
A Protein-Based, Long-Acting HIV-1 Fusion Inhibitor with an Improved Pharmacokinetic Profile

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
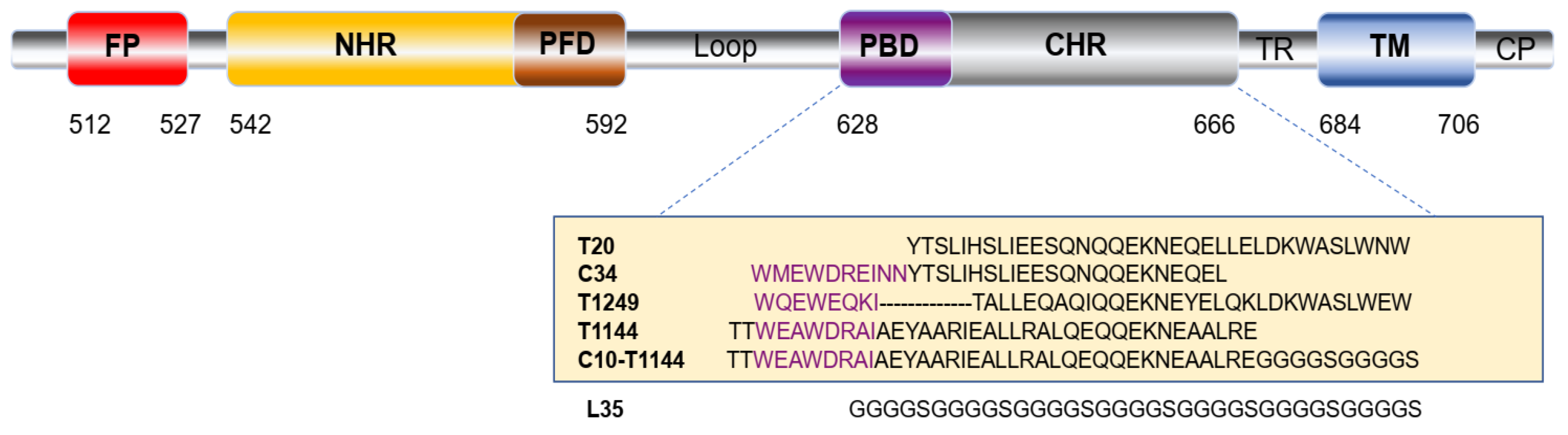
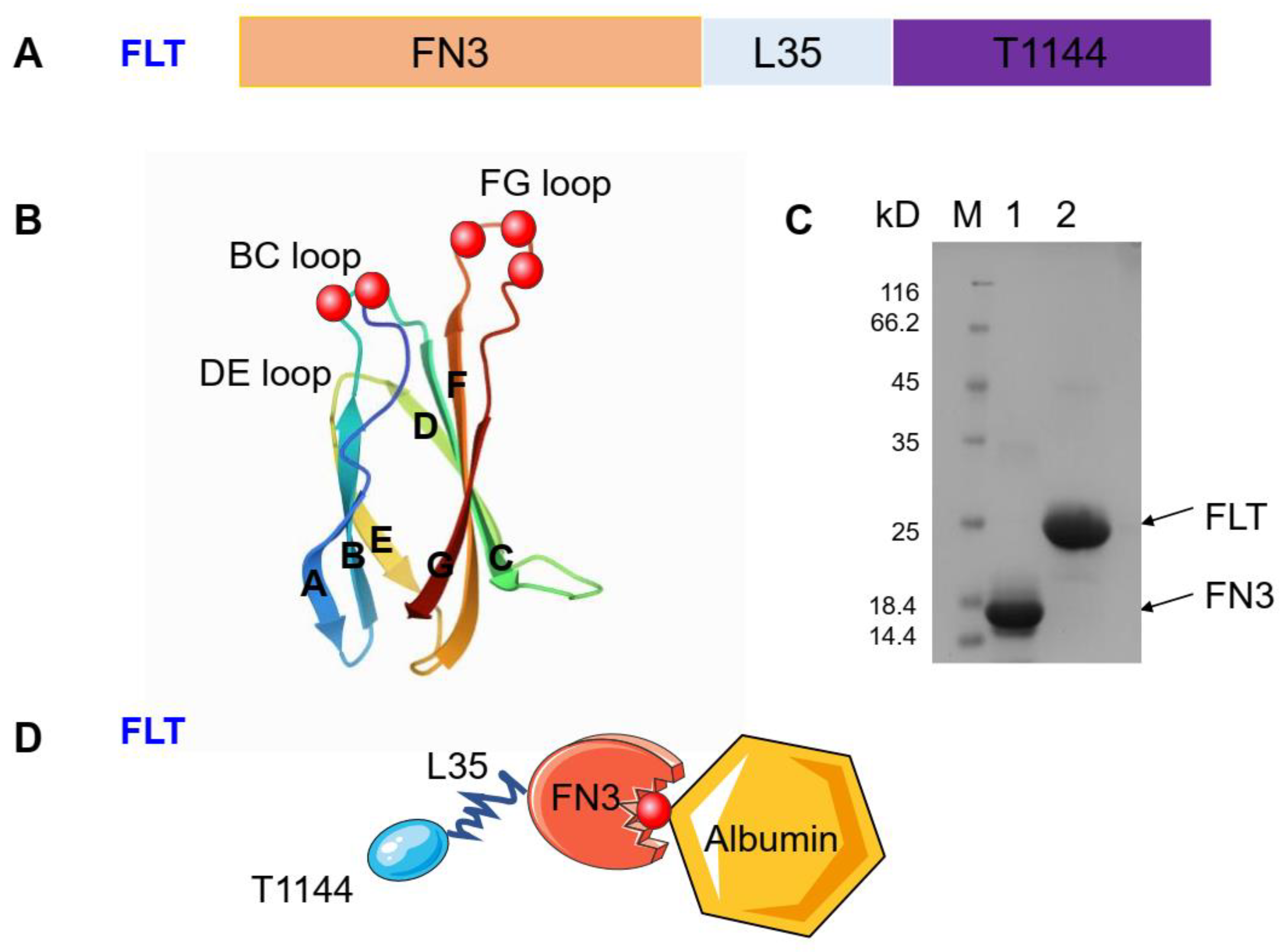
2.1. Construction and Expression of FLT
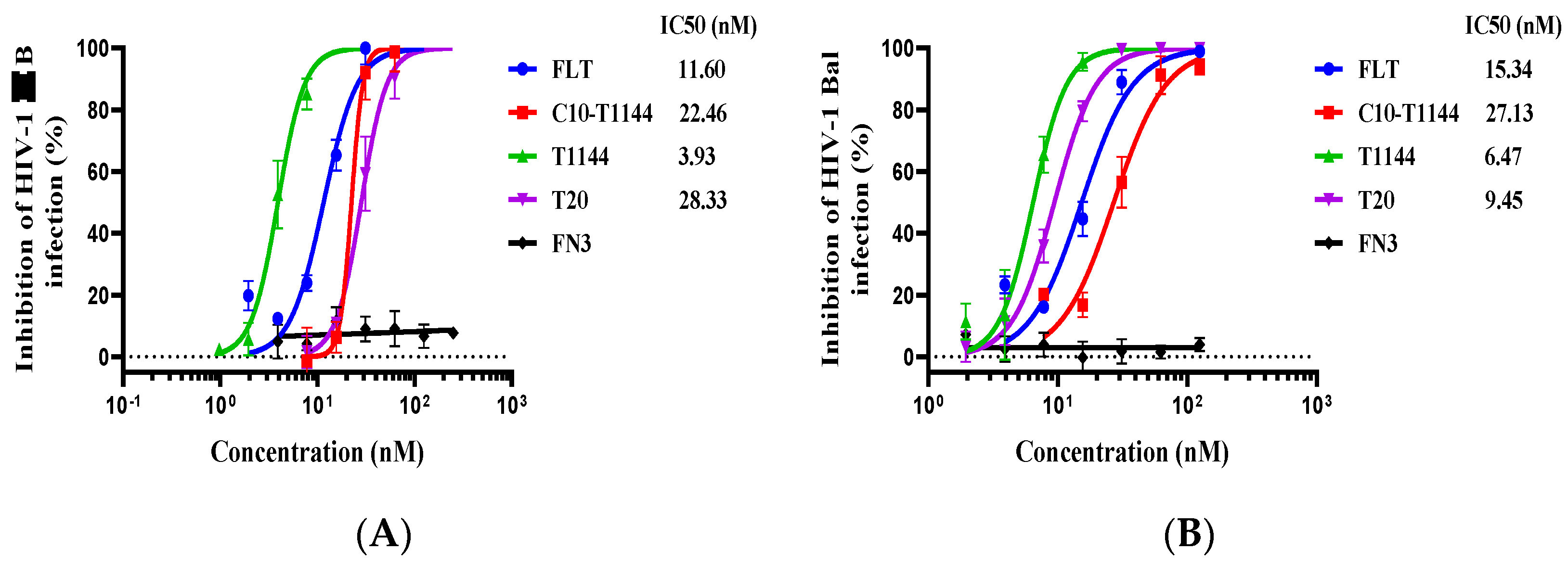
2.2. FLT Exhibited Potent Inhibitory Activity against Infection by Laboratory-Adapted HIV-1 Strains and Divergent Primary HIV-1 Isolates
