
In Vitro and In Vivo Antiviral Studies of New Heteroannulated 1,2,3-Triazole Glycosides Targeting the Neuraminidase of Influenza A Viruses


 , , , ,
, , , ,  , ,
, , 
Abstract
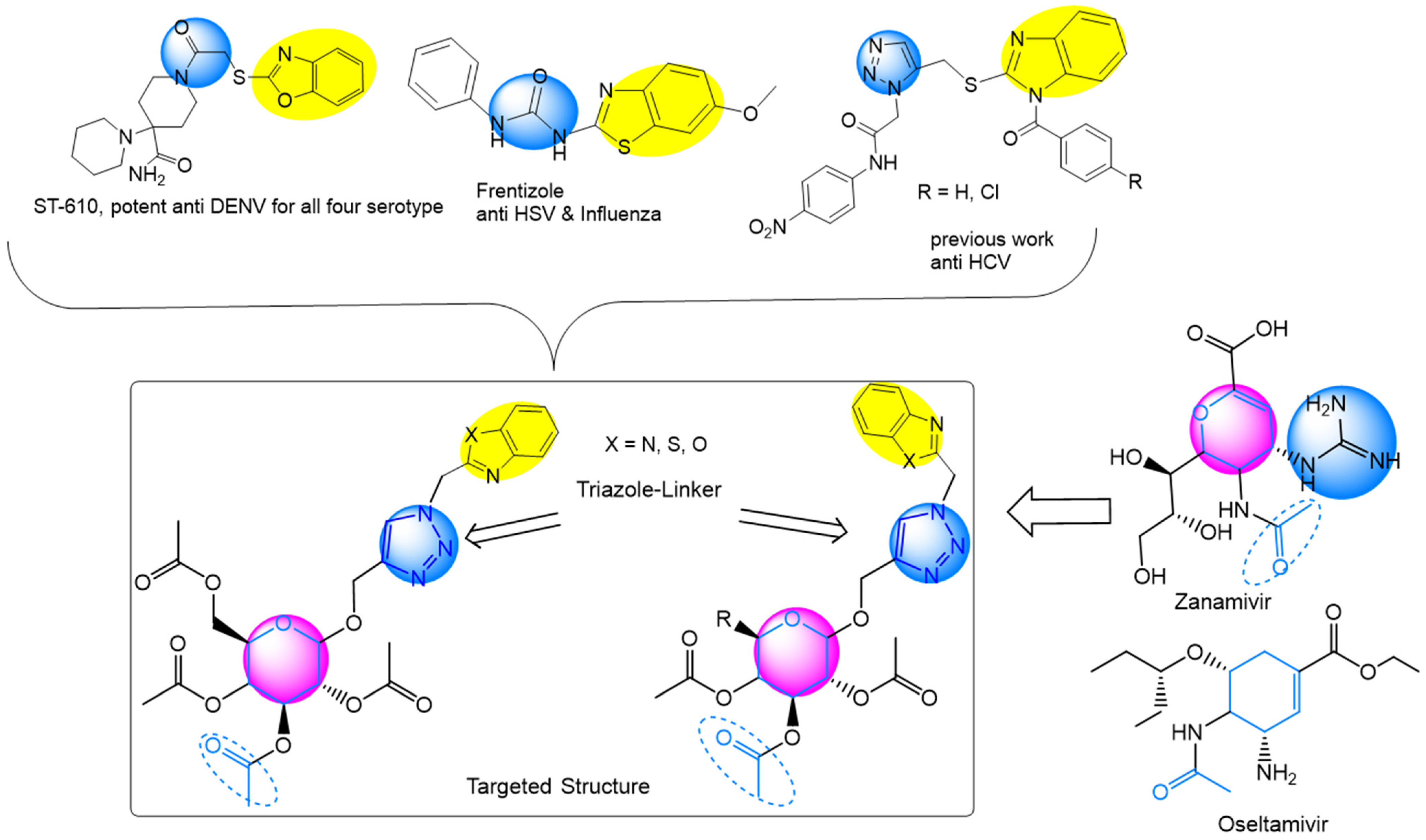
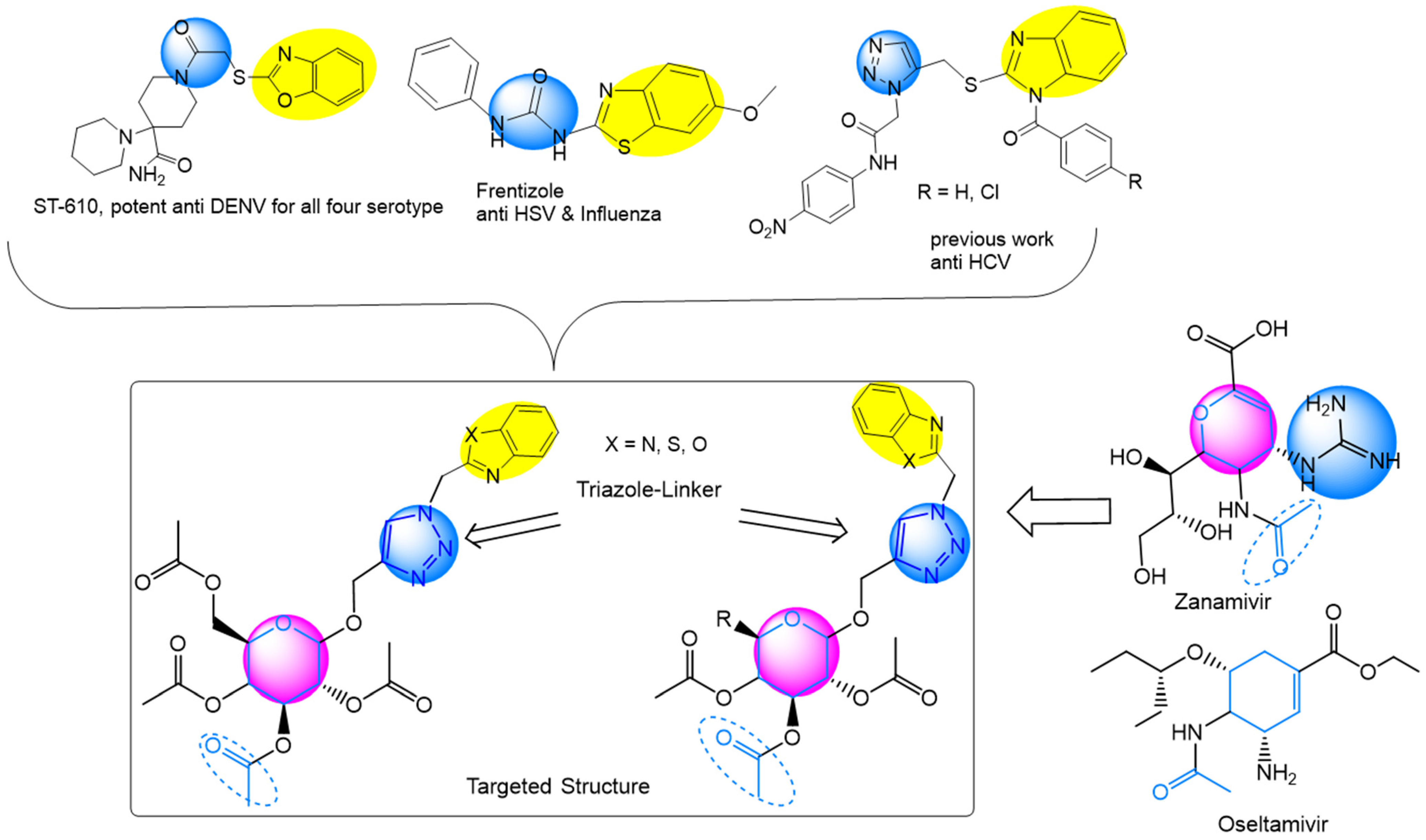
:1. Introduction
2. Results
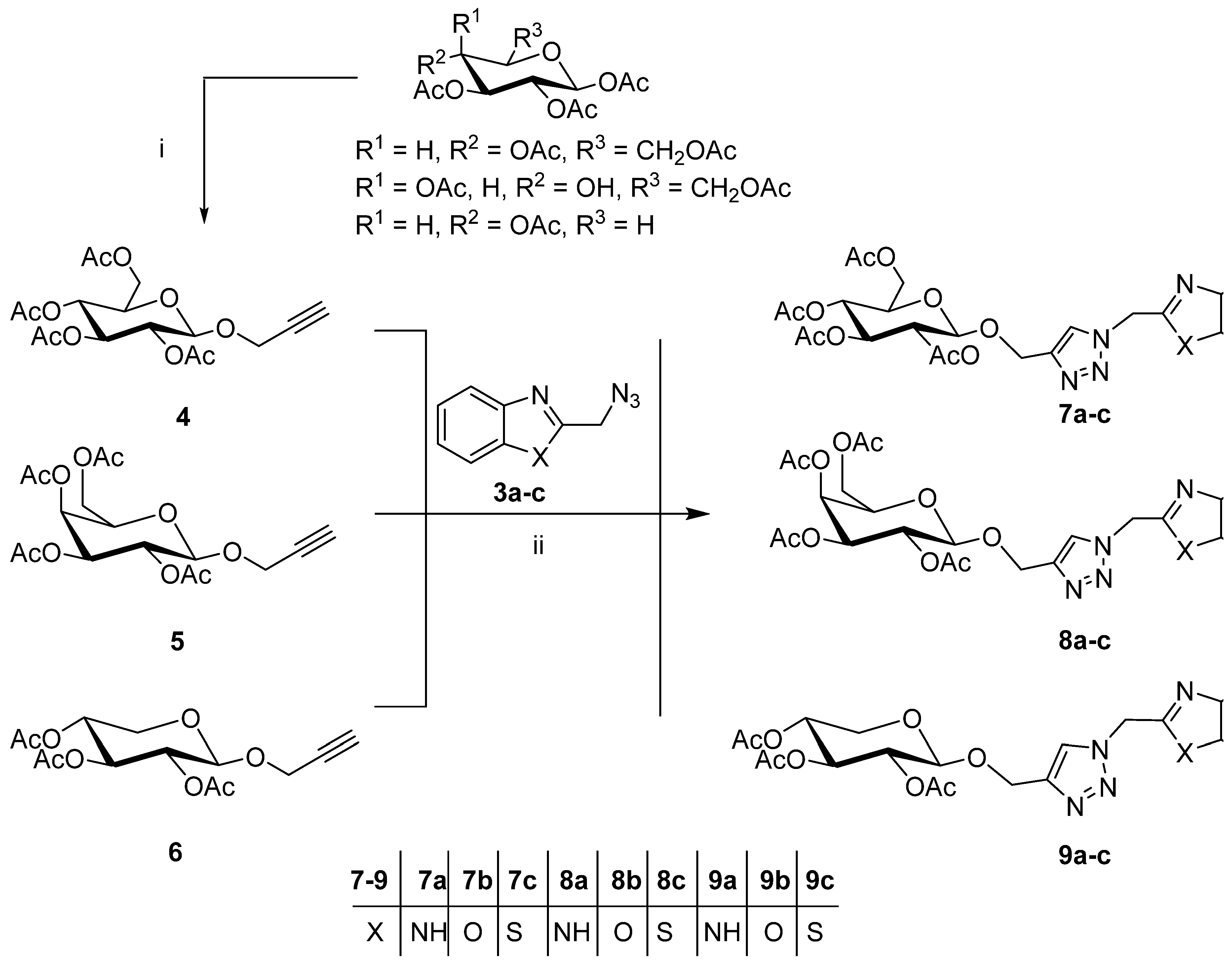
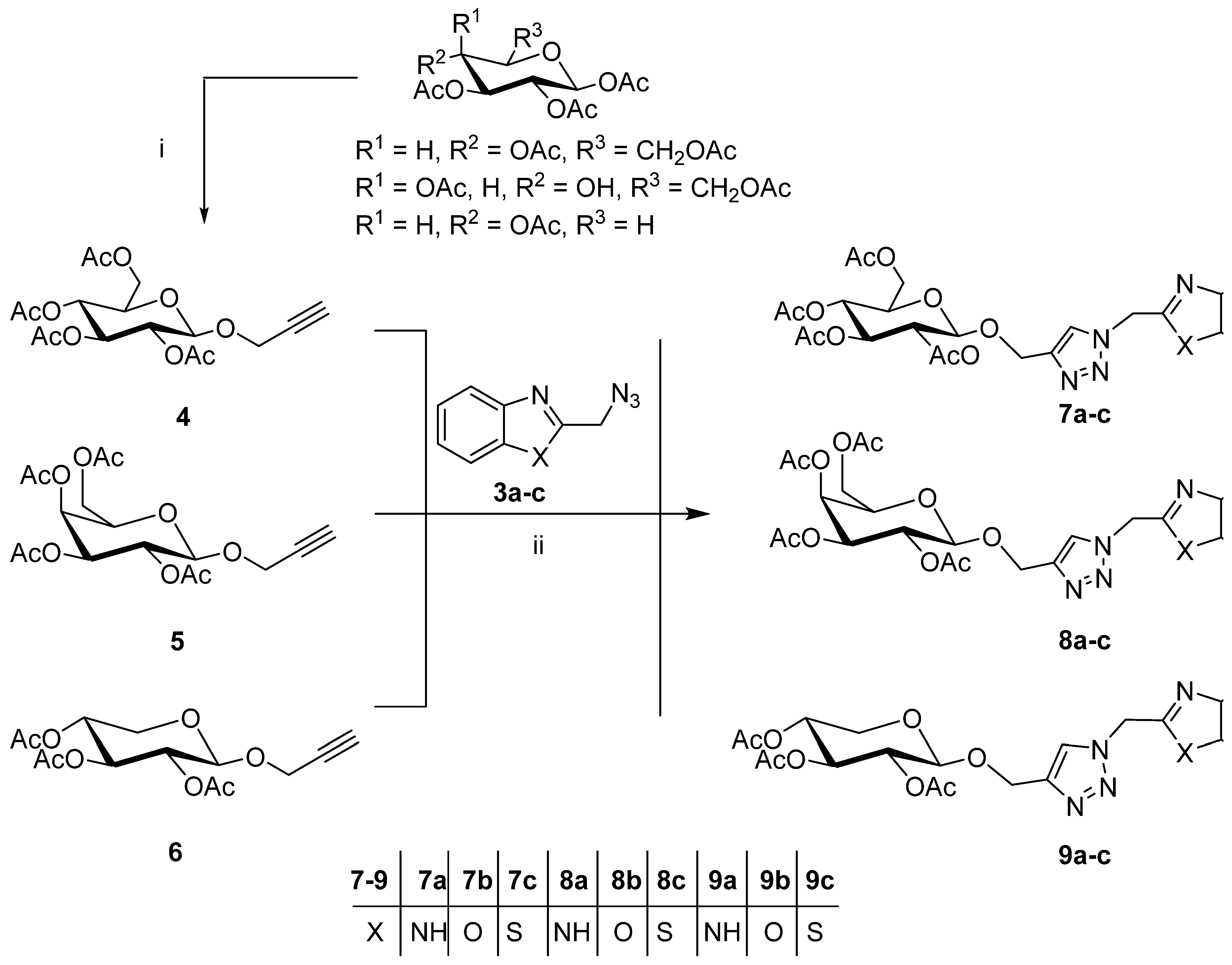
2.1. Synthesis of the Designed Compounds
2.2. Antiviral Efficacy of Synthetic Compounds
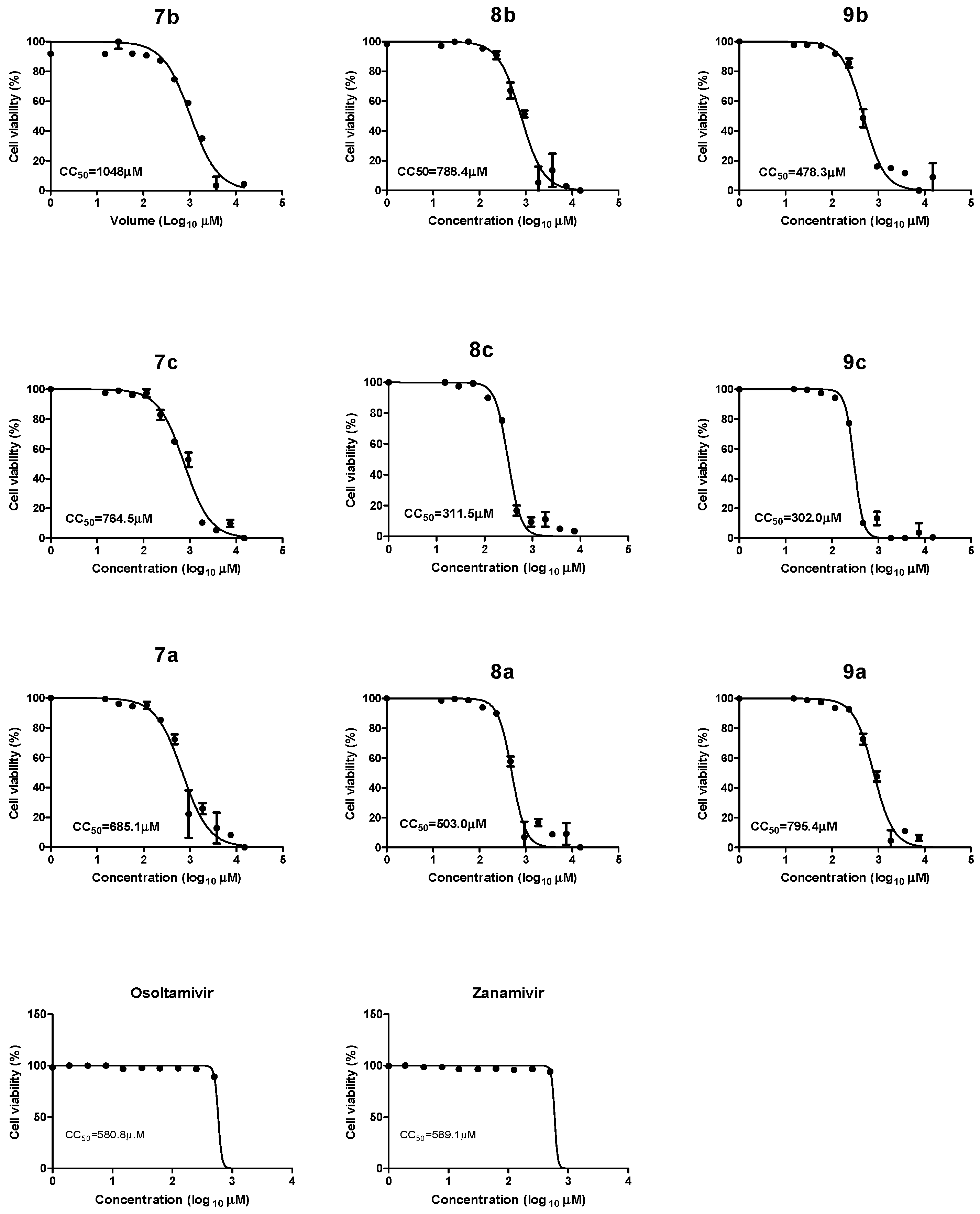
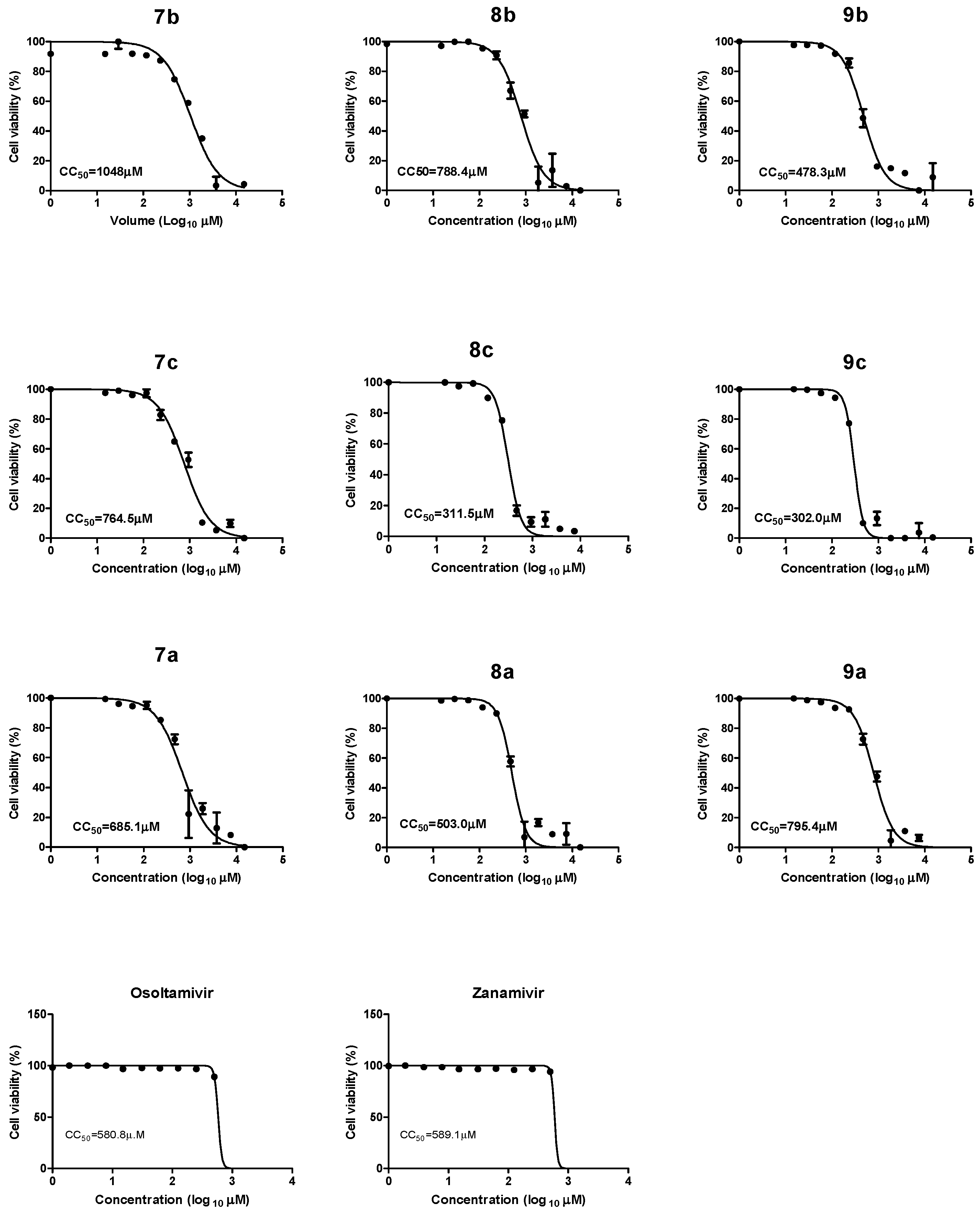
2.2.1. Cytotoxicity of Synthetic Compounds, Oseltamivir, and Zanamivir
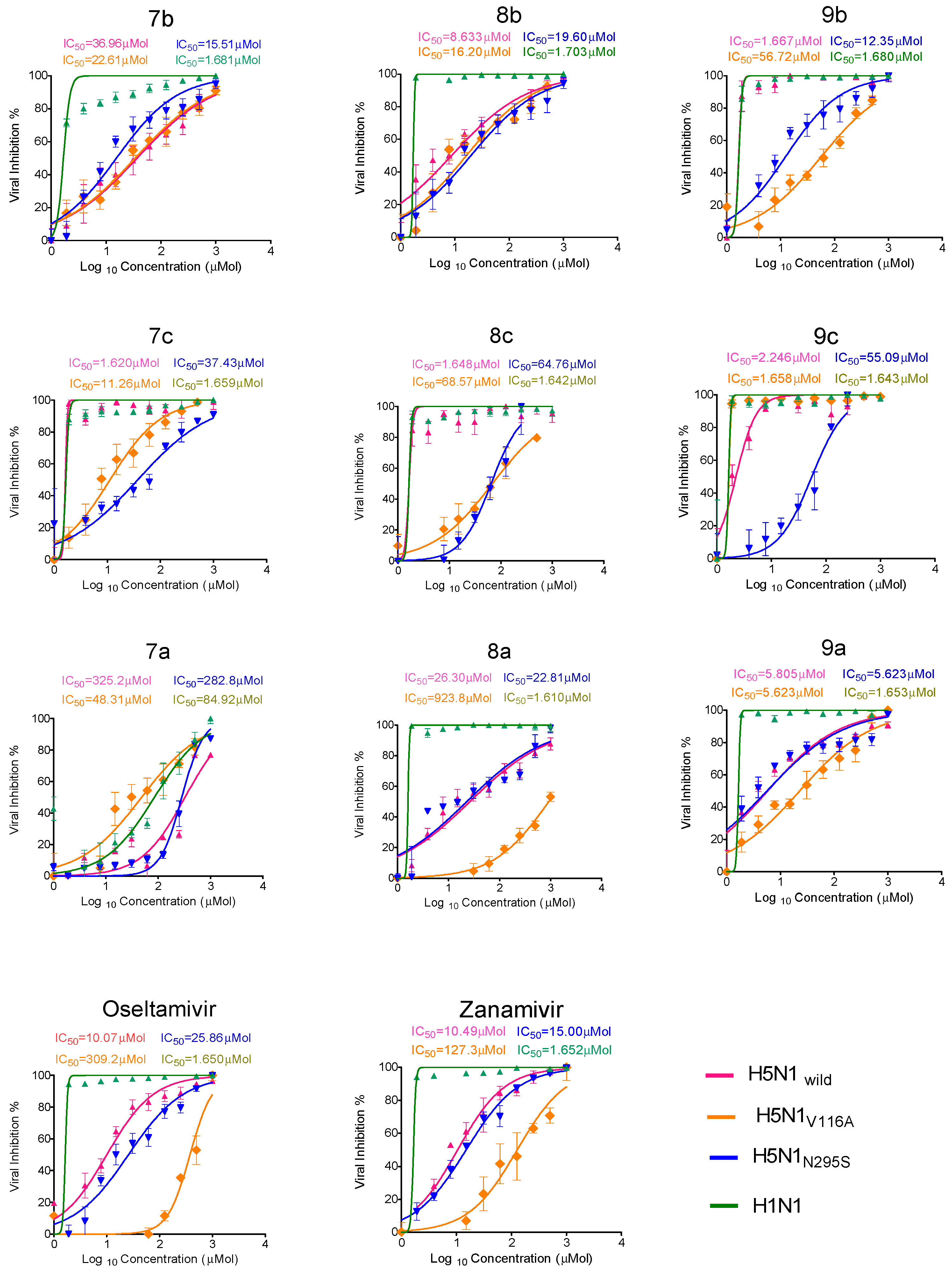
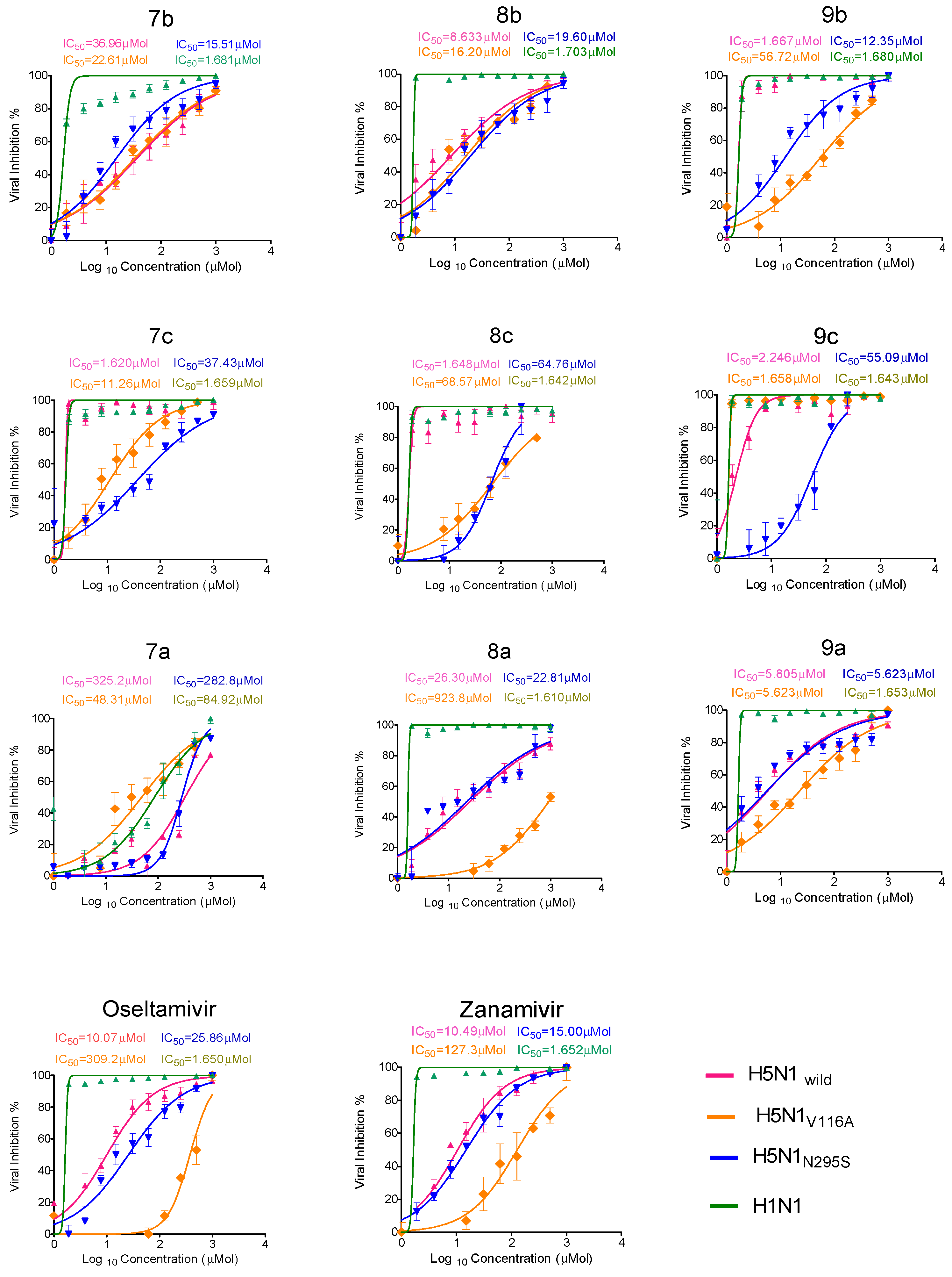
2.2.2. Antiviral Properties
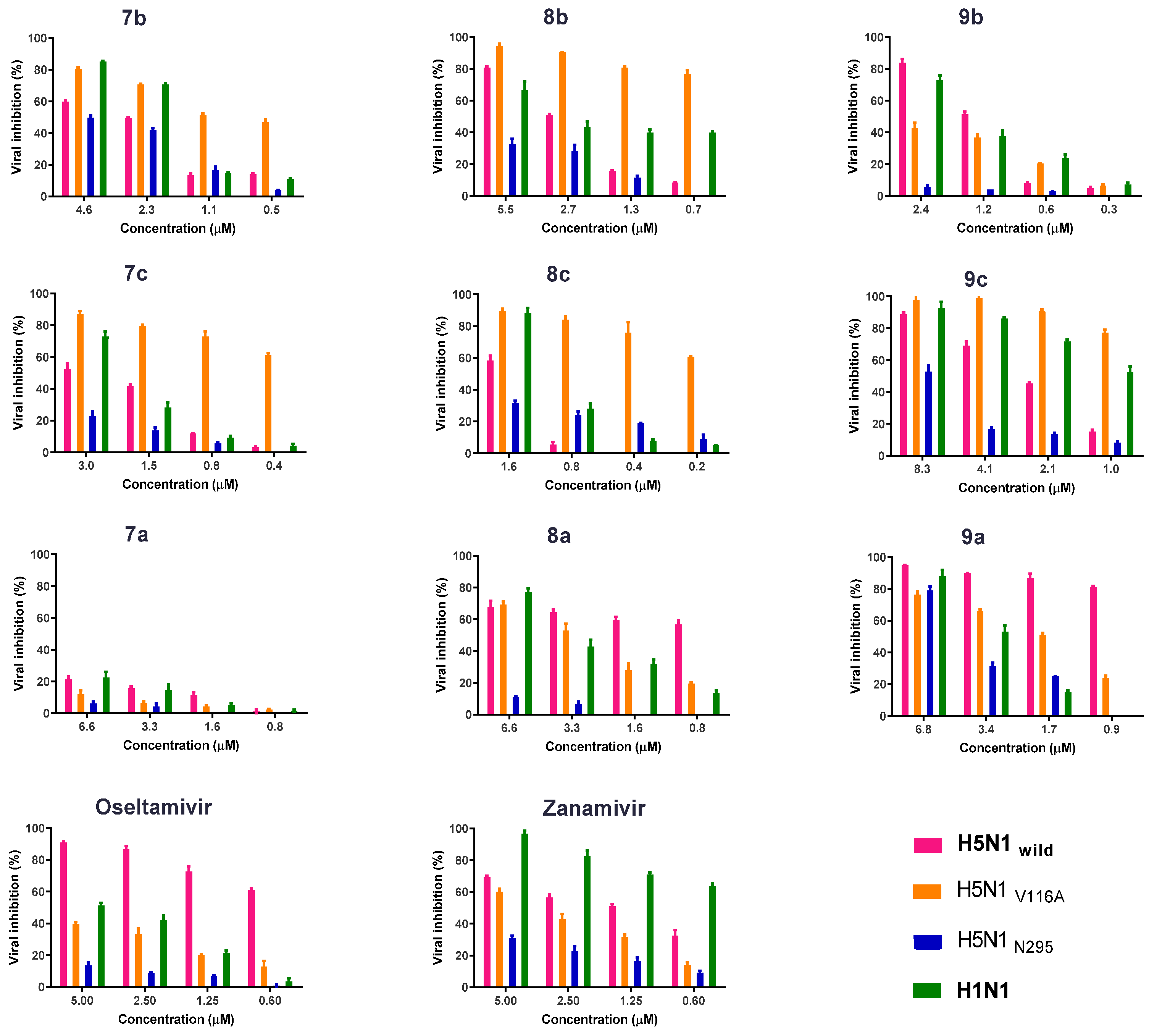
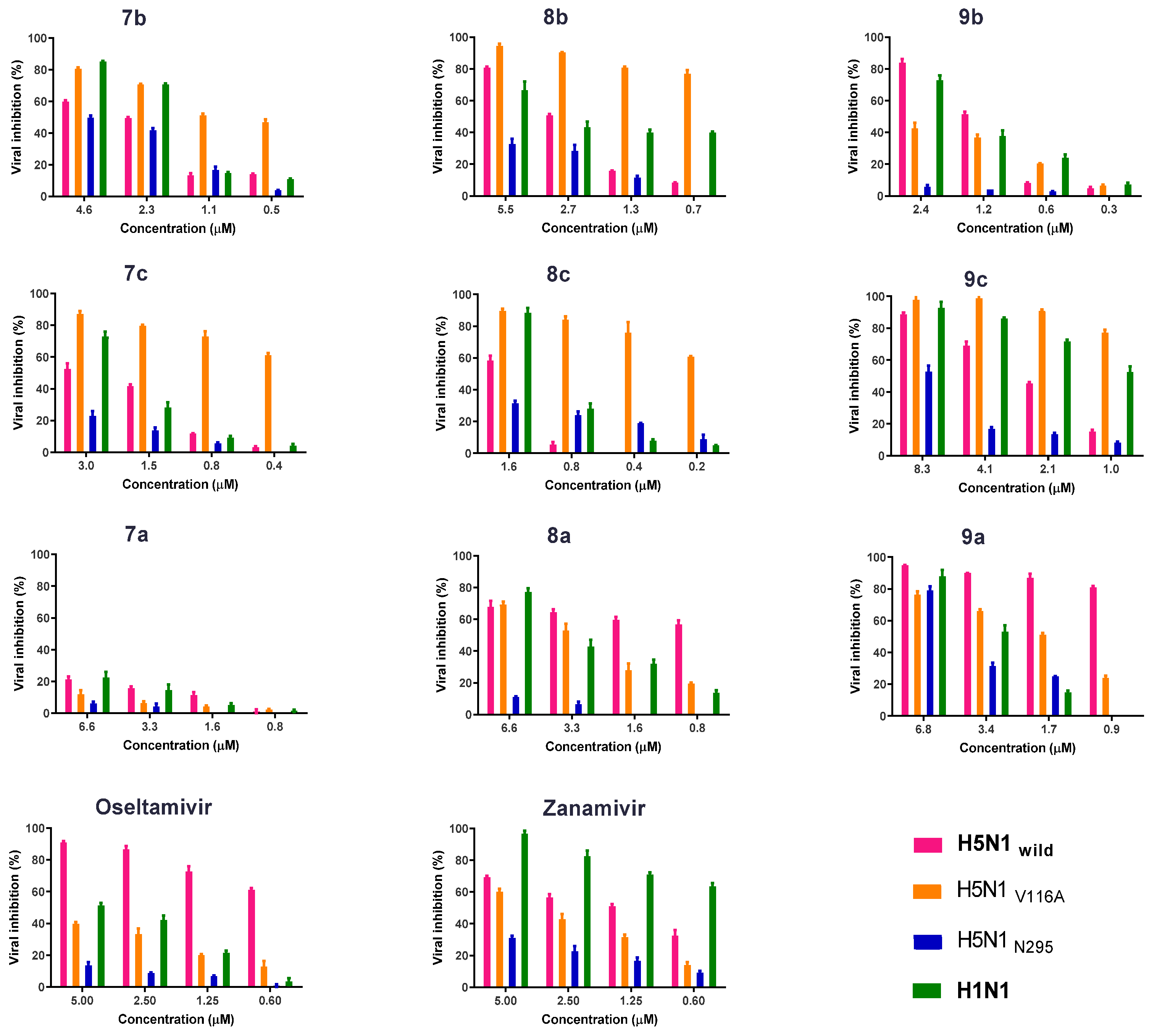
2.2.3. Antiviral Properties According to the Plaque Reduction Assay
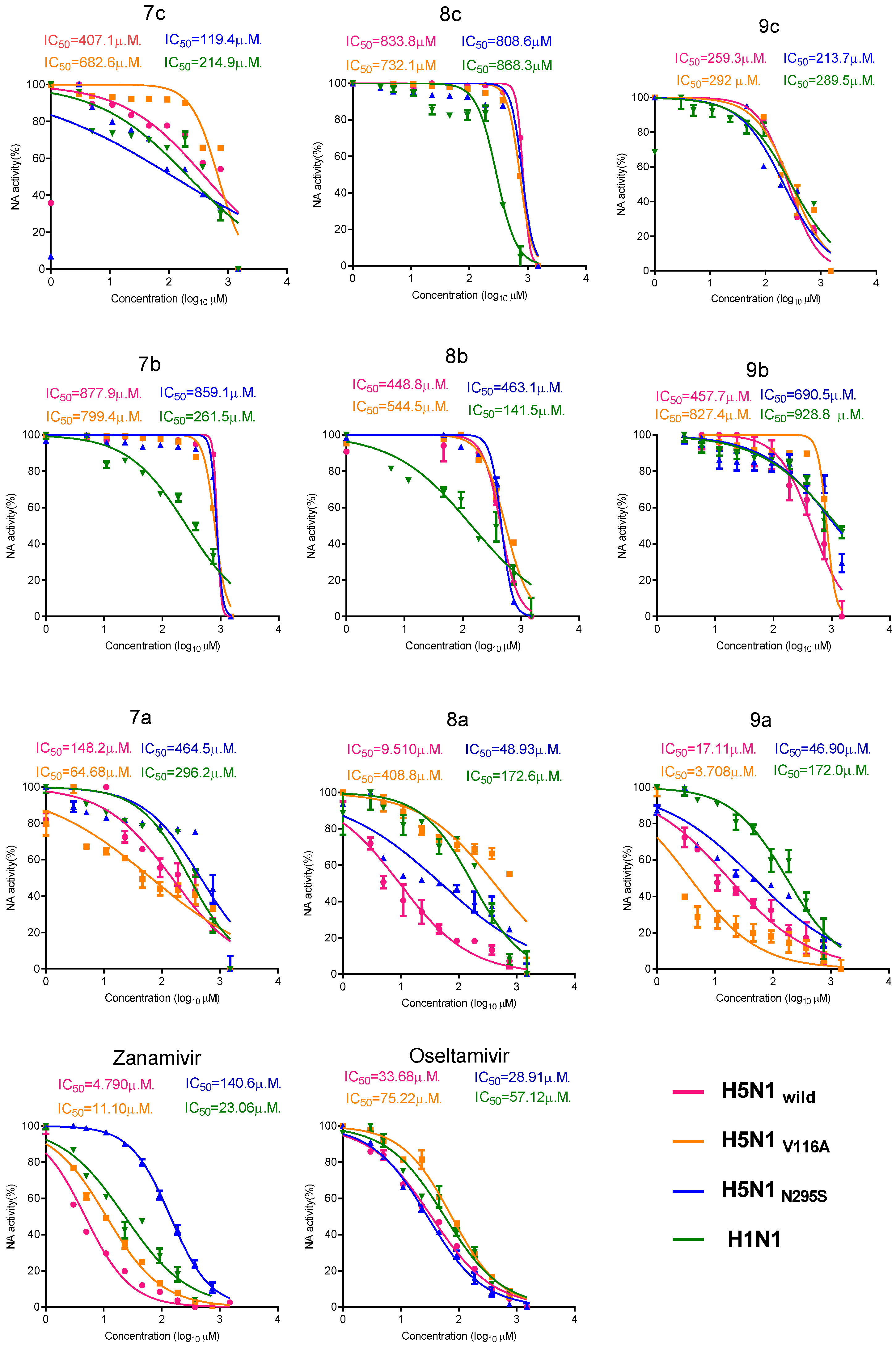
2.2.4. Colorimetric NA Inhibition Assay
2.2.5. Biological Susceptibility Testing of the Developed Compounds on the Propagation of Viruses with MOI 0.005 by HA Titration
2.2.6. In Vivo Study Safety of the Synthetic Compounds
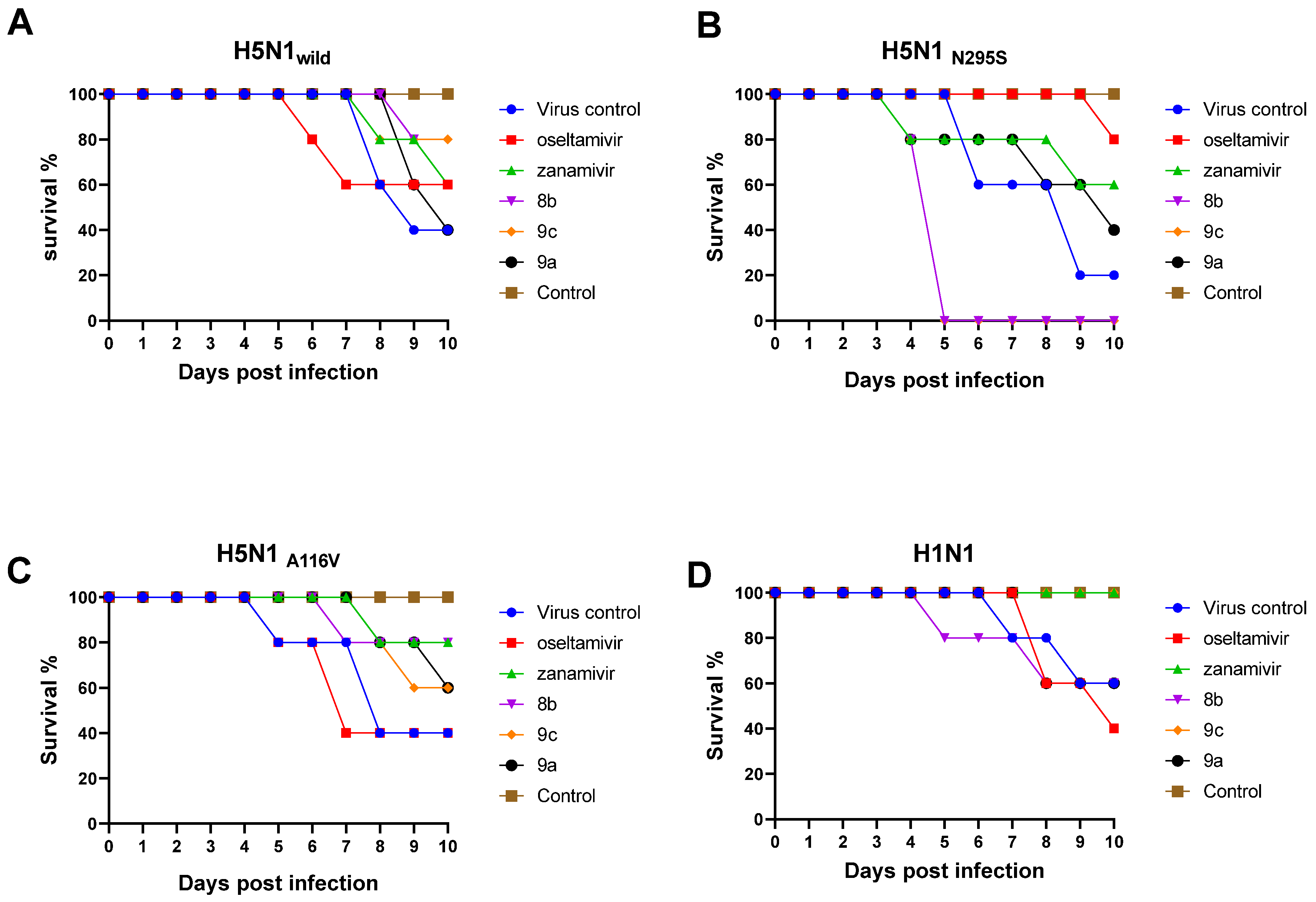
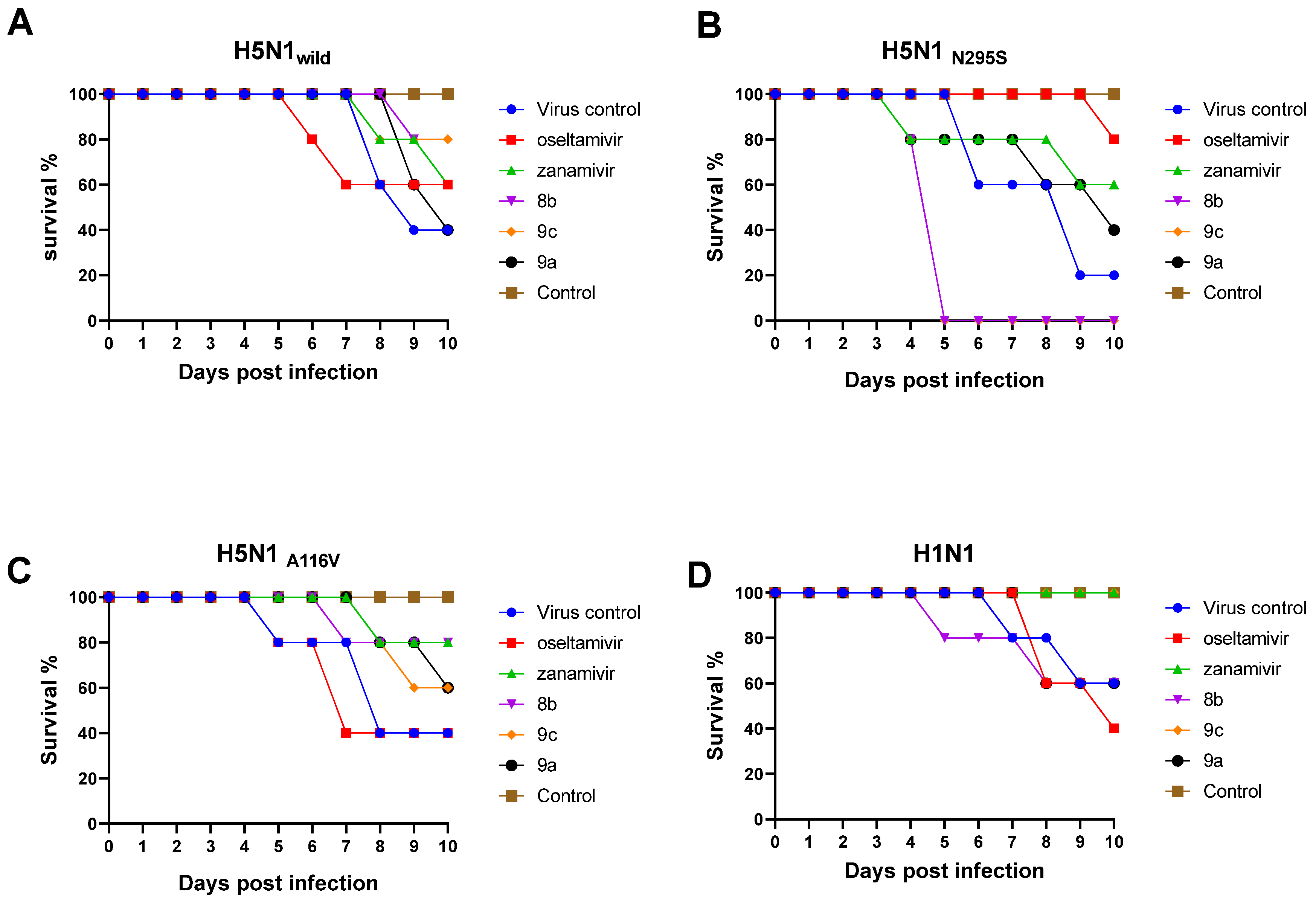
2.2.7. Antiviral Efficacy of the New Synthetic Compounds in Mice
Mortality and Body Weight Loss of the Infected Mice
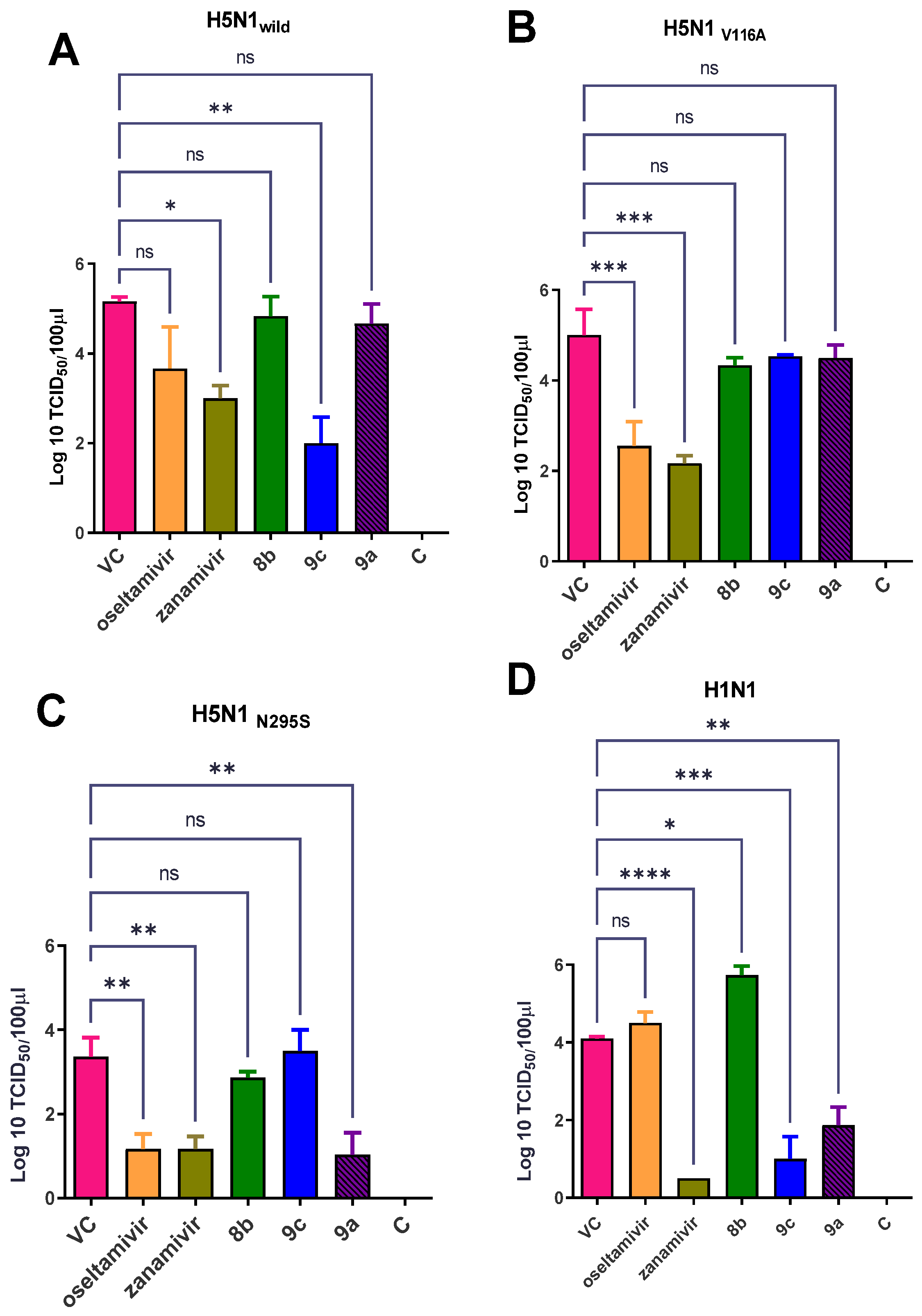
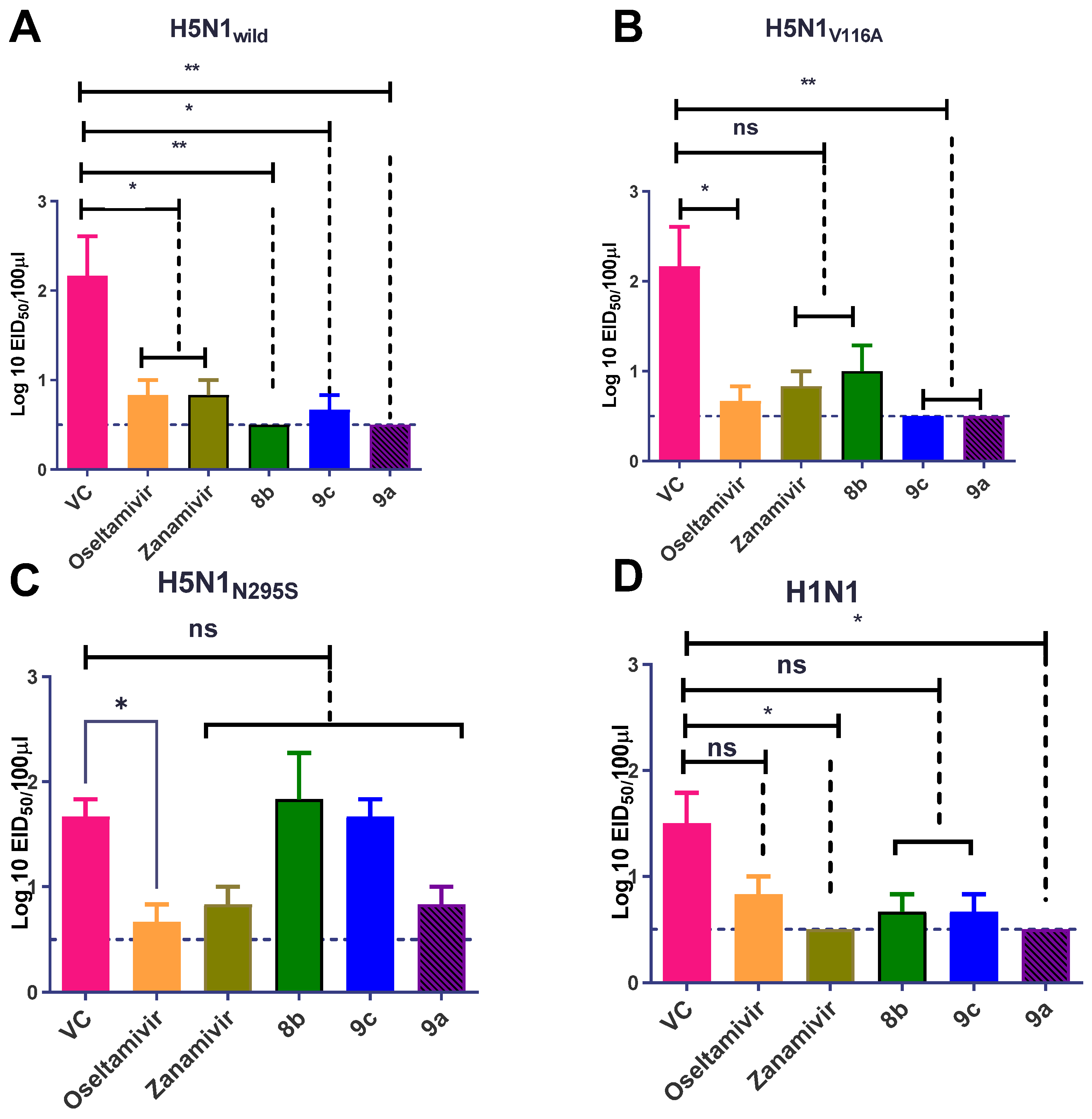
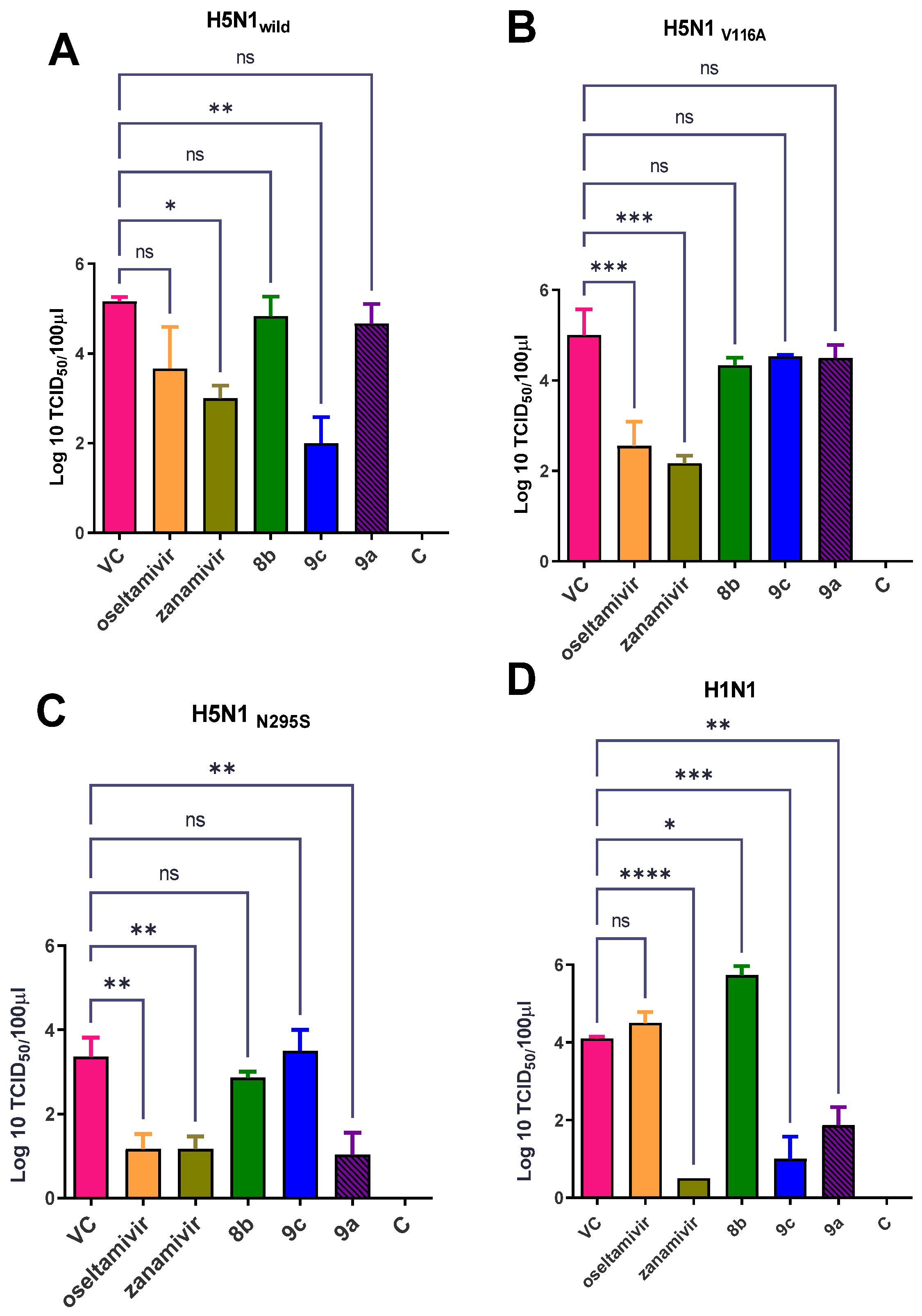
Effect of Synthetic Compounds on a Viral Titer in a Lung and Nasal Wash
2.3. Chemoinformatic Analysis
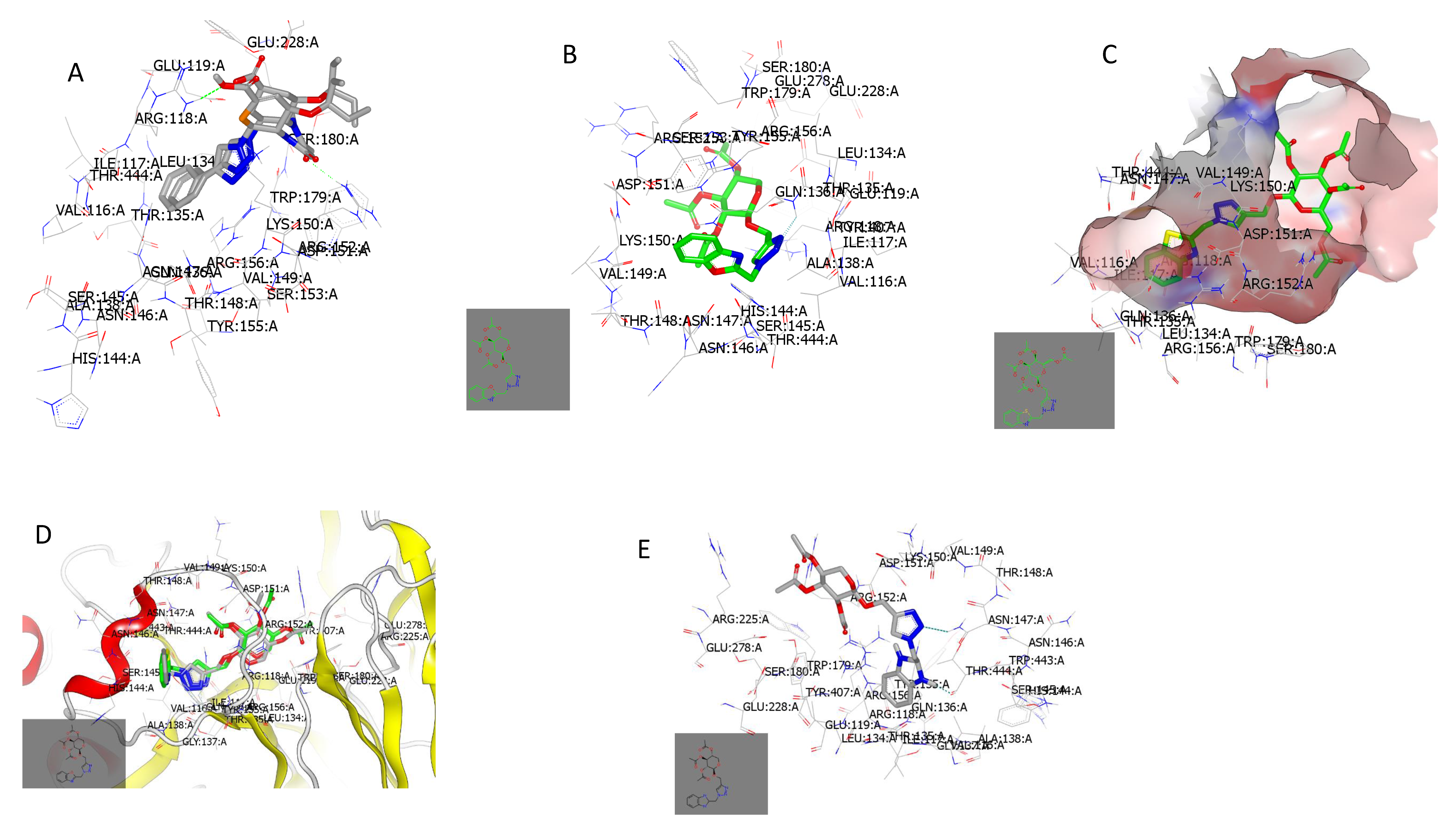
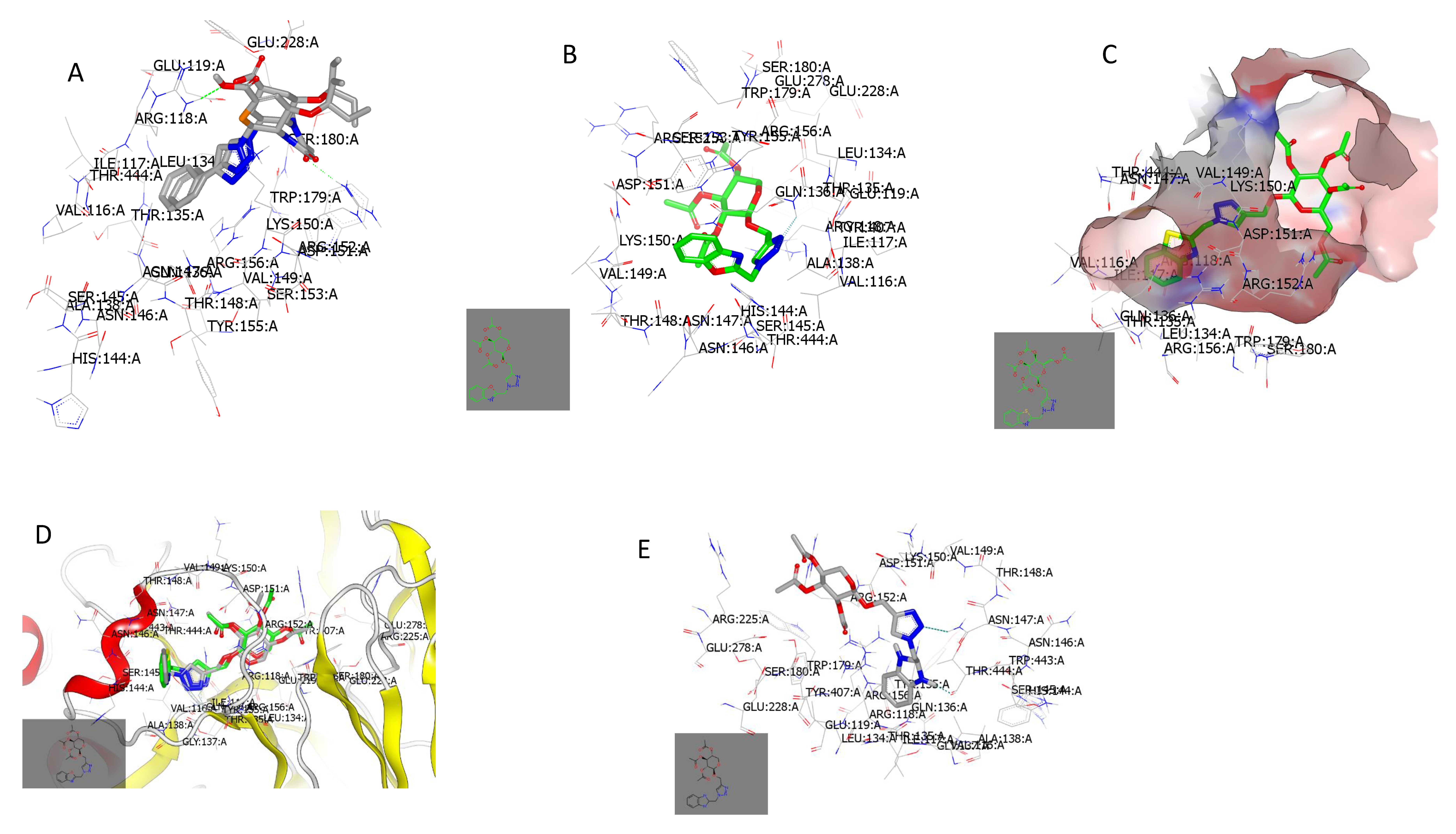
2.3.1. Docking with Neuraminidase PDB ID: 7E6Q
2.3.2. Ligand Efficiency and Lipophilic Efficiency Profile
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. 2-Propynyl 2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosides (4–6)
4.1.2. Bromo-tris-(triphenylphosphine) Copper (I) (CuL3Br)
4.1.3. General Procedure for the Cu(I)-Catalyzed Click Dipolar Coupling of Benzohetero-Azides and Terminal Acetylenic Pyranosyl Sugars
4.1.4. (2R,3R,4S,5R,6R)-2-((1-((1H-Benzo[d]imidazol-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (7a)
