; Gök, Y.; Gülçin, İ. Schiff bases and their amines: Synthesis and discovery of carbonic anhydrase and acetylcholinesterase enzymes inhibitors. Arch. Pharm. 2018, 351, 1800146. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J. Enzym. Inhib. Med. Chem. 2018, 33, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Trah, S.; Lamberth, C. Synthesis of novel 3, 4, 6-trisubstituted quinolines enabled by a Gould-Jacobs cyclization. Tetrahedron Lett. 2017, 58, 794–796. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Youssif, B.G.M.; Abd El Ghany, M.A.; Abdelazeem, A.H.; Ibrahim, H.M.; Moustafa, A.E.G.A.; Treamblu, L.; Bukhari, S.N.A. Synthesis, biological evaluation, docking study and ulcerogenicity profiling of some novel quinoline-2-carboxamides as dual COXs/LOX inhibitors endowed with anti-inflammatory activity. Eur. J. Med. Chem. 2017, 127, 972–985. [Google Scholar] [CrossRef]
- Wang, G.-W.; Jia, C.-S.; Dong, Y.-W. Benign and highly efficient synthesis of quinolines from 2-aminoarylketone or 2-aminoarylaldehyde and carbonyl compounds mediated by hydrochloric acid in water. Tetrahedron Lett. 2006, 47, 1059–1063. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamashita, M.; Sawai, Y.; Nakamoto, K.; Goto, M. Syntheses of metabolites of ethyl 4-(3, 4-dimethoxyphenyl)-6, 7-dimethoxy-2-(1, 2, 4-triazol-1-ylmethyl) quinoline-3-carboxylate (TAK-603). Tetrahedron 2006, 62, 8707–8714. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido [2,3-d]pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Zordok, W.A.; El-Sayed, A.S.A. Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives. Int. J. Mol. Sci. 2021, 22, 10979. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Boztaş, M.; Çetinkaya, Y.; Topal, M.; Gülçin, İ.; Menzek, A.; Şahin, E.; Tanc, M.; Supuran, C.T. Synthesis and Carbonic Anhydrase Isoenzymes I, II, IX, and XII Inhibitory Effects of Dimethoxybromophenol Derivatives Incorporating Cyclopropane Moieties. J. Med. Chem. 2014, 58, 640–650. [Google Scholar] [CrossRef]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. Cancer Cell Cult. 2011, 731, 237–245. [Google Scholar] [CrossRef]
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.; Memon, Z.; Ahmed, N.; Hasnain, S.N. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for Bcl-2-directed therapy? Cancer Manag. Res. 2018, 10, 403–416. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.M.; Ghorab, M.M.; Bua, S.; Supuran, C.T. Iodoquinazolinones bearing benzenesulfonamide as human carbonic anhydrase I, II, IX and XII inhibitors: Synthesis, biological evaluation and radiosensitizing activity. Eur. J. Med. Chem. 2020, 200, 112449. [Google Scholar] [CrossRef]
- Wassel, M.M.S.; Ragab, A.; Elhag Ali, G.A.M.; Mehany, A.B.M.; Ammar, Y.A. Novel adamantane-pyrazole and hydrazone hybridized: Design, synthesis, cytotoxic evaluation, SAR study and molecular docking simulation as carbonic anhydrase inhibitors. J. Mol. Struct. 2021, 1223, 128966. [Google Scholar] [CrossRef]