2.3. FLT Effectively Inhibited Infection by Drug-Resistant HIV-1 Strains
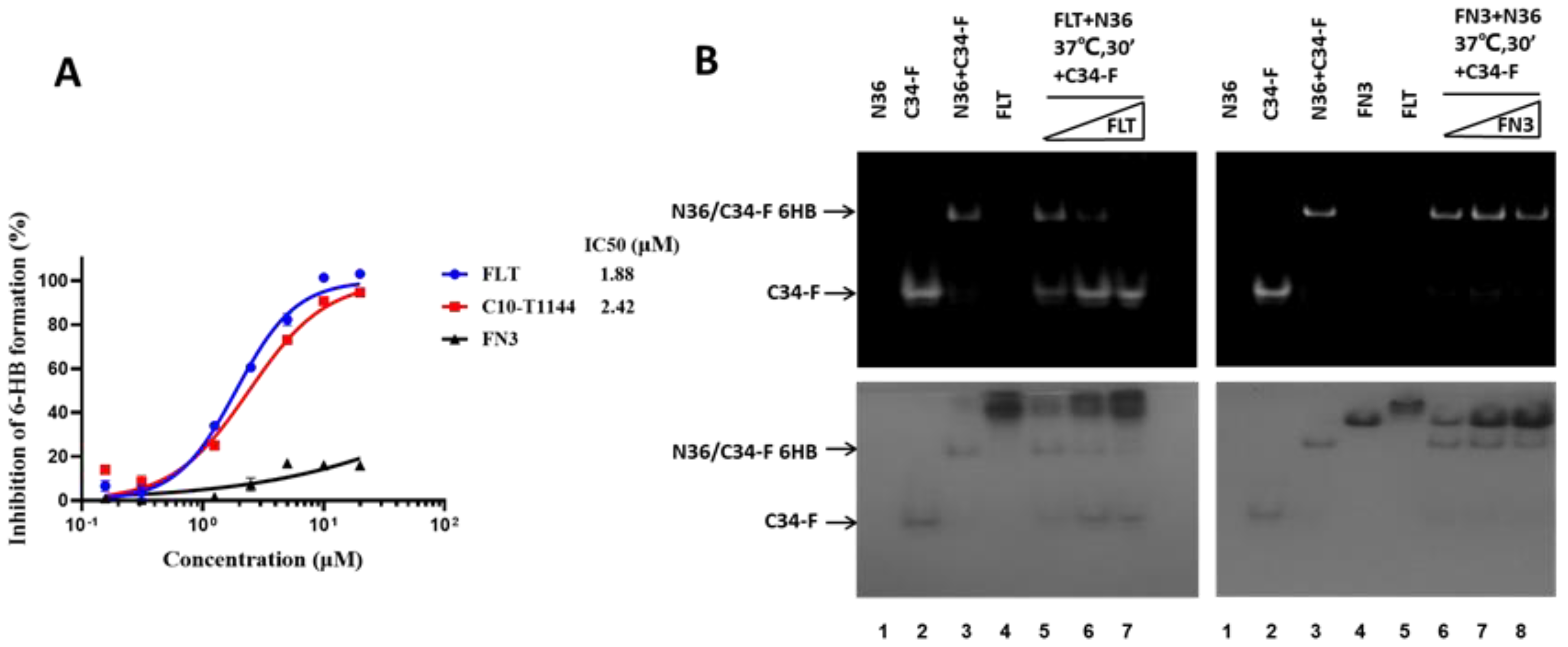
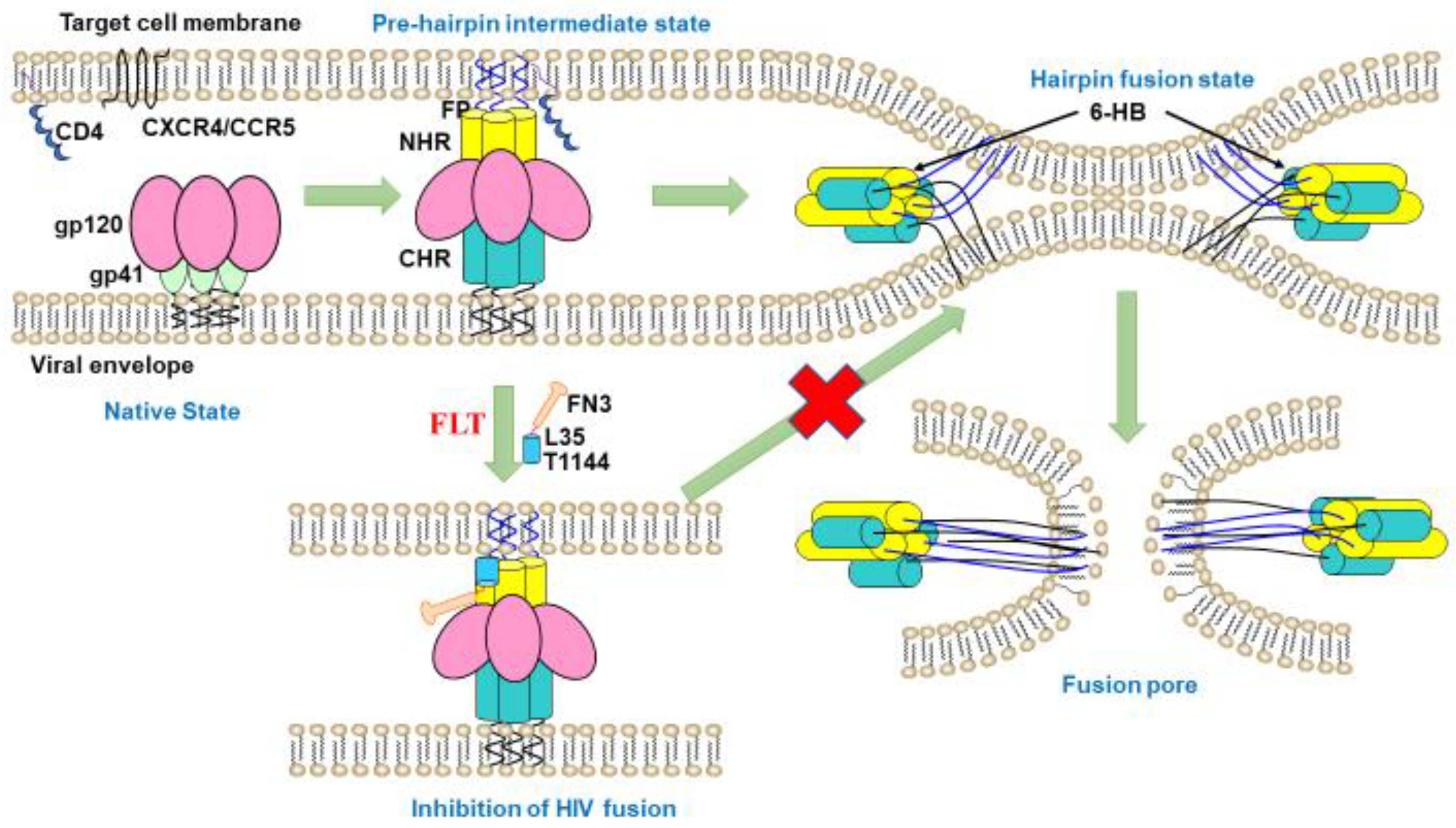
2.4. FLT Interfered with gp41 Six-Helix Bundle (6-HB) Formation
2.5. FLT Bound to HSA as Measured by Isothermal Titration Calorimetry (ITC)
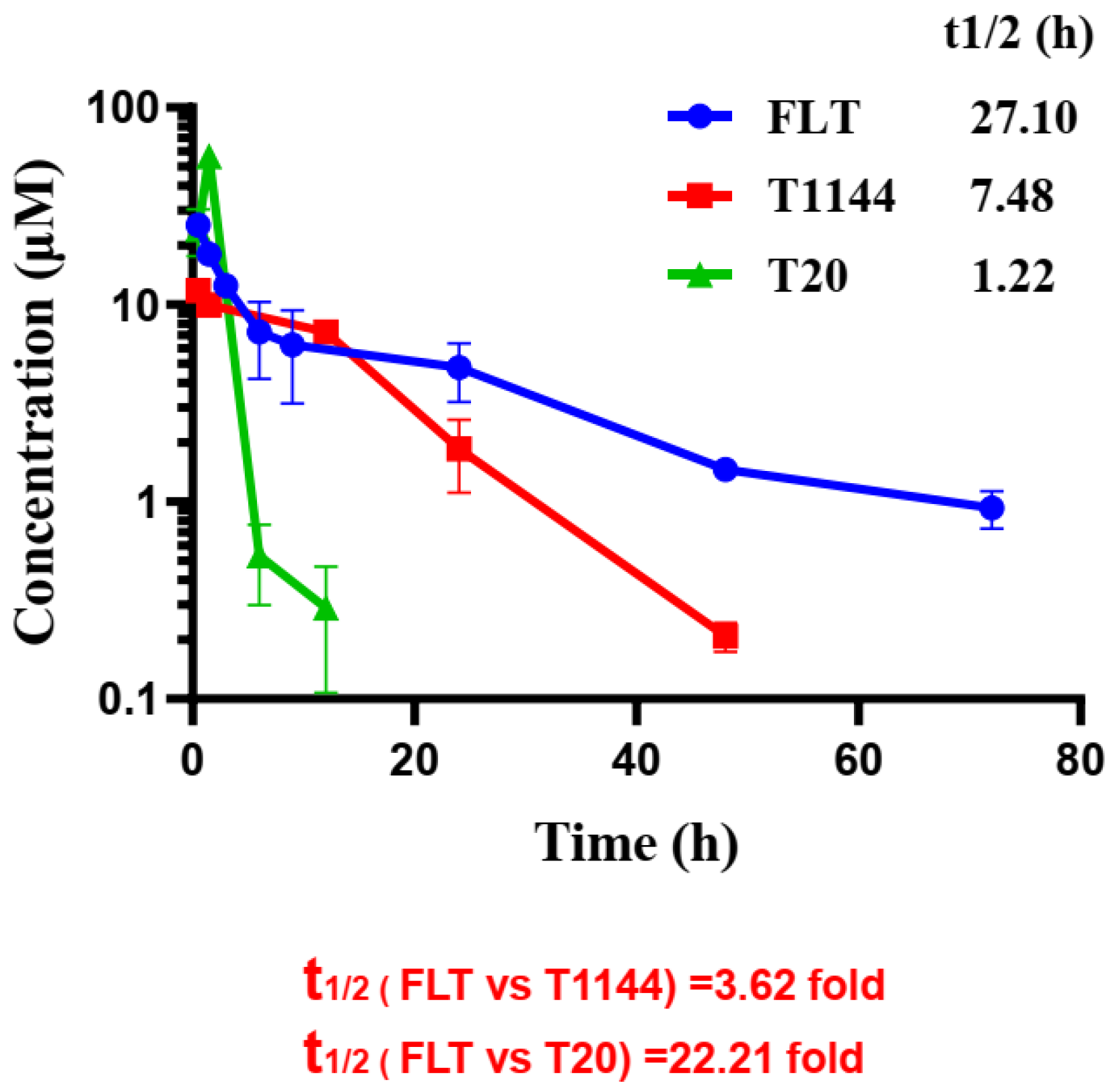
2.6. FLT Displayed Improved Pharmacokinetic Profiles in Rats