4.1.5. (2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-((1-(benzo[d]oxazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (7b)
4.1.6. (2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-((1-(benzo[d]thiazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (7c)
4.1.7. (2R,3R,4S,5S,6R)-2-((1-((1H-Benzo[d]imidazol-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (8a)
4.1.8. (2R,3S,4S,5R,6R)-2-(Acetoxymethyl)-6-((1-(benzo[d]oxazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (8b)
4.1.9. (2R,3S,4S,5R,6R)-2-(Acetoxymethyl)-6-((1-(benzo[d]thiazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (8c)
4.1.10. (2R,3R,4S,5R,)-2-((1-((1H-Benzo[d]imidazol-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (9a)
4.1.11. (2R,3R,4S,5R)-2-((1-(Benzo[d]oxazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (9b)
4.1.12. (2R,3R,4S,5R)-2-((1-(Benzo[d]thiazol-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (9c)
4.2. Cells, Wild-Type Viruses, and Antiviral Compounds
4.3. Generation of NAI-Resistant H5N1 Viruses
4.4. MTT Assay
4.5. Crystal Violet Assay
4.6. Plaque Reduction Assay
4.7. Colorimetric NA Inhibition Assay
4.8. Biological Susceptibility Testing of the Developed Compounds
4.9. Animal Experiments: In Vivo Study of the Synthetic Compounds’ Safety
4.10. Chemoinformatic Studies
4.10.1. Molecular Docking
4.10.2. Physiochemical Parameter and Lipophilicity Calculations
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abed, Y.; Baz, M.; Boivin, G. Impact of neuraminidase mutations conferring influenza resistance to neuraminidase inhibitors in the N1 and N2 genetic backgrounds. Antivir. Ther. 2006, 11, 971–976. [Google Scholar]
- Prachanronarong, K.L.; Canale, A.S.; Liu, P.; Somasundaran, M.; Hou, S.; Poh, Y.-P.; Han, T.; Zhu, Q.; Renzette, N.; Zeldovich, K.B.; et al. Mutations in Influenza A Virus Neuraminidase and Hemagglutinin Confer Resistance against a Broadly Neutralizing Hemagglutinin Stem Antibody. J. Virol. 2019, 93, e01639-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alame, M.M.; Massaad, E.; Zaraket, H. Peramivir: A Novel Intravenous Neuraminidase Inhibitor for Treatment of Acute Influenza Infections. Front. Microbiol. 2016, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Chou, J.J. Influenza M2 proton channels. Biochim. Biophys. Acta 2011, 1808, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef] [PubMed]
