
Pan-Phosphodiesterase Inhibitors Attenuate TGF-β-Induced Pro-Fibrotic Phenotype in Alveolar Epithelial Type II Cells by Downregulating Smad-2 Phosphorylation

 , , , ,
, , , , 
Abstract

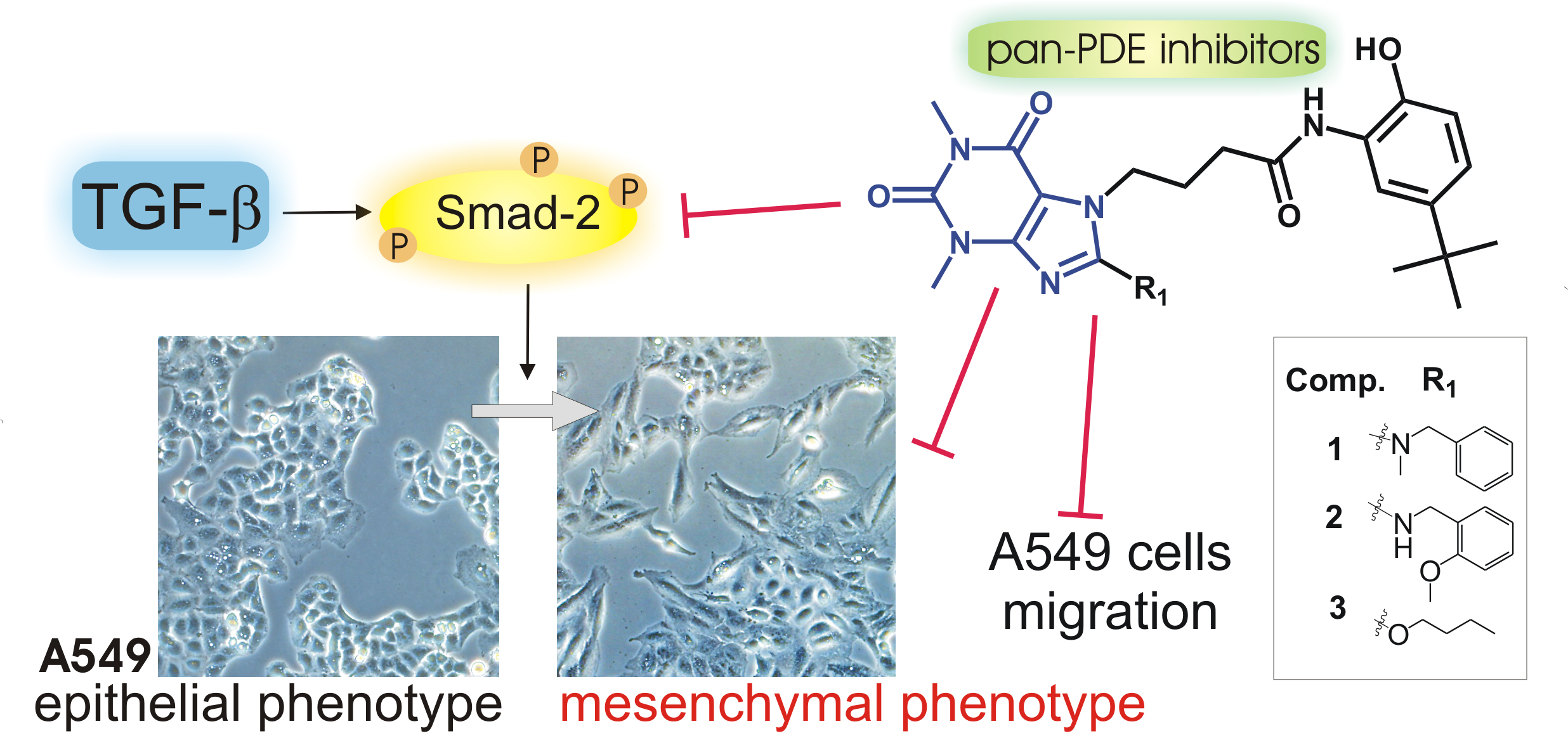{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Pan-PDE Inhibitors Slightly Modulate Basic A549 Cells’ Functions
2.2. TGF-β-Induced Mesenchymal-like Phenotype in A549 Cells Is Diminished by Pan-PDE Inhibitors
2.3. TGF-β-Induced A549 Cells’ Migration Is Decreased in Response to Pan-PDE Inhibitors
2.4. Pan-PDE Inhibitors Activate CREB Phosphorylation in TGF-β-Induced A549 Cells
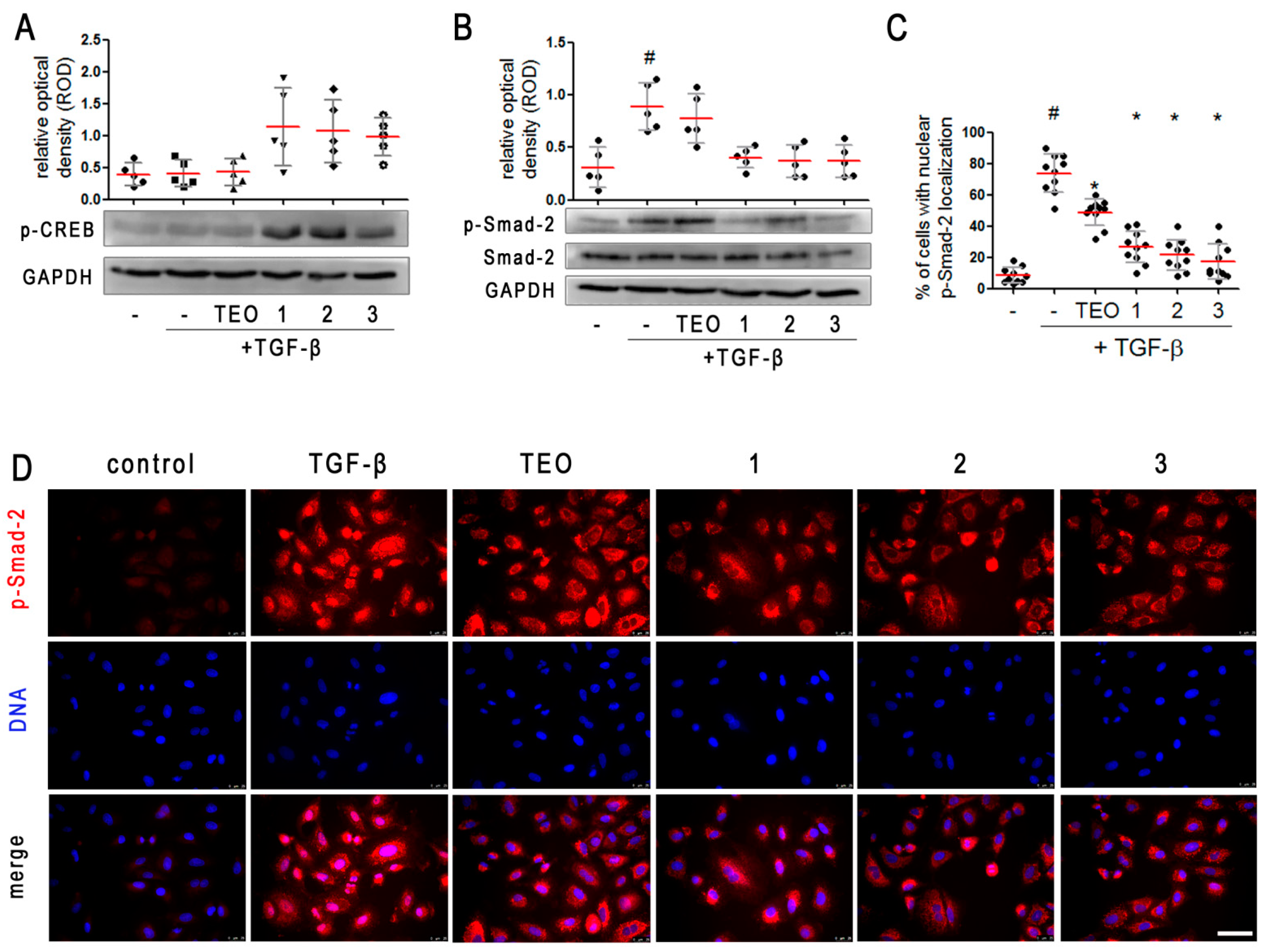
2.5. Pan-PDE Inhibitors Attenuate Smad-2 Signaling in TGF-β-Induced Alveolar Epithelial Type II Cells
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Culture
4.3. Cell Morphology, Viability, and Proliferation
4.4. Migration Assays
4.5. Immunofluorescence
4.6. ELISA
4.7. Western Blot Analysis
4.8. Gelatin Zymography
4.9. RNA Extraction and Reverse Transcription-Quantitative PCR
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 911–921. [Google Scholar] [CrossRef]
- Nishioka, M.; Venkatesan, N.; Dessalle, K.; Mogas, A.; Kyoh, S.; Lin, T.-Y.; Nair, P.; Baglole, C.J.; Eidelman, D.H.; Ludwig, M.S.; et al. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir. Res. 2015, 16, 72. [Google Scholar] [CrossRef]
- Habiel, D.M.; Espindola, M.S.; Jones, I.C.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. CCR10+ epithelial cells from idiopathic pulmonary fibrosis lungs drive remodeling. JCI Insight 2018, 3, e122211. [Google Scholar] [CrossRef]
- Paw, M.; Wnuk, D.; Jakieła, B.; Bochenek, G.; Sładek, K.; Madeja, Z.; Michalik, M. Responsiveness of human bronchial fibroblasts and epithelial cells from asthmatic and non-asthmatic donors to the transforming growth factor-β1 in epithelial-mesenchymal trophic unit model. BMC Mol. Cell Biol. 2021, 22, 19. [Google Scholar] [CrossRef]
- Reeves, S.R.; Barrow, K.A.; Kolstad, T.K.; White, M.P.; Rich, L.M.; Wight, T.N.; Debley, J.S. Fibroblast gene expression following asthmatic bronchial epithelial cell conditioning correlates with epithelial donor lung function and exacerbation history. Sci. Rep. 2018, 8, 15768. [Google Scholar] [CrossRef]