- Höpp, M.; Erxleben, A.; Rombeck, I.; Lippert, B. The Uracil C(5) Position as a Metal Binding Site: Solution and X-ray Crystal Structure Studies of PtII and HgII Compounds. Inorg. Chem. 1996, 35, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Zamora, F.; Amo-Ochoa, P.; Lippert, B. Pyrimidine Nucleobases as Versatile and Multidentate Ligands for Heavy Metal Ions. Significance of Metal Binding to the C(5) Sites of Uracil and Cytosine. In Cytotoxic, Mutagenic and Carcinogenic Potential of Heavy Metals Related to Human Environment; Springer: Dordrecht, The Netherlands, 1997; Volume 26, pp. 511–520. [Google Scholar] [CrossRef]
- Patil, Y.P.; Nethaji, M. Synthesis and crystal structure of copper (II) uracil ternary polymeric complex with 1,10-phenanthroline along with the Hirshfeld surface analysis of the metal binding sites for the uracil ligand. J. Mol. Struct. 2015, 1081, 14–21. [Google Scholar] [CrossRef]
- Siters, K.E.; Sander, S.A.; Morrow, J.R. Selective Binding of Zn2+ Complexes to Non-Canonical Thymine or Uracil in DNA or RNA. Prog. Inorg. Chem. 2014, 59, 245–298. [Google Scholar] [CrossRef]
- Xia, C.-Q.; Jiang, N.; Zhang, J.; Chen, S.-Y.; Lin, H.-H.; Tan, X.-Y.; Yue, Y.; Yu, X.-Q. The conjugates of uracil–cyclen Zn(II) complexes: Synthesis, characterization, and their interaction with plasmid DNA. Bioorg. Med. Chem. 2006, 14, 5756–5764. [Google Scholar] [CrossRef]
- El-kalyoubi, S.; Agili, F.; Youssif, S. Novel 2-Thioxanthine and Dipyrimidopyridine Derivatives: Synthesis and Antimicrobial Activity. Molecules 2015, 20, 19263–19276. [Google Scholar] [CrossRef]
- Singh, M.K.; Chandra, A.; Singh, B.; Singh, R.M. Synthesis of diastereomeric 2,4-disubstituted pyrano [2,3-b]quinolines from 3-formyl-2-quinolones through O–C bond formation via intramolecular electrophilic cyclization. Tetrahedron Lett. 2007, 48, 5987–5990. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Insuasty, D.; Lima, S.; Gutiérrez, L.; Nogueras, M.; Marchal, A.; Abonia, R.; Andújar, S.; Spiegel, S.; Cobo, J.; et al. Design of new quinolin-2-one-pyrimidine hybrids as sphingosine kinases inhibitors. Bioorg. Chem. 2020, 94, 103414. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | Ki (µM) | ||||||
|---|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | hCA I | hCA II | hCA IX | hCA XII | |
| 10a | 0.97 | 0.52 | 0.25 | 0.32 | 0.53 | 0.29 | 0.14 | 0.17 |
| 10b | 1.01 | 1.14 | 0.63 | 0.35 | 0.56 | 0.63 | 0.35 | 0.19 |
| 10c | 1.76 | 0.66 | 0.40 | 0.49 | 0.97 | 0.37 | 0.22 | 0.27 |
| 10d | 0.23 | 0.26 | 0.22 | 0.19 | 0.13 | 0.14 | 0.12 | 0.11 |
| 10e | 1.25 | 1.21 | 1.37 | 0.59 | 0.69 | 0.67 | 0.76 | 0.33 |
| 10f | 0.33 | 0.79 | 0.27 | 0.17 | 0.18 | 0.44 | 0.14 | 0.09 |
| 10g | 2.16 | 3.36 | 1.08 | 0.33 | 1.20 | 1.86 | 0.60 | 0.18 |
| 10h | 3.23 | 4.50 | 0.60 | 0.40 | 1.78 | 2.49 | 0.33 | 0.22 |
| 10i | 2.11 | 1.07 | 0.91 | 0.23 | 1.17 | 0.59 | 0.51 | 0.13 |
| 10j | 0.89 | 2.60 | 0.87 | 0.62 | 0.49 | 1.44 | 0.48 | 0.34 |
| 10k | 2.12 | 6.57 | 1.28 | 0.54 | 1.17 | 3.63 | 0.71 | 0.30 |
| 10l | 0.25 | 1.88 | 0.14 | 0.19 | 0.14 | 1.04 | 0.08 | 0.11 |
| AAZ | 0.76 | 0.39 | 0.15 | 0.23 | 0.42 | 0.22 | 0.08 | 0.13 |