- Abdelwhab, E.-S.; Veits, J.; Mettenleiter, T. Biological fitness and natural selection of amantadine resistant variants of avian influenza H5N1 viruses. Virus Res. 2016, 228, 109–113. [Google Scholar] [CrossRef]
- Gubareva, L.; Mohan, T. Antivirals Targeting the Neuraminidase. Cold Spring Harb. Perspect. Med. 2022, 12, a038455. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Fry, A.M.; Gubareva, L.V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 2012, 17, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Yen, H.-L.; Hoffmann, E.; Taylor, G.; Scholtissek, C.; Monto, A.S.; Webster, R.G.; Govorkova, E.A. Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J. Virol. 2006, 80, 8787–8795. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Pandey, A.; Manvati, S. Coumarin: An emerging antiviral agent. Heliyon 2020, 6, e03217. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Kumar, I.; Rana, S.; Cho, J.W.; Rode, C.V. Synthesis of hybrid 1,2,3-triazolo-δ-lactams/lactones using Huisgen [3+2] cycloaddition ‘click-chemistry’ in water. Tetrahedron Asymmetry 2010, 21, 352–355. [Google Scholar] [CrossRef]
- Genin, M.J.; Allwine, D.A.; Anderson, D.J.; Barbachyn, M.R.; Emmert, D.E.; Garmon, S.A.; Graber, D.R.; Grega, K.C.; Hester, J.B.; Hutchinson, D.K.; et al. Substituent effects on the antibacterial activity of nitrogen-carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms Haemophilus influenzae and Moraxella catarrhalis. J. Med. Chem. 2000, 43, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Kolb, H.C.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zheng, M.; Tang, W.; He, P.-L.; Zhu, W.; Li, T.; Zuo, J.-P.; Liu, H.; Jiang, H. Syntheses of triazole-modified zanamivir analogues via click chemistry and anti-AIV activities. Bioorgan. Med. Chem. Lett. 2006, 16, 5009–5013. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. C-Glycoside Clustering on Calix[4]arene, Adamantane, and Benzene Scaffolds through 1,2,3-Triazole Linkers. J. Org. Chem. 2006, 71, 7546–7557. [Google Scholar] [CrossRef]