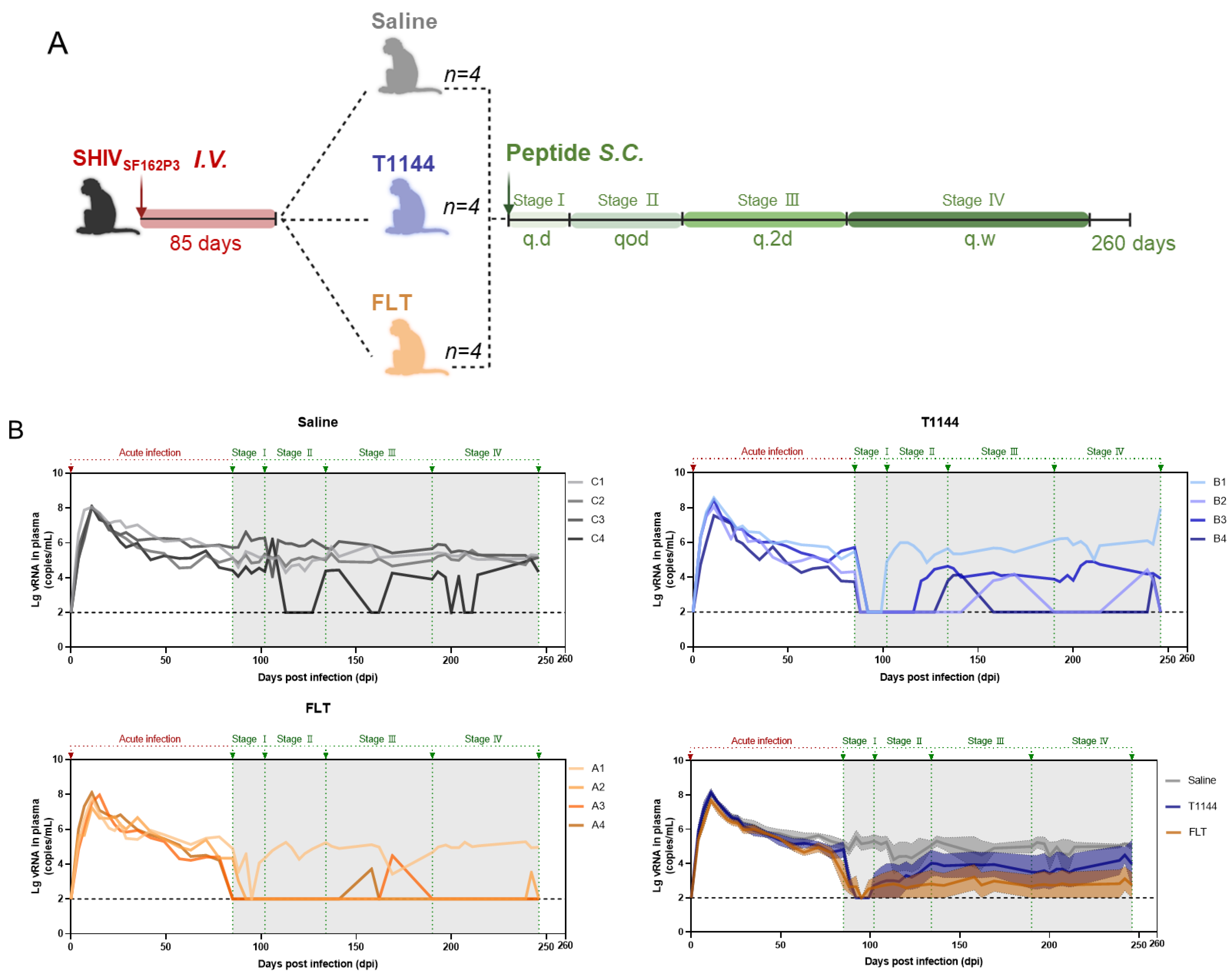
2.7. FLT Exhibited In Vivo Efficacy against Chronic SHIV Infection in Nonhuman Primates (NHPs)
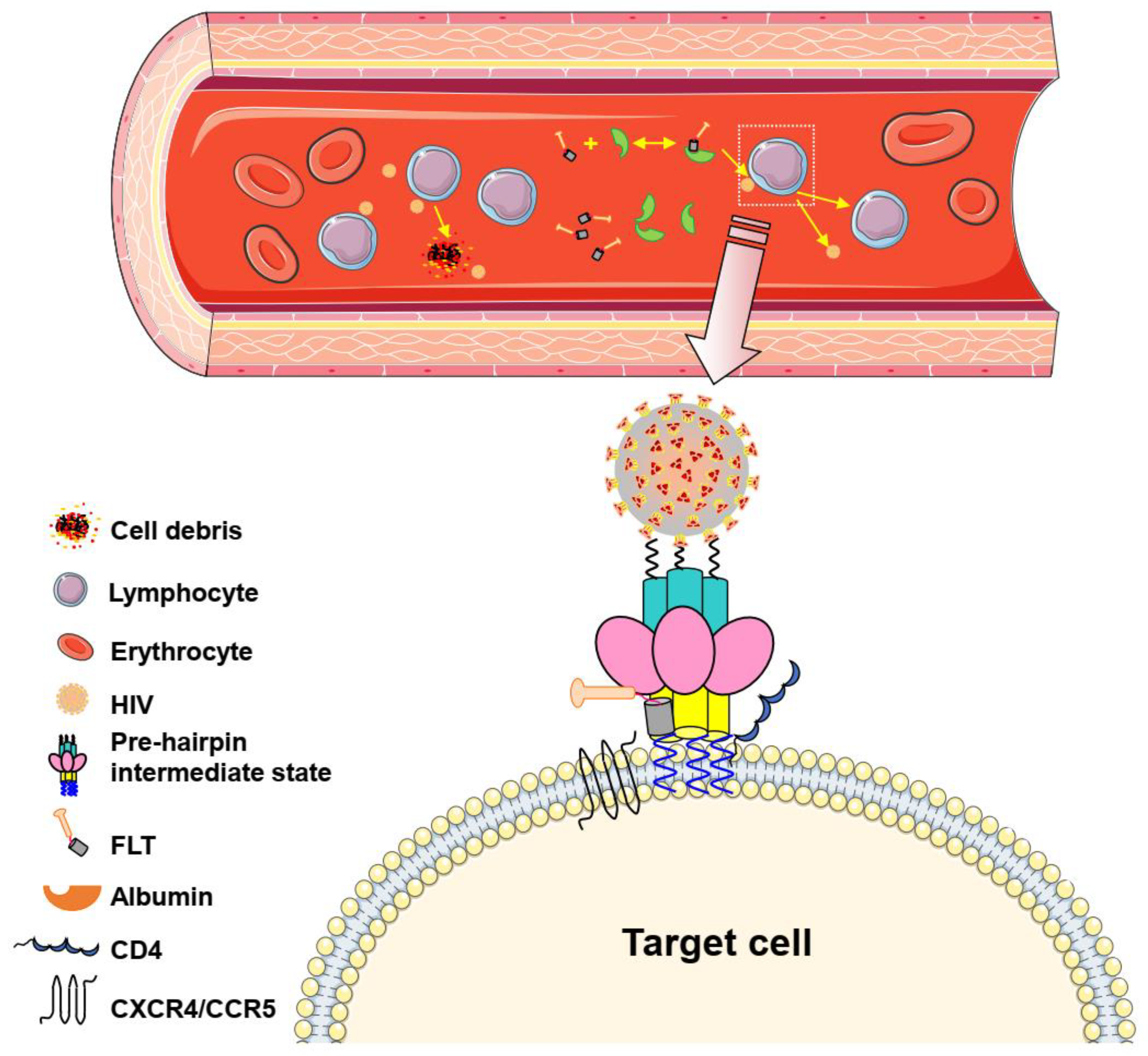
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Construction of the Expression Vectors
4.3. Expression and Purification of the Peptides
4.4. Inhibition of HIV-1 Infection
4.5. Fluorescence Native Polyacrylamide Gel Electrophoresis (FN-PAGE)
4.6. Detection of Inhibition of 6-HB Formation by ELISA
4.7. Isothermal Titration Calorimetry (ITC)
4.8. Pharmacokinetic Study
4.9. Rhesus Monkey Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dybul, M.; Attoye, T.; Baptiste, S.; Cherutich, P.; Dabis, F.; Deeks, S.G.; Dieffenbach, C.; Doehle, B.; Goodenow, M.M.; Jiang, A.; et al. The case for an HIV cure and how to get there. Lancet HIV 2021, 8, e51–e58. [Google Scholar] [CrossRef]
- Wymant, C.; Bezemer, D.; Blanquart, F.; Ferretti, L.; Gall, A.; Hall, M.; Golubchik, T.; Bakker, M.; Ong, S.H.; Zhao, L.; et al. A highly virulent variant of HIV-1 circulating in the Netherlands. Science 2022, 375, 540–545. [Google Scholar] [CrossRef] [PubMed]
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 17 February 2022).