- Reeves, S.R.; Kang, I.; Chan, C.K.; Barrow, K.A.; Kolstad, T.K.; White, M.P.; Ziegler, S.F.; Wight, T.N.; Debley, J.S. Asthmatic bronchial epithelial cells promote the establishment of a Hyaluronan-enriched, leukocyte-adhesive extracellular matrix by lung fibroblasts. Respir. Res. 2018, 19, 146. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Z.; Pan, H.-Y.; Wang, D.-X.; Deng, Z.-T.; Ye, X.-L. TGF-beta1 induces human bronchial epithelial cell-to-mesenchymal transition in vitro. Lung 2009, 187, 187–194. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Cattani-Cavalieri, I.; Musheshe, N.; Nikolaev, V.O.; Schmidt, M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 2019, 197, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Page, C.P. Phosphodiesterase Inhibitors for the Treatment of Asthma and Chronic Obstructive Pulmonary Disease. Int. Arch. Allergy Immunol. 2014, 165, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E. Inhaled Phosphodiesterase 4 (PDE4) Inhibitors for Inflammatory Respiratory Diseases. Front. Pharmacol. 2020, 11, 259. [Google Scholar] [CrossRef]
- Kawamatawong, T. Phosphodiesterase-4 Inhibitors for Non-COPD Respiratory Diseases. Front. Pharmacol. 2021, 12, 518345. [Google Scholar] [CrossRef]
- Matera, M.G.; Ora, J.; Cavalli, F.; Rogliani, P.; Cazzola, M. New Avenues for Phosphodiesterase Inhibitors in Asthma. J. Exp. Pharmacol. 2021, 13, 291–302. [Google Scholar] [CrossRef]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Kolosionek, E.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Guenther, A.; Grimminger, F.; Seeger, W.; Banat, G.A.; Schermuly, R.T.; Pullamsetti, S.S. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: The role of phosphodiesterase 4. Mol. Biol. Cell 2009, 20, 4751–4765. [Google Scholar] [CrossRef] [PubMed]
- Chłoń-Rzepa, G.; Ślusarczyk, M.; Jankowska, A.; Gawalska, A.; Bucki, A.; Kołaczkowski, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Zygmunt, M.; et al. Novel amide derivatives of 1,3-dimethyl-2,6-dioxopurin-7-yl-alkylcarboxylic acids as multifunctional TRPA1 antagonists and PDE4/7 inhibitors: A new approach for the treatment of pain. Eur. J. Med. Chem. 2018, 158, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Bucki, A.; Gawalska, A.; Kołaczkowski, M.; Pawłowski, M. Novel butanehydrazide derivatives of purine-2,6-dione as dual PDE4/7 