- Hotha, S.; Kashyap, S. “Click Chemistry” Inspired Synthesis of pseudo-Oligosaccharides and Amino Acid Glycoconjugates. J. Org. Chem. 2006, 71, 364–367. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K.; Smith, G.F. Heterocyclic Chemistry, 3rd ed.; Chapman and Hall: London, UK, 1995; pp. 72–119. [Google Scholar]
- Roth, H.J.; Kleemann, A.; Beisswenger, T.; Cooke, M.D.; Sammes, P.G. (Eds.) Pharmaceutical Chemistry, Drug Synthesis; Prentice Hall Europe: London, UK, 1988; Volume 1, p. 407. [Google Scholar]
- Gudmundsson, K.S.; Freeman, G.A.; Drach, J.C.; Townsend, L.B. Synthesis of Fluorosugar Analogues of 2,5,6-Trichloro-1-(β-d-ribofuranosyl)benzimidazole as Antivirals with Potentially Increased Glycosidic Bond Stability. J. Med. Chem. 2000, 43, 2473–2478. [Google Scholar] [CrossRef]
- Zacny, V.L.; Gershburg, E.; Davis, M.G.; Biron, K.K.; Pagano, J.S. Inhibition of Epstein-Barr Virus Replication by a Benzimidazole l-Riboside: Novel Antiviral Mechanism of 5,6-Dichloro-2-(Isopropylamino)-1-β-l-Ribofuranosyl-1H-Benzimidazole. J. Virol. 1999, 73, 7271–7277. [Google Scholar] [CrossRef] [Green Version]
- Underwood, M.R.; Harvey, R.J.; Stanat, S.C.; Hemphill, M.L.; Miller, T.; Drach, J.C.; Townsend, L.B.; Biron, K.K. Inhibition of Human Cytomegalovirus DNA Maturation by a Benzimidazole Ribonucleoside Is Mediated through the UL89 Gene Product. J. Virol. 1998, 72, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Takashima, H.; Ubasawa, M.; Sekiya, K.; Inouye, N.; Baba, M.; Shigeta, S.; Walker, R.T.; De Clercq, E.; Miyasaka, T. Synthesis and Antiviral Activity of 6-Benzyl Analogs of 1-[(2-Hydroxyethoxy)methyl]-5-(phenylthio)thymine (HEPT) as Potent and Selective Anti-HIV-1 Agents. J. Med. Chem. 1995, 38, 2860–2865. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Adachi, S.; Chao, Q.; Rose, D.W.; Carrera, C.J.; Cottam, H.B.; Carson, D.A.; Leoni, L.M. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. J. Am. Soc. Hematol. 2000, 96, 3537–3543. [Google Scholar]