- AIDS by the Numbers. Available online: https://www.unaids.org/en (accessed on 17 February 2022).
- FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 17 February 2022).
- Lu, L.; Su, S.; Yang, H.; Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell 2021, 184, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Yi, H.A.; Fochtman, B.C.; Rizzo, R.C.; Jacobs, A. Inhibition of HIV entry by targeting the envelope transmembrane subunit gp41. Curr. HIV Res. 2016, 14, 283–294. [Google Scholar] [CrossRef]
- Patel, I.H.; Zhang, X.; Nieforth, K.; Salgo, M.; Buss, N. Pharmacokinetics, pharmacodynamics and drug interaction potential of enfuvirtide. Clin. Pharmacokinet. 2005, 44, 175–186. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, X.; Chong, H.; Zhu, Y.; Wei, H.; Wu, X.; He, J.; Wang, X.; He, Y. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: Implications for viral entry and inhibition. J. Virol. 2017, 91, e00831-17. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.; Wu, X.; Su, Y.; He, Y. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 2016, 30, 1187–1196. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Chong, H.H.; Yu, D.W.; Guo, Y.; Zhou, Y.S.; He, Y.X. Design and characterization of cholesterylated peptide HIV-1/2 fusion inhibitors with extremely potent and long-lasting antiviral activity. J. Virol. 2019, 93, e02312–e02318. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV Fusion Inhibitor C34, the Anti-HIV Drug Fuzeon (T-20) Inhibits HIV-1 Entry by Targeting Multiple Sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Deeks, S.G.; Hoh, R.; Beatty, G.; Kuritzkes, B.A.; Martin, J.N.; Kuritzkes, D.R. Rapid emergence of enfuvirtide re-sistance in HIV-1-infected patients: Results of a clonal analysis. J. Acquir. Immune Defic. Syndr. 2006, 43, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvir-tide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.; Cai, L.; Lu, H.; Qi, Z.; Jiang, S. Combinations of the First and Next Generations of Human Immunodeficiency Virus (HIV) Fusion Inhibitors Exhibit a Highly Potent Synergistic Effect against Enfuvirtide-Sensitive and Resistant HIV Type 1 Strains. J. Virol. 2009, 83, 7862–7872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.I.; Castillo-Mancilla, J.R.; Tashima, K.T.; Landovitz, R.L. Advances in Long-Acting Agents for the Treatment of HIV Infection. Drugs 2020, 80, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Yang, B.; Lim, S.I.; Kim, J.C.; Tae, G.; Kwon, I. Site-Specific Albumination as an Alternative to PEGylation for the Enhanced Serum Half-Life in Vivo. Biomacromolecules 2016, 17, 1811–1817. [Google Scholar] [CrossRef]
- Hamburger, A.E.; Kim, S.; Welch, B.D.; Kay, M.S. Steric Accessibility of the HIV-1 gp41 N-trimer Region. J. Biol. Chem. 2005, 280, 12567–12572. [Google Scholar] [CrossRef] [Green Version]
- Blassel, L.; Zhukova, A.; Villabona-Arenas, C.J.; Atkins, K.E.; Hué, S.; Gascuel, O. Drug resistance mutations in HIV: New bioinformatics approaches and challenges. Curr. Opin. Virol. 2021, 51, 56–64. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Zhang, L.; Debnath, A.K. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J. Virol. Methods 1999, 80, 85–96. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Q.; Jiang, S. Determination of the HIV-1 gp41 fusogenic core conformation modeled by synthetic peptides: Applicable for identification of HIV-1 fusion inhibitors. Peptides 2003, 24, 1303–1313. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Notaril, S.; Narciso, P.; Bolli, A.; Fasano, M.; Ascenzi, P. Binding of Anti-HIV drugs to human serum albumin. Acquir. Immune Defic. Syndr. 2004, 56, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Bumbaca, B.; Li, Z.; Shah, D.K. Pharmacokinetics of protein and peptide conjugates. Drug Metab. Pharmacokinet. 2019, 34, 42–54. [Google Scholar] [CrossRef]
- Michot, N.; Guyochin, A.; Cinier, M.; Savignard, C.; Kitten, O.; Pascual, M.H.; Pouzieux, S.; Ozoux, M.L.; Verdier, P.; Vicat, P.; et al. Albumin Binding Nanofitins, a new scaffold to extend half-life of Biologics—A case study with exenatide peptide. Peptides 2022, 152, 170760. [Google Scholar] [CrossRef]