| Compound | I/IX | II/IX | I/XII | II/XII |
|---|---|---|---|---|
| 10a | 3.88 | 2.09 | 3.05 | 1.65 |
| 10b | 1.60 | 1.81 | 2.88 | 3.24 |
| 10c | 4.37 | 1.65 | 3.62 | 1.37 |
| 10d | 1.09 | 1.21 | 1.24 | 1.37 |
| 10e | 0.91 | 0.88 | 2.12 | 2.06 |
| 10f | 1.23 | 2.99 | 1.95 | 4.72 |
| 10g | 1.99 | 3.10 | 6.47 | 10.05 |
| 10h | 5.39 | 7.52 | 8.00 | 11.16 |
| 10i | 2.31 | 1.17 | 9.07 | 4.60 |
| 10j | 1.02 | 2.98 | 1.43 | 4.18 |
| 10k | 1.66 | 5.14 | 3.96 | 12.26 |
| 10l | 1.75 | 13.20 | 1.29 | 9.75 |
| AAZ | 5.12 | 2.61 | 3.31 | 1.69 |
| Compound | MCF-7 | A549 | Cytotoxicity IC50 µM | |||
|---|---|---|---|---|---|---|
| Bcl2 nm/mL | Bax pg/mL | Bcl2 nm/mL | Bax pg/mL | MCF7 | A549 | |
| 10d | 2.587 ± 0.03 | 403.1 ± 4.69 | 2.841 ± 0.01 | 278.6 ± 9.14 | 2.87 ± 0.05 | 11.83 ± 0.22 |
| 10l | 3.296 ± 0.09 | 337.2 ± 7.55 | 4.605 ± 0.28 | 208.4 ± 4.07 | 4.08 ± 0.08 | 26.10 ± 0.56 |
| Staurosporine | 2.829 ± 0.07 | 381.8 ± 11.4 | 3.78 ± 0.14 | 310.5 ± 9.7 | 6.92 ± 0.18 | 6.06 ± 0.17 |
| control | 7.727 ± 0.2 | 62.86 ± 4.7 | 8.63 ± 0.16 | 47.72 ± 2.31 | - | - |
| Compound | MR | TPSA | Log P | GI Absorption | BBB Permeant | CYP1A2 Inhibitor | Lipinski #Violations | Bioavailability Score | PAINS #Alerts | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|---|---|---|
| 10a | 83.91 | 136.96 | 0.72 | High | No | No | 0 | 0.55 | 0 | 2.84 |
| 10b | 113.3 | 126.1 | 2.15 | High | No | No | 0 | 0.55 | 0 | 3.27 |
| 10c | 93.62 | 126.1 | 1.26 | High | No | No | 0 | 0.55 | 0 | 3.01 |
| 10d | 88.81 | 126.1 | 0.92 | High | No | No | 0 | 0.55 | 0 | 2.89 |
| 10e | 93.38 | 141.12 | 1.65 | High | No | No | 0 | 0.55 | 0 | 2.93 |
| 10f | 98.58 | 126.1 | 1.61 | High | No | No | 0 | 0.55 | 0 | 3.13 |
| 10g | 98.34 | 141.12 | 1.99 | Low | No | No | 0 | 0.55 | 0 | 3.05 |
| 10h | 90.4 | 146.19 | 0.84 | Low | No | No | 0 | 0.55 | 0 | 2.86 |
| 10i | 119.79 | 135.33 | 2.14 | High | No | No | 0 | 0.55 | 0 | 3.34 |
| 10j | 100.11 | 135.33 | 1.25 | High | No | No | 0 | 0.55 | 0 | 3.07 |
| 10k | 95.3 | 135.33 | 0.95 | High | No | No | 0 | 0.55 | 0 | 2.95 |
| 10l | 99.87 | 150.35 | 1.67 | Low | No | No | 0 | 0.55 | 0 | 2.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kalyoubi, S.A.; Taher, E.S.; Ibrahim, T.S.; El-Behairy, M.F.; Al-Mahmoudy, A.M.M. Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors. Pharmaceuticals 2022, 15, 494. https://doi.org/10.3390/ph15050494
El-Kalyoubi SA, Taher ES, Ibrahim TS, El-Behairy MF, Al-Mahmoudy AMM. Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors. Pharmaceuticals. 2022; 15(5):494. https://doi.org/10.3390/ph15050494
Chicago/Turabian StyleEl-Kalyoubi, Samar A., Ehab S. Taher, Tarek S. Ibrahim, Mohammed Farrag El-Behairy, and Amany M. M. Al-Mahmoudy. 2022. "Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors" Pharmaceuticals 15, no. 5: 494. https://doi.org/10.3390/ph15050494
APA StyleEl-Kalyoubi, S. A., Taher, E. S., Ibrahim, T. S., El-Behairy, M. F., & Al-Mahmoudy, A. M. M. (2022). Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors. Pharmaceuticals, 15(5), 494. https://doi.org/10.3390/ph15050494