- Ramiz, M.M.; El-Sayed, W.A.; Hagag, E.; Abdel-Rahman, A.A.H. Synthesis and antiviral activity of new substituted pyrimidine glycosides. J. Heterocycl. Chem. 2011, 48, 1028–1038. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Rashad, A.E.; Awad, S.M.; Ali, M.M. Synthesis and in vitro antitumor activity of new substituted thiopyrimidine acyclic nucleosides and their thioglycoside analogs. Nucleosides Nucleotides Nucleic Acids 2009, 28, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, A.A.-H.; Zeid, I.F.; Barakat, H.A.; El-Sayed, W.A. Anti-hepatitis B virus activity of new substituted pyrimidine acyclic nucleoside analogues. Z. Nat. C J. Biosci. 2009, 64, 767–772. [Google Scholar] [CrossRef]
- Byrd, C.M.; Dai, D.; Grosenbach, D.W.; Berhanu, A.; Jones, K.F.; Cardwell, K.B.; Schneider, C.; Wineinger, K.A.; Page, J.M.; Harver, C.; et al. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob. Agents Chemother. 2013, 57, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, C.M.; Grosenbach, D.W.; Berhanu, A.; Dai, D.; Jones, K.F.; Cardwell, K.B.; Schneider, C.; Yang, G.; Tyavanagimatt, S.; Harver, C.; et al. Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob. Agents Chemother. 2013, 57, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Asiri, Y.I.; Alsayari, A.; Muhsinah, A.B.; Mabkhot, Y.N.; Hassan, M.Z. Benzothiazoles as potential antiviral agents. J. Pharm. Pharmacol. 2020, 72, 1459–1480. [Google Scholar] [CrossRef]
- Kanwal, A.; Ahmad, M.; Aslam, S.; Naqvi, S.A.R.; Saif, M.J. Recent Advances in Antiviral Benzimidazole Derivatives: A Mini Review. Pharm. Chem. J. 2019, 53, 179–187. [Google Scholar] [CrossRef]
- Hurt, A.C.; Deng, Y.M.; Ernest, J.; Caldwell, N.; Leang, L.; Iannello, P.; Komadina, N.; Shaw, R.; Smith, D.; Dwyer, D.E.; et al. Oseltamivir-resistant influenza viruses circulating during the first year of the influenza A(H1N1) 2009 pandemic in the Asia-Pacific region, March 2009 to March 2010. Eurosurveillance 2011, 16, 19770. [Google Scholar] [CrossRef]