- Rondon, A.; Mahri, S.; Morales-Yanez, F.; Dumoulin, M.; Vanbever, R. Protein engineering strategies for improved pharma-cokinetics. Adv. Funct. Mater. 2021, 31, 202101633. [Google Scholar] [CrossRef]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-covalent albumin-binding ligands for extending the circulating half-life of small bio-therapeutics. MedChemComm 2019, 10, 1068–1081. [Google Scholar] [CrossRef]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Hantschel, O.; Biancalana, M.; Koide, S. Monobodies as enabling tools for structural and mechanistic biology. Curr. Opin. Struct. Biol. 2020, 60, 167–174. [Google Scholar] [CrossRef]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Main, A.L.; Harvey, T.S.; Baron, M.; Boyd, J.; Campbell, I.D. The three-dimensional structure of the tenth type III module of fibronectin: An insight into RGD-mediated interactions. Cell 1992, 71, 671–678. [Google Scholar] [CrossRef]
- Gapizov, S.S.; Petrovskaya, L.E.; Shingarova, L.N.; Kryukova, E.A.; Boldyreva, E.F.; Lukashev, E.P.; Yakimov, S.A.; Svirshchevskaya, E.V.; Dolgikh, D.A.; Kirpichnikov, M.P. Fusion with an albumin-binding domain improves pharmacokinetics of an alphavbeta3-integrin binding fibronectin scaffold protein. Biotechnol. Appl. Biochem. 2019, 66, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Fang, P.; Kay, B.K. Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase. New Biotechnol. 2012, 29, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.J.; McGinnis, J.E.; Martinez, M.J.; Wang, G.L.; Zhou, J.; Simmons, E.; Amet, T.; Abdeen, S.J.; Van Huysse, J.W.; Bowsher, R.R.; et al. FN3-based monobodies selective for the receptor binding domain of the SARS-CoV-2 spike protein. New Biotechnol. 2021, 62, 79–85. [Google Scholar] [CrossRef]
- Wojcik, J.; Hantschel, O.; Grebien, F.; Kaupe, I.; Bennett, K.L.; Barkinge, J.; Jones, R.B.; Koide, A.; Superti-Furga, G.; Koide, S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010, 17, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Pan, C.; Li, Y.; Lu, H.; He, W.; Jiang, S. A bivalent recombinant protein inactivates HIV-1 by targeting the gp41 pre-hairpin fusion intermediate induced by CD4 D1D2 domains. Retrovirology 2012, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Schneider, E.L.; Ashley, G.W.; Dillen, L.; Stoops, B.; Austin, N.E.; Malcolm, B.A.; Santi, D.V. Half-life extension of the HIV-fusion inhibitor peptide TRI-1144 using a novel linker technology. Eur. J. Pharm. Biopharm. 2015, 93, 254–259. [Google Scholar] [CrossRef]
- Peters, T., Jr. Serum albumin. Adv. Protein. Chem. 1985, 37, 161–245. [Google Scholar]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin (R) domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Yao, C.; Zhao, Q.-X.; Cai, W.-P.; Wang, M.; Lu, H.-Z.; Chen, Y.-Y.; Liu, L.; Wang, H.; He, Y.; et al. Efficacy and safety of the long-acting fusion inhibitor albuvirtide in antiretroviral-experienced adults with human immunodeficiency virus-1: Interim analysis of the randomized, controlled, phase 3, non-inferiority TALENT study. Chin. Med. J. 2020, 133, 2919–2927. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Yao, C.; Wang, L.; Min, W.J.; Xu, J.H.; Xiao, J.H.; Huang, M.X.; Chen, B.; Liu, B.; Li, X.L.; et al. An albu-min-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob. Agents Chemother. 2010, 54, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millham, L.R.; Scott, J.A.; Sax, P.E.; Shebl, F.M.; Reddy, K.P.; Losina, E.; Walensky, R.P.; Freedberg, K.A. Clinical and Economic Impact of Ibalizumab for People with Multidrug-Resistant HIV in the United States. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, T.; Kobayashi, M.; Seki, T.; Miki, S.; Wakasa-Morimoto, C.; Suyama-Kagitani, A.; Kawauchi-Miki, S.; Taishi, T.; Kawasuji, T.; Johns, B.A.; et al. Antiviral Characteristics of GSK1265744, an HIV Integrase Inhibitor Dosed Orally or by Long-Acting Injection. Antimicrob. Agents Chemother. 