inhibitors with potential anti-inflammatory activity: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 146, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Jankowska, A.; Ślusarczyk, M.; Jakieła, B.; Plutecka, H.; Pociecha, K.; Świerczek, A.; Popiół, J.; Koczurkiewicz-Adamczyk, P.; Wyska, E.; et al. Synthesis and in vitro evaluation of anti-inflammatory, antioxidant, and anti-fibrotic effects of new 8-aminopurine-2,6-dione-based phosphodiesterase inhibitors as promising anti-asthmatic agents. Bioorg. Chem. 2021, 117, 105409. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ellen, E.; Świerczek, A.; Pociecha, K.; Koczurkiewicz, P.; Piska, K.; Gawędzka, A.; Wyska, E.; et al. Novel phosphodiesterases inhibitors from the group of purine-2,6-dione derivatives as potent modulators of airway smooth muscle cell remodelling. Eur. J. Pharmacol. 2019, 865, 172779. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Ferdek, P.E.; Kusiak, A.A.; Świerczek, A.; Pociecha, K.; Koczurkiewicz-Adamczyk, P.; Wyska, E.; et al. A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling. Int. J. Mol. Sci. 2020, 21, 4008. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef]
- James, A.L.; Wenzel, S. Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 2007, 30, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.; O’Reilly, P.; Antony, V.B.; Gaggar, A.; Thannickal, V.J. Matrix Remodeling in Pulmonary Fibrosis and Emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 54, 751–760. [Google Scholar] [CrossRef]
- Doherty, T.; Broide, D. Cytokines and growth factors in airway remodeling in asthma. Curr. Opin. Immunol. 2007, 19, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The Cytokine Network in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Maher, T.M.; Strek, M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Gomer, R.H. New approaches to modulating idiopathic pulmonary fibrosis. Curr. Allergy Asthma Rep. 2013, 13, 607–612. [Google Scholar] [CrossRef][Green Version]