- Srivastava, S.; Pandeya, S.N.; Yadav, M.K.; Singh, B.K. Synthesis and Analgesic Activity of Novel Derivatives of 1,2-Substituted Benzimidazoles. J. Chem. 2013, 2013, 694295. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.-B.; Zhang, A.-L.; Tang, J.-J.; Gao, J.-M. Synthesis and Antifungal Activity of 2-Chloromethyl-1H-benzimidazole Derivatives against Phytopathogenic Fungi In Vitro. J. Agric. Food Chem. 2013, 61, 2789–2795. [Google Scholar] [CrossRef] [PubMed]
- Peprah, K.; Zhu, X.Y.; Eyunni, S.V.K.; Etukala, J.R.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Structure–activity relationship studies of SYA 013, a homopiperazine analog of haloperidol. Bioorgan. Med. Chem. 2012, 20, 1671–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Li, Z.; Fang, Q.; Feng, C.; Zhang, H.; Guo, W.; Wang, H.; Gu, G.; Tian, Y.; Liu, P.; et al. Discovery and Extensive in Vitro Evaluations of NK-HDAC-1: A Chiral Histone Deacetylase Inhibitor as a Promising Lead. J. Med. Chem. 2012, 55, 3066–3075. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, V.; King, M.; Chaumontet, M.; Nothisen, M.; Gabillet, S.; Buisson, D.; Puente, C.; Wagner, A.; Taran, F. Copper-chelating azides for efficient click conjugation reactions in complex media. Angew. Chem. 2014, 126, 5982–5986. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harbor Protocols 2016, 2016. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef]
- Mereyala, H.B.D.; Gurrala, S.R. A highly diastereoselective, practical synthesis of allyl, propargyl 2,3,4,6-tetra-O-acetyl-²-d-gluco,#2-d-galactopyranosides and allyl, propargyl heptaacetyl02-d-lactosides. Carbohydr. Res. 1998, 307, 351–354. [Google Scholar]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.C.; Selnick, H.; Wolanski, B.; Tomassini, J.E. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996, 40, 1304–1307. [Google Scholar] [CrossRef] [Green Version]
- Tumpey, T.M.; García-Sastre, A.; Mikulasova, A.; Taubenberger, J.K.; Swayne, D.E.; Palese, P.; Basler, C.F. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA 2002, 99, 13849–13854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarbet, E.B.; Vollmer, A.H.; Hurst, B.L.; Barnard, D.L.; Furuta, Y.; Smee, D.F. In vitro activity of favipiravir and neuraminidase inhibitor combinations against oseltamivir-sensitive and oseltamivir-resistant pandemic influenza A (H1N1) virus. Arch. Virol. 2014, 159, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