2015, 59, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Pu, J.; Su, S.; Hua, C.; Su, X.; Wang, Q.; Jiang, S.; Lu, L. Revisiting the mechanism of enfuvirtide and designing an analog with improved fusion inhibitory activity by targeting triple sites in gp41. AIDS 2019, 33, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Rasquinha, G.; Du, L.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules 2019, 24, 1134. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Bi, W.; Xu, W.; Cheng, L.; Xue, J.; Wang, Q.; Yu, F.; Xia, S.; Wang, Q.; Li, G.; Qin, C.; et al. IgG Fc-binding motif-conjugated HIV-1 fusion inhibitor exhibits improved potency and in vivo half-life: Potential application in combination with broad neu-tralizing antibodies. PLoS Pathog. 2019, 15, e1008082. [Google Scholar] [CrossRef]
- Xue, J.; Cong, Z.; Xiong, J.; Wang, W.; Jiang, H.; Chen, T.; Wu, F.; Liu, K.; Su, A.; Ju, B.; et al. Repressive Effect of Primary Virus Replication on Superinfection Correlated with Gut-Derived Central Memory CD4+ T Cells in SHIV-Infected Chinese Rhesus Macaques. PLoS ONE 2013, 8, e72295. [Google Scholar] [CrossRef]
- Xue, J.; Chong, H.; Zhu, Y.; Zhang, J.; Tong, L.; Lu, J.; Chen, T.; Cong, Z.; Wei, Q.; He, Y. Efficient treatment and pre-exposure prophylaxis in rhesus macaques by an HIV fusion-inhibitory lipopeptide. Cell 2022, 185, 131–144.e18. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 Clinical Isolates | IC50 (nM) | ||
|---|---|---|---|
| FN3 | FLT | Enfuvirtide (T20) | |
| HIV-1 96USSN20 (X4/R5, A) | >128 | 65.3 ± 2.6 | 55.3 ± 2.4 |
| HIV-1 96USNG17 (X4, A) | >128 | 9.8 ± 0.9 | 49.3 ± 4.9 |
| HIV-1 90US_873 (R5, B) | >128 | 6.5 ± 0.2 | 50.1 ± 2.0 |
| HIV-1 BZ167 (X4, B) | >128 | 8.8 ± 0.3 | 53.9 ± 4.3 |
| HIV-1 SE364 (R5, C) | >128 | 12.5 ± 0.5 | 45.2 ± 3.2 |
| HIV-1 PBL288 (R5, C) | >128 | 9.3 ± 0.5 | 46.9 ± 2.1 |
| HIV-1 92UG001 (X4/R5, D) | >128 | 11.7 ± 0.8 | 77.1 ± 6.8 |
| HIV-1 J32228M4 (R5, D) | >128 | 6.4 ± 0.3 | 21.1 ± 1.1 |
| HIV-1 DJ263 (R5, CRF02_AG) | >128 | 7.5 ± 0.3 | 55.6 ± 1.2 |
| HIV-1 CAM1475MV (R5, CRF02_AG) | >128 | 10.2 ± 0.6 | 61.2 ± 1.3 |
| HIV-1 Drug-Resistant Strains | IC50 (nM) | ||
|---|---|---|---|
| FN3 | FLT | Enfuvirtide (T20) | |
| T20-resistant strains | |||
| HIV NL4-3 (T20-sensitive strain) | >128 | 24.2 ± 2.0 | 20.8 ± 5.6 |
| HIV NL4-3 N42T, N43K | >128 | 28.1 ± 1.8 | >128 |
| HIV NL4-3 V38E, N42S | >128 | 25.9 ± 1.0 | >128 |
| HIV NL4-3 V38A, N42T | >128 | 19.4 ± 1.5 | >128 |
| NRTI/NNRTI-resistant strains | |||
| HIV-1 A018A (NRTI -sensitive) | >128 | 24.7 ± 1.7 | 37.9 ± 2.6 |
| HIV-1 A012 (NRTI-resistant) | >128 | 28.6 ± 9.0 | 27.6 ± 5.1 |
| HIV-1 IIIB A17 (NNRTI-resistant) | >128 | 23.2 ± 1.9 | 40.2 ± 4.6 |
| Integrase inhibitor-resistant strains | |||
| HIV-1 NL4-3 CA138763 | >128 | 20.2 ± 0.6 | 31.2 ± 1.3 |
| HIV-1 NL4-3 CA138764 | >128 | 22.3 ± 2.1 | 35.3 ± 1.4 |
| HIV-1 NL4-3 CA138767 | >128 | 20.7 ± 0.3 | 34.6 ± 2.5 |
| Peptide | T1/2 (h) | Tmax (h) | Cmax (μg/mL) | AUC (μg/mL/h) | CL (mL/h) |
|---|---|---|---|---|---|
| FLT | 27.09 ± 6.9 | 0.5 | 863.2 ± 73.5 | 10624.1 ± 1583.2 | 0.063 ± 0.004 |
| T1144 | 7.48 ± 0.4 | 0.5 | 147.4 ± 2.1 | 2338.9 ± 127.0 | 0.349 ± 0.019 |
| T20 | 1.22 ± 0.2 | 1.5 | 305.0 ± 20.9 | 967.3 ± 83.4 | 0.855 ± 0.075 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Cong, Z.; Duan, Q.; Wang, Q.; Su, S.; Wang, R.; Lu, L.; Xue, J.; Jiang, S. A Protein-Based, Long-Acting HIV-1 Fusion Inhibitor with an Improved Pharmacokinetic Profile. Pharmaceuticals 2022, 15, 424. https://doi.org/10.3390/ph15040424
Xu W, Cong Z, Duan Q, Wang Q, Su S, Wang R, Lu L, Xue J, Jiang S. A Protein-Based, Long-Acting HIV-1 Fusion Inhibitor with an Improved Pharmacokinetic Profile. Pharmaceuticals. 2022; 15(4):424. https://doi.org/10.3390/ph15040424
Chicago/Turabian StyleXu, Wei, Zhe Cong, Qianyu Duan, Qian Wang, Shan Su, Rui Wang, Lu Lu, Jing Xue, and Shibo Jiang. 2022. "A Protein-Based, Long-Acting HIV-1 Fusion Inhibitor with an Improved Pharmacokinetic Profile" Pharmaceuticals 15, no. 4: 424. https://doi.org/10.3390/ph15040424
APA StyleXu, W., Cong, Z., Duan, Q., Wang, Q., Su, S., Wang, R., Lu, L., Xue, J., & Jiang, S. (2022). A Protein-Based, Long-Acting HIV-1 Fusion Inhibitor with an Improved Pharmacokinetic Profile. Pharmaceuticals, 15(4), 424. https://doi.org/10.3390/ph15040424