- Berair, R.; Brightling, C.E. Asthma therapy and its effect on airway remodelling. Drugs 2014, 74, 1345–1369. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.P.; Deshpande, D.A.; Penn, R.B. New targets for resolution of airway remodeling in obstructive lung diseases. F1000Research 2018, 7, 680. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiró, T.; Serrano, A.; Guijarro, R.; Zaragozá, C.; Tenor, H.; Cortijo, J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm. Pharmacol. Ther. 2014, 28, 138–148. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Artigues, E.; Aparicio, J.; Tenor, H.; Sanz, C.; Cortijo, J. Simvastatin Increases the Ability of Roflumilast N-oxide to Inhibit Cigarette Smoke-Induced Epithelial to Mesenchymal Transition in Well-differentiated Human Bronchial Epithelial Cells in vitro. COPD 2015, 12, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Chen, W.-J.; Liaw, S.-F.; Lin, M.-W.; Lin, S.-C. Effects of aminophylline on airway epithelial-mesenchymal transition in brown Norway rats after repeated allergen challenge. Exp. Lung Res. 2019, 45, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Contreras, S.; Ribera, P.; Morell, A.; Serrano, A.; Milara, J.; Milara, J. Roflumilast N- oxide combined with sildenafil reverses cellular remodeling on IPF models. Eur. Respir. J. 2017, 50, OA4833. [Google Scholar]
- Wright, L.C.; Seybold, J.; Robichaud, A.; Adcock, I.M.; Barnes, P.J. Phosphodiesterase expression in human epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 1998, 275, L694–L700. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Jahn, H.U.; Seybold, J.; Neurohr, C.; Barnes, P.J.; Hippenstiel, S.; Kraemer, H.J.; Suttorp, N. Identification and function of cyclic nucleotide phosphodiesterase isoenzymes in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 20, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Cattani-Cavalieri, I.; Valença, S.S.; Musheshe, N.; Schmidt, M. Function of cAMP scaffolds in obstructive lung disease: Focus on epithelial-to-mesenchymal transition and oxidative stress. Br. J. Pharmacol. 2019, 176, 2402–2415. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Vang, A.G.; Basole, C.; Dong, H.; Nguyen, R.K.; Housley, W.; Guernsey, L.; Adami, A.J.; Thrall, R.S.; Clark, R.B.; Epstein, P.M.; et al. Differential Expression and Function of PDE8 and PDE4 in Effector T cells: Implications for PDE8 as a Drug Target in Inflammation. Front. Pharmacol. 2016, 7, 259. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Schmidt, M.; Gosens, R. PDE8: A Novel Target in Airway Smooth Muscle. Am. J. Respir. Cell Mol. Biol. 2018, 58, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.R.; Lee, J.M.; Seo, H.J.; Park, J.S.; Lee, J.H.; Park, S.W.; Jang, A.S.; Kim, D.J.; Koh, E.S.; Uh, S.T.; et al. Apolipoprotein A1 Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition of Alveolar Epithelial Cells. Tuberc. Respir. Dis. 2016, 79, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Shen, J. Hesperidin inhibits the epithelial to mesenchymal transition induced by transforming growth factor-β1 in A549 cells through Smad signaling in the cytoplasm. Braz. J. Pharm. Sci. 2019, 55, e18172. [Google Scholar] [CrossRef]
- Ji, Y.; Dou, Y.-N.; Zhao, Q.-W.; Zhang, J.-Z.; Yang, Y.; Wang, T.; Xia, Y.-F.; Dai, Y.; Wei, Z.-F. Paeoniflorin suppresses TGF-β mediated epithelial-mesenchymal transition in pulmonary fibrosis through a Smad-dependent pathway. Acta Pharmacol. Sin. 2016, 37, 794–804. [Google Scholar] [CrossRef]
- Kang, H.; Lee, M.; Jang, S.-W. Celastrol inhibits TGF-β1-induced epithelial-mesenchymal transition by inhibiting Snail and regulating E-cadherin expression. Biochem. Biophys. Res. Commun. 2013, 437, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Ihara, H.; Mitsuishi, Y.; Kato, M.; Takahashi, F.; Tajima, K.; Hayashi, T.; Hidayat, M.; Winardi, W.; Wirawan, A.; Hayakawa, D.; et al. Nintedanib inhibits epithelial-mesenchymal transition in A549 alveolar epithelial cells through regulation of the TGF-β/Smad pathway. Respir. Investig. 2020, 58, 275–284. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Z.; Jia, Q.; Bo, C.; Shao, H.; Zhang, Z. Pirfenidone inhibits epithelial-mesenchymal transition and pulmonary fibrosis in the rat silicosis model. Toxicol. Lett. 2019, 300, 59–66. [Google Scholar] [CrossRef]
- Fehrholz, M.; Speer, C.P.; Kunzmann, S. Caffeine and rolipram affect Smad signalling and TGF-β1 stimulated CTGF and transgelin expression in lung epithelial cells. PLoS ONE 2014, 9, e97357. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Dua, K.; Adcock, I.M.; Horvat, J.C.; Kim, R.Y.; Hansbro, P.M. Cellular mechanisms underlying steroid-resistant asthma. Eur. Respir. Rev. 2019, 28, 190096. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.; Plumb, J.; Kaur, M.; Ray, D.; Singh, D. Additive anti-inflammatory effects of corticosteroids and phosphodiesterase-4 inhibitors in COPD CD8 cells. Respir. Res. 2016, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.L.; Milara, J.; Lluch, J.; De Diego, A.; Sanz, C.; Cortijo, J. Phosphodiesterase-4 inhibition improves corticosteroid insensitivity in pulmonary endothelial cells under oxidative stress. Allergy 2013, 68, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Lluch, J.; Almudever, P.; Freire, J.; Xiaozhong, Q.; Cortijo, J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2014, 134, 314–322.e9. [Google Scholar] [CrossRef]
- Pawłowski, M.; Chłoń-Rzepa, G.; Obniska, J.; Zejc, A.; Charakchieva-Minol, S.; Mokrosz, M.J. Synthesis 5HT1A and 5-HT2A receptor affinity of new 1-phenylpiperazinylpropyl derivatives of purine-2,6- and pyrrolidine-2,5-diones. Il Farmaco 2000, 55, 461–468. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ferreira, B.; Koczurkiewicz-Adamczyk, P.; Pękala, E.; Wyska, E.; Pociecha, K.; Gosens, R. Pan-Phosphodiesterase Inhibitors Attenuate TGF-β-Induced Pro-Fibrotic Phenotype in Alveolar Epithelial Type II Cells by Downregulating Smad-2 Phosphorylation. Pharmaceuticals 2022, 15, 423. https://doi.org/10.3390/ph15040423
Wójcik-Pszczoła K, Chłoń-Rzepa G, Jankowska A, Ferreira B, Koczurkiewicz-Adamczyk P, Pękala E, Wyska E, Pociecha K, Gosens R. Pan-Phosphodiesterase Inhibitors Attenuate TGF-β-Induced Pro-Fibrotic Phenotype in Alveolar Epithelial Type II Cells by Downregulating Smad-2 Phosphorylation. Pharmaceuticals. 2022; 15(4):423. https://doi.org/10.3390/ph15040423
Chicago/Turabian StyleWójcik-Pszczoła, Katarzyna, Grażyna Chłoń-Rzepa, Agnieszka Jankowska, Bruno Ferreira, Paulina Koczurkiewicz-Adamczyk, Elżbieta Pękala, Elżbieta Wyska, Krzysztof Pociecha, and Reinoud Gosens. 2022. "Pan-Phosphodiesterase Inhibitors Attenuate TGF-β-Induced Pro-Fibrotic Phenotype in Alveolar Epithelial Type II Cells by Downregulating Smad-2 Phosphorylation" Pharmaceuticals 15, no. 4: 423. https://doi.org/10.3390/ph15040423
APA StyleWójcik-Pszczoła, K., Chłoń-Rzepa, G., Jankowska, A., Ferreira, B., Koczurkiewicz-Adamczyk, P., Pękala, E., Wyska, E., Pociecha, K., & Gosens, R. (2022). Pan-Phosphodiesterase Inhibitors Attenuate TGF-β-Induced Pro-Fibrotic Phenotype in Alveolar Epithelial Type II Cells by Downregulating Smad-2 Phosphorylation. Pharmaceuticals, 15(4), 423. https://doi.org/10.3390/ph15040423