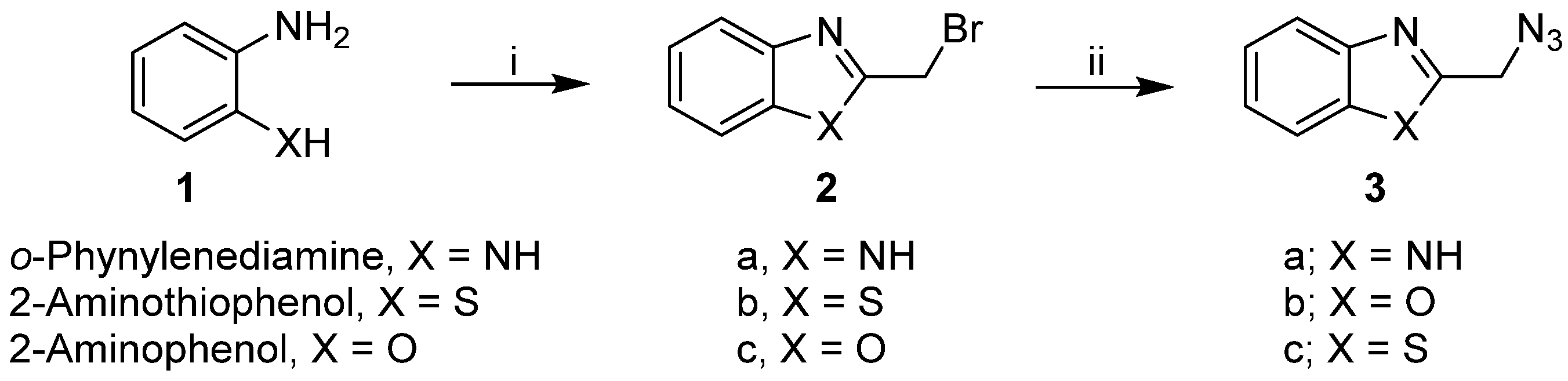

{kind=link}
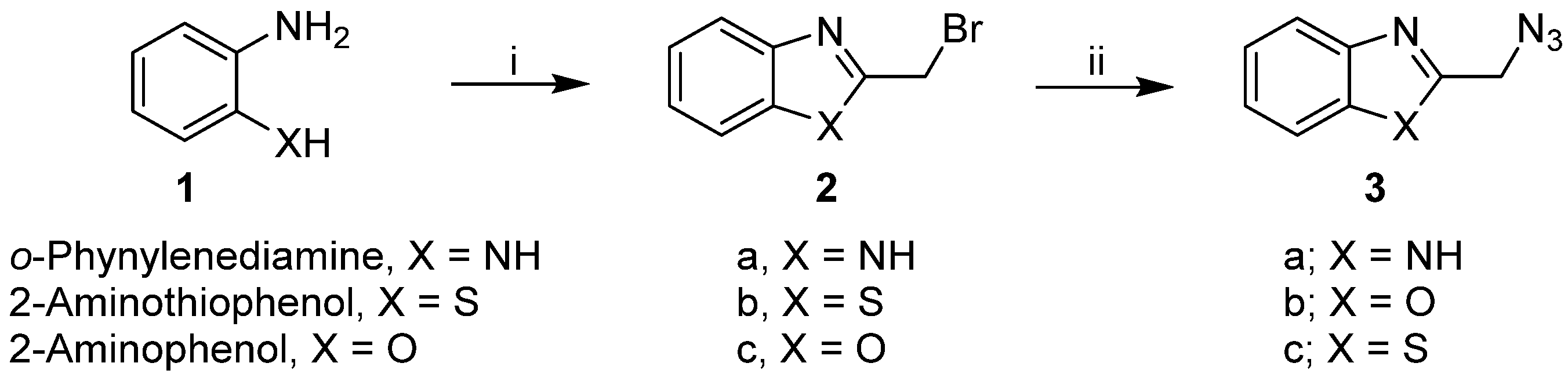{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | CC50 (µM) | IC50 (50% Inhibitory Concentration) (µM) | SI (Selectivity Index) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H5N1wild | H5N1V116A | H5N1N295S | H1N1 | H5N1wild | H5N1V116A | H5N1N295S | H1N1 | ||
| 7b | 1048 | 36.96 | 22.6 | 15.51 | 1.68 | 28.3 | 46.3 | 67.5 | 623.8 |
| 8b | 788.4 | 8.633 | 16.2 | 19.6 | 1.7 | 91.3 | 48.6 | 40.2 | 463.7 |
| 9b | 478.3 | 1.667 | 56.7 | 12.3 | 1.68 | 286.4 | 8.4 | 38.8 | 284.7 |
| 7c | 764.5 | 1.62 | 11.26 | 37.4 | 1.65 | 472.5 | 67.9 | 20.4 | 463.9 |
| 8c | 311.5 | 1.648 | 68.57 | 64.7 | 1.64 | 189 | 4.5 | 4.8 | 189.9 |
| 9c | 302.0 | 2.246 | 1.65 | 55.09 | 1.64 | 134.4 | 183 | 5.4 | 184.1 |
| 7a | 685.1 | 325 | 48.31 | 282.8 | 84.9 | 2.1 | 14.1 | 2.4 | 8 |
| 8a | 503 | 26.3 | 923.8 | 22.81 | 1.61 | 19.1 | 0.54 | 22.0 | 312.4 |
| 9a | 795.4 | 5.805 | 5.623 | 5.623 | 1.65 | 137 | 141.5 | 141.45 | 482 |
| zanamivir | 589.1 | 10.4 | 127.3 | 15.00 | 1.652 | 56.6 | 4.6 | 39.2 | 356.59 |
| oseltamivir | 580.8 | 10.1 | 309.2 | 25.8 | 1.650 | 57.5 | 1.878 | 22.5 | 352 |
| Type of Analysis | ALT | AST | Creatinine | Urea | |
|---|---|---|---|---|---|
| Compound Code | Control | 6.5 ± 0.5 | 12.83 ± 1.81 | 0.30 ± 0.15 | 5.64 ± 2.17 |
| 8b | 1st # | 10.66 ± 4.32 | 15 ± 3.41 | 0.17 ± 0.04 | 9.00 ± 3.81 |
| 2nd | 8.20 ± 2.62 | 15.77 ± 0.9944 | 0.13 ± 0.04 | 6.66 ± 1.54 | |
| 3rd | 6.72 ± 1.47 | 16 ± 5.099 | 0.10 ± 0.02 | 7.32 ± 3.90 | |
| 9a | 1st | 9.64 ± 3.64 | 20 ± 6.957 | 0.13 ± 0.04 | 7.32 ± 2.09 |
| 2nd | 10.64 ± 1.38 * | 13.77 ± 1.298 | 0.13 ± 0.04 | 7.32 ± 0.48 | |
| p * = 0.0085 | |||||
| 3rd | 8.67 ± 2.16 | 15.33 ± 0.9686 | 0.17 ± 0.04 | 4.64 ± 1.12 | |
| 9c | 1st | 7.60 ± 1.56 | 11.93 ± 3.765 | 0.14 ± 0.05 | 7.90 ± 3.13 |
| 2nd | 7.80 ± 2.07 | 9.689 ± 2.922 | 0.14 ± 0.05 | 3.64 ± 1.49 | |
| p * = 0.0395 | |||||
| 3rd | 5.54 ± 1.22 | 12.33 ± 3.169 | 0.13 ± 0.04 | 4.88 ± 2.13 | |
| Compound | M.wt | Molecular Formula | NHA | cLogP | IC50 µM | pIC50 | LE | LLE |
|---|---|---|---|---|---|---|---|---|
| 8b | 560.52 | C25H28N4O11 | 40 | 1.73 | 6.863 | 8.16 | 0.28 | 6.43 |
| 9b | 488.45 | C22H24N4O9 | 35 | 1.67 | 1.679 | 8.77 | 0.34 | 7.10 |
| 8c | 576.58 | C25H28N4O10S | 40 | 1.86 | 1.790 | 8.74 | 0.30 | 6.88 |
| 9c | 504.51 | C22H24N4O8S | 35 | 1.80 | 2.280 | 8.64 | 0.33 | 6.84 |
| 9a | 487.47 | C22H25N5O8 | 35 | 1.24 | 2.75 | 8.56 | 0.34 | 7.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutkat, O.; Kandeil, A.; Moatasim, Y.; Elshaier, Y.A.M.M.; El-Sayed, W.A.; Gaballah, S.T.; El Taweel, A.; Kamel, M.N.; El Sayes, M.; Ramadan, M.A.; et al. In Vitro and In Vivo Antiviral Studies of New Heteroannulated 1,2,3-Triazole Glycosides Targeting the Neuraminidase of Influenza A Viruses. Pharmaceuticals 2022, 15, 351. https://doi.org/10.3390/ph15030351
Kutkat O, Kandeil A, Moatasim Y, Elshaier YAMM, El-Sayed WA, Gaballah ST, El Taweel A, Kamel MN, El Sayes M, Ramadan MA, et al. In Vitro and In Vivo Antiviral Studies of New Heteroannulated 1,2,3-Triazole Glycosides Targeting the Neuraminidase of Influenza A Viruses. Pharmaceuticals. 2022; 15(3):351. https://doi.org/10.3390/ph15030351
Chicago/Turabian StyleKutkat, Omnia, Ahmed Kandeil, Yassmin Moatasim, Yaseen A. M. M. Elshaier, Wael A. El-Sayed, Samir T. Gaballah, Ahmed El Taweel, Mina Nabil Kamel, Mohamed El Sayes, Mohammed A. Ramadan, and et al. 2022. "In Vitro and In Vivo Antiviral Studies of New Heteroannulated 1,2,3-Triazole Glycosides Targeting the Neuraminidase of Influenza A Viruses" Pharmaceuticals 15, no. 3: 351. https://doi.org/10.3390/ph15030351
APA StyleKutkat, O., Kandeil, A., Moatasim, Y., Elshaier, Y. A. M. M., El-Sayed, W. A., Gaballah, S. T., El Taweel, A., Kamel, M. N., El Sayes, M., Ramadan, M. A., El-Shesheny, R., Abdel-Megeid, F. M. E., Webby, R., Kayali, G., & Ali, M. A. (2022). In Vitro and In Vivo Antiviral Studies of New Heteroannulated 1,2,3-Triazole Glycosides Targeting the Neuraminidase of Influenza A Viruses. Pharmaceuticals, 15(3), 351. https://doi.org/10.3390/ph15030351